

Abstract
Amyloid aggregates, found in patients that suffer from Alzheimer's disease, are composed of fibril-forming peptides in a β-sheet conformation. One of the most abundant components in amyloid aggregates is the β-amyloid peptide 1–42 (Aβ 1–42). Membrane alterations may proceed to cell death by either an oxidative stress mechanism, caused by the peptide and synergized by transition metal ions, or through formation of ion channels by peptide interfacial self-aggregation. Here we demonstrate that Langmuir films of Aβ 1–42, either in pure form or mixed with lipids, develop stable monomolecular arrays with a high surface stability. By using micropipette aspiration technique and confocal microscopy we show that Aβ 1–42 induces a strong membrane destabilization in giant unilamellar vesicles composed of palmitoyloleoyl-phosphatidylcholine, sphingomyelin, and cholesterol, lowering the critical tension of vesicle rupture. Additionally, Aβ 1–42 triggers the induction of a sequential leakage of low- and high-molecular-weight markers trapped inside the giant unilamellar vesicles, but preserving the vesicle shape. Consequently, the Aβ 1–42 sequence confers particular molecular properties to the peptide that, in turn, influence supramolecular properties associated to membranes that may result in toxicity, including: 1), an ability of the peptide to strongly associate with the membrane; 2), a reduction of lateral membrane cohesive forces; and 3), a capacity to break the transbilayer gradient and puncture sealed vesicles.
INTRODUCTION
Alzheimer's disease is characterized by the presence of amyloid plaques surrounded by dead and damaged neurons in the brain (Selkoe, 1991; Matsuzaki and Horikiri, 1999). One of the most abundant components of these plaques is the β-amyloid peptide 1–42 (Aβ 1–42). This peptide is the product of the proteolytic cleavage of the Amyloid Precursor Protein (Kang et al., 1987; Selkoe, 1993). In the core of the plaque the amyloid peptides adopt an antiparallel β-sheet conformation (McLaurin et al., 1998). Aβ 1–42 is toxic to neuronal cells in culture when they are exposed to the aggregated form of the peptide (Yankner et al., 1990; Lambert et al., 1998). Although the etiologic role of amyloid peptide (Aβ 1–42) in Alzheimer's disease is accepted (Kang et al., 1987; Selkoe, 1993), the molecular mechanism of neurotoxicity remains unclear and is under debate.
A number of observations indicate that the primary target of amyloid peptides is the cell membrane of neurons. It was previously reported that amyloid peptides alter important physical and biological properties of these membranes (Arispe et al., 1993; Pillot et al., 1996; Choo-Smith et al., 1997) and interact with specific lipids (Terzi et al., 1997; Ji et al., 2002a,b; Curtain et al., 2003; Lau et al., 2003). In addition, several studies reported in the last few years demonstrate that Alzheimer's peptides form ion pores in membrane bilayers (Lin et al., 2001; Kagan et al., 2002; Arispe, 2004; Micelli et al., 2004). Important membrane-peptide interactions, including peptide insertion, cytotoxicity, and pore formation, have been described when cholesterol is present in model membranes and correlated with the presence of transient lipid domains or lipid rafts (Arispe and Doh, 2002; Ji et al., 2002b; Curtain et al., 2003; Lau et al., 2003; Tashima et al., 2004). Sphingomyelin-cholesterol (SM/Chol) lipid bilayers are known to form rafts or liquid-ordered/liquid-disordered domains that are representative of a native cellular membrane (Dietrich et al., 2001). Recently, cholesterol (Chol) was associated with the membrane release of β-amyloid peptide from brain lipid bilayers (Tashima et al., 2004).
In this work, we have analyzed the surface properties and lipid-peptide interactions of Aβ 1–42 using the monolayer technique. By micropipette aspiration we have quantitatively measured a membrane destabilization effect when giant unilamellar vesicles (GUVs) are exposed to Aβ 1–42 peptide. Also, we have developed a new method using confocal microscopy to directly observe the lytic or membrane-disruptive action of peptides. This new approach employs GUVs filled with different molecular size fluorophores and allows us to monitor membrane integrity and vesicle leakage after peptide addition to the freestanding membranes.
The data reported in our study demonstrate that: 1), Aβ 1–42 forms highly stable films at the air-water interface; 2), the behavior of peptide-lipid monolayers depends on the lipid composition; 3), the presence of Aβ 1–42 peptide destabilizes lipid bilayers, as indicated by a reduction of the critical lysis tension of GUVs; and 4), the peptide strongly alters the permeability of bilayers in GUVs, followed by the leakage of the fluorescent contents. The results support the idea that the destructive action of toxic accumulated Aβ 1–42 peptide over neurons may primarily be originated by an irreversible damage at the cell membrane level.
MATERIALS AND METHODS
Chemicals
1,2-di-palmitoyl-sn-glycero-3-phosphocholine (DPPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol (Chol), and bovine brain sphingomyelin (SM) were purchased from Avanti Polar Lipids (Birmingham, AL) and used without further purification. Alexa488-dextran and Alexa546-maleimide were obtained from Molecular Probes (Eugene, OR).
Aβ 1–42 was prepared by continuous flow Fmoc-solid phase peptide synthesis as previously described (Tickler et al., 2001). After global cleavage and deprotection in a mixture of trifluoroacetic acid/H2O/ethanedithiol/triethylsilane (94:2.5:2.5:1 v/v, 10 ml), the crude lyophilized Aβ 1–42 (∼20 mg) was then dissolved in 0.1% aq. trifluoroacetic acid solution (10 ml) and 1 ml aliquots were reacted with hydrogen peroxide (250 μL) for 10 min at room temperature, then immediately purified by RP-HPLC and lyophilized. Analysis by matrix-assisted laser desorption ionization time of flight mass spectrometry MH+ found 4517 (expected 4515), and amino-acid analysis showed good correlation with expected values.
Monolayer studies
Monolayer experiments were performed at room temperature. The subphase was NaCl 145 mM at the specified pH. Pure peptide monolayers were formed by direct spreading from DMSO/chloroform/methanol (50:33:17, v/v) solutions by using a microsyringe. The total surface area of the Teflon trough was 80 cm2 with a 75-ml volume of the subphase. Spreading solvent was allowed to evaporate for at least 5 min before compression was started, at a rate of 43 cm2/min. For lipid-peptide mixed monolayers, peptide and lipid were premixed in the desired proportions from their respective pure solution, and then directly spread on the surface. The surface pressure (Π, Wilhelmy method via platinized-Pt plate), the area enclosing the monolayer, and the surface potential (ΔV, via milliVoltmeter with air-ionizing 241Am plate and calomel-electrode pair) were automatically measured (with the control unit Monofilmmeter with Film Lift, Mayer Feintechnique, Gottingen, Germany). The data were continuously and simultaneously recorded with a double-channel X-YY recorder.
The calculation of the expected cross-sectional molecular area of an α-helix parallel or perpendicular to the air-water interface can be calculated assuming an ideal all-α-helix conformation. The peptide length is estimated considering an average of 0.15 nm of axial rise per residue for an α-helix structure. A diameter of an ideal α-helix with hydrophobic amino acids is estimated in 1.5 nm—0.5 nm for the helix backbone and 1.0 nm for the lateral side chains of the amino acid surrounding the helix backbone (Ambroggio et al., 2004).
The behavior of mixed lipid-peptide monolayers was analyzed by comparing the experimental force-area curve with the theoretical isotherm for the corresponding films in which no interactions between the film-forming molecules are assumed (Gaines, 1966; Fidelio et al., 1981). The immiscible behavior between the film-forming molecules in the mixed monolayer was determined according to the surface phase rule (Gaines, 1966; Fidelio et al., 1981). The collapse pressure is the highest lateral pressure by which a monolayer can be compressed without detectable expulsion of the molecules to form a bulk phase (Gaines, 1966). In practice, the collapse pressure of a compression isotherm is taken at the point of surface pressure where we observe the first discontinuity in the lateral pressure-area isotherm. This was estimated by taking the tangent of each point (derivative) of the curve. Briefly, if one collapse point is observed for a mixed film (in which individual components have differentiated collapse pressures) the surface phase rule determines lateral miscibility in the film. Conversely, if two collapse points are well determined in the isotherm, a lateral inhomogeneity (with some film-forming amphiphile molecules separating out from the rest of film components when they are under compression) is expected (Gaines, 1966).
Vesicle preparation, confocal microscopy, and micropipette aspiration techniques
For confocal microscopy experiments, phospholipid stock solutions were prepared in chloroform at concentrations of 0.2 mg/ml. In our experiments we formed giant unilamellar vesicles (GUVs) with POPC and POPC/SM/Chol (1:1:1 molar ratio).
The electroformation method, developed by Angelova and co-workers (Angelova and Dimitrov, 1986; Angelova et al., 1992) was used to prepare giant vesicles. GUVs were formed in a temperature-controlled chamber as described previously (Bagatolli and Gratton, 1999, 2000). GUVs were prepared using the following steps: 3 μl of lipid stock solution were spread on each of the two sample chamber platinum wires. The chamber was then put into a vacuum overnight to remove any remaining trace of organic solvent. The chamber and a solution of sucrose 150 mM, Alexa488-Dextran 1 μM, and Alexa546-maleimide 10 μM with an overall osmolarity of 150 mOsM (measured with an Advanced Instruments Model 3D3 Osmometer; Advanced Instruments, Norwood, MA) were separately equilibrated to temperatures above the lipid mixture phase transition(s) and then 0.4 ml of sucrose solution containing the fluorophores was added to cover the platinum wires. Immediately after buffer addition, the platinum wires were connected to a function generator (Digimess FG 100; Grundig Instruments, Nurnberg, Germany) and a low-frequency alternating field (sinusoidal wave function with a frequency of 10 Hz and amplitude of 1 V) was applied for 120 min. The AC field was turned off and the GUVs were collected from the chamber. To remove nontrapped fluorescent probes, the vesicles were washed several times with iso-osmolar glucose solution, leaving the sucrose-dye-loaded GUVs (higher density) to precipitate at the bottom of a 15-ml centrifuge tube and then discarding the supernatant.
Aliquots of giant vesicles suspended in glucose (0.3 ml) were added to an eight-well plastic chamber (Lab-Tek Brand Products, Naperville, IL). Due to the density difference between the sugar solutions inside and outside of vesicles, GUVs precipitate at the bottom of the chamber, which facilitates the observation in the inverted confocal microscope. The chamber was located in an inverted confocal microscope (Zeiss LSM 510 META; Carl Zeiss, Jena, Germany) for observation. The excitation wavelengths were 488 nm (for Alexa488-Dextran) and 543 nm (for Alexa546). An iso-osmolar solution of peptide then was injected into the chamber and a time-series scan was started to follow the peptide-vesicle interaction.
All leakage experiments where quantitatively analyzed by using MetaMorph software (Universal Imaging, Molecular Devices, Downingtown, PA) by measuring the change in fluorescence intensity for each probe inside the GUVs after exposure to Aβ 1–42 peptide.
The micropipette technique allows the direct physical manipulation of individual GUVs and the controlled application of mechanical stresses to the lipid bilayer; in this manner, material properties of the bilayer such as the critical lysis tension can be measured (Evans and Needham, 1987; Evans and Rawicz, 1990). Sucrose-filled vesicles were diluted in an iso-osmolar glucose solution and added to a custom-designed micromanipulation chamber (chamber A, volume ∼400 μl) pretreated with bovine serum albumin (BSA, 1 mg/ml) to prevent lipid adhesion to the glass (before the addition of GUVs and peptide solution, the BSA was flushed out of the chamber several times with BSA-free glucose solution). A small aliquot (∼1–2 μl) of vesicle suspension were added to the glucose-filled chamber, and after a few minutes the vesicles precipitated at the bottom of the chamber. The chamber was mounted on the modified stage of a Zeiss Axiovert S100 inverted microscope, and the vesicles were observed with Hoffman Modulation Contrast optics (40× objective magnification, 100× total magnification; W. Nuhsbaum Inc., McHenry, IL). The micropipette was connected to a water-filled manometer with in-line pressure transducers (Validyne Engineering, Los Angeles, CA) that allowed accurate measurements of applied suction pressures at the pipette tip. All experiments were continuously recorded to video and analyzed separately. Initially, as a control, a test vesicle was aspirated using a 5-μm diameter micropipette and the suction pressure was steadily increased until the vesicle lysed. The lysis tension (τlyse) was then calculated using the relation below (Evans and Needham, 1987),
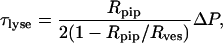 |
(1) |
where ΔP is the suction pressure applied, and Rpip and Rves are the pipette inner radius and vesicle spherical radius, respectively.
After the control, vesicles of the appropriate size and unilamellarity were selected, gently aspirated at a sublytic level of applied tension, and transferred to an adjacent test chamber containing peptide in equiosmotic glucose solution (chamber B). The transfer was effected by inserting the vesicle (and the micropipette supporting it) inside the bore of a large (∼100–200 μm) transfer pipette, filled with glucose solution, which acted as a protective sheath for the vesicle during transfer from one chamber to another. The vesicle was held under a constant suction pressure. If peptides interact and destabilize the membrane by insertion, then a membrane projection into the micropipette may be seen and a new τlyse can be calculated.
The peptide solutions were prepared by dilution of Aβ 1–42 from a concentrated DMSO solution to a glucose solution. The final osmolarity of the sample was measured and adjusted to the same values as for the vesicle and chamber solutions, to prevent unwanted water efflux or influx through the bilayer from occurring during the experiment.
The experiments were carried out at room temperature. At this temperature GUVs composed of POPC/SM/Chol (1:1:1 molar ratio) display liquid ordered-liquid disordered phase coexistence (not shown) as previously reported for DOPC/SM/Chol (1:1:1 molar ratio) mixtures (Dietrich et al., 2001).
RESULTS
Pure peptide monolayers
Pure Aβ 1–42 monolayers show a collapse pressure (stability of the film against lateral compression) of ∼33 mN/m, a surface potential at maximal packing (at collapse pressure) of ∼260 mV and a molecular area of 2.30 nm2/molecule, deduced from the compression isotherms of the film (Fig. 1).
FIGURE 1.

Surface behavior of pure Aβ 1–42 peptide monolayers. Lateral pressure area (solid line) and surface potential area (dashed line) isotherms of Aβ 1–42. The subphase was 145 mM NaCl, pH 6.
Lipid-peptide mixed monolayers
The compression isotherms of Aβ 1–42 mixed with DPPC at different peptide-lipid area ratios are shown in Fig. 2. A surface molecular area analysis is necessary because the area of Aβ 1–42 is more than three times higher than the corresponding molecular area of the lipids, and equimolar proportions of both components will generate films in which 80% of the surface area is covered by the peptide. Two collapse pressures can be seen in the compression isotherms of Aβ 1–42-DPPC mixed monolayer (Fig. 2). The lower collapse pressure (from 30 to 40 mN/m) is indicative of the collapse of the peptide-enriched phase. On the other hand, the higher collapse pressure (∼60 mN/m) indicates the collapse of the lipid-enriched phase (Gaines, 1966; Ambroggio et al., 2004). When the proportion of the peptide was lowered in the mixed interface, the lower collapse pressure went up, indicating a partial miscibility of the peptide with the lipid.
FIGURE 2.

Surface behavior in mixed DPPC-Aβ 1–42 peptide interfaces. Lateral pressure area (solid line) and surface potential area (dashed line) isotherms. The corresponding peptide area as a proportion of the mixed monolayer was 25% (1), 50% (2), and 75% (3). Subphase 145 mM NaCl, pH 6. Arrows indicate the collapse pressure of the peptide-enriched phase. Note the effect of the peptide on the liquid expanded-liquid condensed phase transition of the DPPC-enriched phase (∼10 mN/m).
DPPC has a liquid-expanded to liquid-condensed phase-transition at ∼10 mN/m, as is observed in Fig. 2 (Gaines, 1966). This transition is still present when the lipid was mixed with Aβ 1–42 but it becomes more distorted as the peptide proportion is increased (Fig. 2). At low proportion of peptide, slightly positive deviations from the ideal mixing behavior were observed (<10%; not shown).
A similar behavior was seen in POPC-Aβ 1–42 mixed interfaces. Pure POPC monolayers are liquid-expanded. A partial miscibility of the peptide was deduced and no obvious deviations from ideal behavior were observed (not shown). Also, a partial miscibility is evidenced, since the first collapsing point is increased as the peptide proportion is lowered in the mixed interface.
Compression isotherms for Aβ 1–42 mixed with POPC/Chol (7:3 molar ratio), SM, SM/Chol (7:3 molar ratio), and POPC/SM/Chol (1:1:1 molar ratio) were analyzed. The collapse pressure of the peptide-enriched phase depends on the lipid composition (Table 1) and was in the range of between 32 and 43 mN/m when it was analyzed at the same lipid/peptide area proportion. Slight deviations from ideal behavior were observed at the lipid/peptide proportions studied.
TABLE 1.
Collapse pressure of the Aβ 1–42 enriched phase in lipid-peptide monolayers
| Lipid (molar ratio) | Collapse pressure (mN/m)* |
|---|---|
| DPPC | 34 |
| POPC | 35 |
| POPC/Chol (7:3) | 33 |
| SM | 43 |
| SM/Chol (7:3) | 39 |
| SM/POPC/Chol (1:1:1) | 32 |
Standard deviation: ±1 mN/m.
Micropipette manipulation
Videomicrographs of vesicles composed of POPC and POPC/SM/Chol (1:1:1 molar ratio) are shown in Fig. 3 (top and bottom, respectively). Vesicle diameters were in the range of 25–35 μm. A suction pressure was applied to the vesicles with a micropipette and the critical tension of lysis (τlyse) was measured for vesicles exposed to an Aβ 1–42 peptide solution. These critical tension values were compared with those obtained for GUVs without peptide exposition (Fig. 3).
FIGURE 3.

Videomicrographs of GUVs tested by micromanipulation after exposure to Aβ 1–42 peptide. Giant unilamellar vesicle composed of POPC (top) and composed of POPC/SM/Chol (1:1:1 molar ratio, bottom) held under constant, low membrane tension exposed to 5 μM Aβ 1–42 peptide. (A) Before and (B) after peptide exposure. τlyse is the critical tension to lyse the vesicle (mN/m) and is shown in the figure to denote the change in the membrane physical properties under the different conditions. It is the average tension when the vesicle breaks as the suction pressure was increased. Videomicrographs were taken optimizing the brightness and contrast setup to set the best condition for vesicle vision. The size bar at bottom-right of each panel indicates the scale of the pictures (5 μm). Inserted bars indicate the diameter of GUVs and the projection toward the inner of the suction pipette after peptide interaction.
Pure POPC vesicles had a τlyse of ∼8.2 ± 0.1 mN/m (Fig. 3 A, top); after peptide exposure this pressure decreased drastically to 0.3 mN/m (Fig. 3 B, top). A significant feature was the loss of the optical contrast between the sucrose solution inside of the vesicle and the glucose solution outside of the vesicle; this change in appearance indicates that the bilayer becomes leaky, and the mixing of the two sugar solutions occurs.
Vesicles composed of POPC/SM/Chol (1:1:1 molar ratio) had a higher τlyse than POPC vesicles (Fig. 3 A, bottom). The average τlyse was 12 ± 1 mN/m. The higher value relative to pure POPC bilayers is consistent with the known effect of cholesterol on the mechanical properties of fluid phase bilayer vesicles (Needham and Nunn, 1990). When the POPC/SM/Chol vesicles were exposed to 5 μM solution of Aβ 1–42, the critical tension of lysis was lower, falling to 4 mN/m, and a membrane projection into the micropipette is observed (Fig. 3 B, bottom). A loss of the sucrose-glucose phase contrast was also visually observed (as it was previously indicated for pure POPC system; not quite well appreciated from Fig. 3).
For both POPC and POPC/SM/Chol, there was a lag time in the peptide-vesicle interaction of ∼10 s after vesicle exposure to Aβ 1–42. This time is the time elapsed before any perceptible change in tension can be measured after peptide exposure.
Confocal microscopy
The incorporation of both fluorophores Alexa488-Dextran (MW 10,000) and Alexa546-maleimide (MW ∼1300) inside of GUVs is useful to directly observe any effect on membrane permeability in a continuous manner (leakage of probes from the vesicles).
Fig. 4 (top) shows a time-series after addition of amyloid peptide into the chamber containing POPC GUVs. Initially we observed some lysis of vesicles, although a high population of POPC GUVs remained filled with the probes. After 50 s the loss of the small Alexa546-maleimide fluorophore (red) was evident from the images. This last event occurs without significant leakage of the high molecular weight probe (Alexa488-dextran fluorophore, green). A similar effect was observed when GUVs composed of POPC/SM/Chol (1:1:1 molar ratio) loaded with both fluorophores were exposed to Aβ 1–42 (Fig. 4 bottom). It is noteworthy that the GUVs containing cholesterol display some budding process in the absence of peptide in agreement with that reported by Baumgart and co-workers for DOPC/SM/Chol mixtures (Baumgart et al., 2003).
FIGURE 4.

Direct visualization of lytic action of Aβ 1–42 peptide. Confocal microscopy of POPC (top) and POPC/SM/Chol (1:1:1 molar ratio, bottom) GUVs, filled with Alexa488-Dextran (green) and Alexa546-Maleimide (red), exposed to Aβ 1–42 7.5 μM. Effect of lipid-peptide interaction from the time, 0 s: injection of the peptide. Arrows indicate the differential leakage of the fluorophores.
The different permeation rates of the two fluorophores are shown in Fig. 5 (A and B). The small fluorophore is firstly released from inside of the vesicles before the loss of the high molecular weight fluorophore. Fig. 5 also shows no significant changes in fluorescence intensity in absence of the peptide (Fig. 5 A, inset). This last observation supports the fact that the probes do not undergo significant photobleaching under our experimental conditions. Therefore, we corroborate that the observed decrease in fluorescence intensity is due to the leakage of the probe(s).
FIGURE 5.

Dye release kinetics from inside GUVs after Aβ 1–42 peptide exposure. Alexa488-Dextran (squares) and Alexa546-Maleimide (circles) leakage from inside POPC (A) and POPC/SM/Chol (1:1:1 molar ratio) (B) GUVs after exposure to Aβ 1–42 7.5 μM. The fluorescence intensity was measured with MetaMorph software by quantifying the average gray value of Alexa488-Dextran and Alexa546-Maleimide of each picture inside the GUVs after exposition to Aβ 1–42 peptide. (A, inset) Alexa488-Dextran (squares) and Alexa546-Maleimide (circles) fluorescence intensity in absence of peptide (see text). Initial rates of leakage (slope of the initial linear behavior of dye leakage after peptide exposure): (A) Alexa488-Dextran: 0.72 AU/s, Alexa546-Maleimide: 1.45 AU/s; and (B) Alexa488-Dextran: 0.52 AU/s, Alexa546-Maleimide: 0.96 AU/s.
These results are also supported by the loss of the sucrose-glucose phase contrast observed in the micropipette aspiration experiments (see above) and denote a considerable modification in the permeability properties of a GUV's membrane.
DISCUSSION
In this report we have shown that amyloid Aβ 1–42 peptide forms insoluble monolayers with high stability against lateral compression, where the collapse pressure for pure peptide films was ∼33 mN/m (Fig. 1). This high stability is in agreement with a peptide monolayer at the air-water interface where the peptide molecules adopts mainly a β-sheet structure (Fidelio et al., 1986a; Maget-Dana et al., 1999; Maget-Dana, 1999). Schladitz et al. (1999) clearly showed that the main secondary structure of Aβ 1–40 peptide (two amino acids <Aβ 1–42 at the carboxyl-terminus) is an antiparallel β-sheet parallel to the air-water interface and part of the sequence is an α-helix perpendicular to the interface in a pure peptide monolayer set at 13 mN/m of surface lateral pressure. We find that Aβ 1–42 peptide has a molecular area of ∼2.3 nm2/molecule (at maximal packing, 33 mN/m). This cross-sectional molecular area observed for the peptide is greater than that theoretically expected for the entire sequence of the peptide adopting an α-helix conformation perpendicular to the interface (∼1.8 nm2, see Materials and Methods) and, for this reason, it can be reasonably speculated that a mixture of conformation and orientation for the peptide is possible (Fidelio et al., 1981, 1986b; Ambroggio et al., 2004). Furthermore, the high collapse pressure and the low surface potential observed for the amyloid peptide monolayer are in keeping with peptides adopting β-sheet structure at the interface (Fidelio et al., 1986a; Maget-Dana at al., 1999; Ambroggio et al., 2004). These data also support that Aβ 1–42 instead adopts a more complex conformation (mixture of secondary structures at the air-water interface) at the interface.
The results from lipid-peptide mixed films indicated that Aβ 1–42 peptide has partial miscibility behavior with the lipids employed. The lateral stability of the mixed interface before the peptide-enriched phase collapses was in the range described previously for biological membranes (Blume, 1979; Seelig, 1987). This behavior suggests that the peptide interacts with the membrane, becoming part of it. This means that the peptide remains in the lipid environment at lateral pressures compatible with natural membranes. The fact that no significant deviations from the ideal mixing behavior are observed in the peptide-lipid monolayer data, led us to consider that the presence of the peptide at the membrane may promote local damage to the lipid interface, altering such supramolecular physical properties as the cohesive forces and membrane permeability.
Another important contribution of this work is the direct analysis of the stability of model membranes, represented by giant unilamellar vesicles, which have been exposed to Aβ 1–42 peptide solution. In the micropipette manipulation experiments (Fig. 3) the effects of peptide exposure were clearly demonstrated. The results showed that Aβ 1–42 peptide disrupts the membrane of GUVs composed of POPC or POPC/SM/Chol. These data demonstrate membrane destabilization resulting from the lipid-peptide interaction, where the critical lysis tension of the vesicles was reduced (for POPC GUVs, τlyse goes from 8.2 to 0.3 mN/m and for POPC/SM/Chol GUVs, τlyse goes from 12 to 4 mN/m), indicating the loss of cohesion between the membrane components—a feature that has not been quantitatively described for Aβ 1–42 previously. Notably this reduction in the critical lysis tension is seen for both POPC bilayers and those with SM/Chol, which better resemble a natural membrane. As mentioned before, when the vesicles were exposed to Aβ 1–42 peptide there was a lag time for action, and the interaction takes place with a loss in the sucrose-glucose phase contrast and a reduction of the cohesive forces. The loss in the phase contrast can be explained by an alteration in the permeability of the membrane due to the protein-lipid interaction. The permeation of the sucrose from inside the vesicles (or the penetration of glucose into the GUVs) could be a result of pore-like structures formed in the membrane. These results are analogous to those reported by Longo et al. (1998) for melittin, a lytic pore-forming peptide, which significantly weakens PC vesicles to yield τlyse with similar values to those obtained in our experiments.
The micropipette results discussed above are in agreement with the data obtained from confocal microscopy. GUVs loaded with two fluorescent probes differing in their molecular sizes have given us the possibility of a direct visualization of the differential leakage when the permeability of the membrane is altered as a consequence of amyloid peptide interaction. Several vesicles were analyzed and, as observed in the micropipette aspiration experiments, the stability of POPC and POPC/SM/Chol GUVs was greatly altered when they were exposed to Aβ 1–42 peptide. Our results showed the lysis of fluorescent-labeled vesicles and differential leakage of the probes when the peptide interacts with the membrane (as well as the loss of the sucrose-glucose phase contrast). The fast loss of the small fluorescence probe and the progressive loss of the larger one from inside of the vesicles without a membrane solubilization effect due to peptide-membrane interaction led us to infer that the amyloid peptide may induce singular lipid-peptide organization allowing, in turn, a differential leakage property at the membrane. The effect of amyloid peptide was seen in vesicles of different composition, although some differences were seen between POPC liposomes compared with those formed by SM/Chol, with somewhat slower rate of dye release for the latter (see legend of Fig. 5). Our results suggest that Aβ 1–42 peptide induces release of fluorescent dye of characteristic size, in agreement with the leakage behavior reported by Pillot and co-workers where small unilamellar vesicles loaded with Calcein were exposed to fragments of Aβ 1–42 peptide (Pillot et al., 1996). Furthermore, using atomic force microscopy, Lin and co-workers have shown that Aβ 1–42 forms hexameric structures in model membranes (Lin et al., 1999, 2001), which would be in accordance with our data.
In summary, our results indicate that amyloid peptide clearly disrupts membrane bilayers. Aβ 1–42 peptide has high surface activity similar to that reported for naturally occurring phospholipids with a liquid-expanded characteristic. The stability and miscibility of the peptide monolayer depends on the type of lipids at the interface. The peptide directly interacts with the lipids, associating with the membrane and altering its mechanical properties by changing the cohesive properties among membrane components and the permeability of the bilayer. Mixed lipid-peptide monolayers confirm the ability of the amyloid peptide to support lateral pressures found in natural membranes. Clearly the peptide-lipid interaction induces changes in membrane permeability and this fact could be a consequence of Aβ 1–42 peptide formation of leaky peptide-lipid supramolecular structures at the membrane. It is noteworthy to emphasize that the Aβ 1–42 amyloid peptide has some particular characteristics in common with other amphipathic peptides: an ability to remain at the interface (high lateral stability), with a marked transbilayer instability effect (measured by its capacity to provoke leakage). A similar behavior was observed for antibiotic peptides isolated from Australian tree frog (Ambroggio et al., 2004). An additional important detail is that the effect was observed with Aβ 1–42 in its soluble form, which may have physiological implications in damaging neuronal membranes and Alzheimer's disease.
Acknowledgments
This work was supported by grants from CONICET, FONCYT, SECYT-UNC, Agencia Córdoba Ciencia, and Ministerio de Salud de la Nacion (Carrillo Oñativia fellowship). G.D.F. is a member of the Career Investigators from CONICET. E.E.A. is a Fellow from CONICET. Research in the laboratory of L.A.B. is funded by a grant from SNF, Denmark (No. 21-03-0569), and the Danish National Research Foundation (which supports the MEMPHYS-Center for Biomembrane Physics).
References
- Ambroggio, E. E., F. Separovic, J. Bowie, and G. D. Fidelio. 2004. Surface behaviour and peptide-lipid interactions of the antibiotic peptides, Maculatin and Citropin. Biochim. Biophys. Acta. 1664:31–37. [DOI] [PubMed] [Google Scholar]
- Angelova, M. I., and D. S. Dimitrov. 1986. Liposome electroformation. Faraday Discuss. Chem. Soc. 81:303–311. [Google Scholar]
- Angelova, M. I., S. Soleau, P. Meleard, J. F. Faucon, and P. Bothorel. 1992. Preparation of giant vesicles by external fields. Kinetics and application. Prog. Colloid Polym. Sci. 89:127–131. [Google Scholar]
- Arispe, N., H. B. Pollard, and E. Rojas. 1993. Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA. 90:10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe, N., and M. Doh. 2002. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer's disease AβP (1–40) and (1–42) peptides. FASEB J. 16:1526–1536. [DOI] [PubMed] [Google Scholar]
- Arispe, N. 2004. Architecture of the Alzheimer's A βP ion channel pore. J. Membr. Biol. 197:33–48. [DOI] [PubMed] [Google Scholar]
- Bagatolli, L. A., and E. Gratton. 1999. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophys. J. 77:2090–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli, L. A., and E. Gratton. 2000. A correlation between lipid domain shape and binary phospholipid mixture composition in free standing bilayers: a two-photon fluorescence microscopy study. Biophys. J. 79:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart, T., S. T. Hess, and W. W. Webb. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425:821–824. [DOI] [PubMed] [Google Scholar]
- Blume, A. 1979. A comparative study of the phase transitions of phospholipid bilayers and monolayers. Biochim. Biophys. Acta. 557:32–44. [DOI] [PubMed] [Google Scholar]
- Choo-Smith, L. P., W. Garzon-Rodriguez, C. G. Glabe, and W. K. Surewicz. 1997. Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272:22987–22990. [DOI] [PubMed] [Google Scholar]
- Curtain, C. C., F. Ali, D. G. Smith, A. I. Bush, C. L. Masters, and K. J. Barnham. 2003. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-β peptide with membrane lipid. J. Biol. Chem. 278:2977–2982. [DOI] [PubMed] [Google Scholar]
- Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., and D. Needham. 1987. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion and colloidal interactions. J. Phys. Chem. 91:4219–4228. [Google Scholar]
- Evans, E., and W. Rawicz. 1990. Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys. Rev. Lett. 64:2094–2097. [DOI] [PubMed] [Google Scholar]
- Fidelio, G. D., B. Maggio, F. A. Cumar, and R. Caputto. 1981. Interaction of glycosphingolipids with melittin and myelin basis protein in monolayers. Biochem. J. 193:643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidelio, G. D., B. M. Austen, D. Chapman, and J. A. Lucy. 1986a. Properties of signal-sequence peptides at an air-water interface. Biochem. J. 238:301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidelio, G. D., B. Maggio, and F. A. Cumar. 1986b. Interaction of melittin with glycosphingolipids and phospholipids in mixed monolayers at different temperatures. Effect of the lipid physical state. Biochim. Biophys. Acta. 862:49–56. [DOI] [PubMed] [Google Scholar]
- Gaines, G. L. 1966. Insoluble Monolayers at Liquid-Gas Interfaces. Interscience, New York.
- Ji, S. R., Y. Wu, and S. F. Sui. 2002a. Study of β-amyloid peptide (Aβ40) insertion into phospholipid membranes using monolayer technique. Biochemistry (Mosc.). 67:1283–1288. [DOI] [PubMed] [Google Scholar]
- Ji, S. R., Y. Wu, and S. F. Sui. 2002b. Cholesterol is an important factor affecting the membrane insertion of β-amyloid peptide (Aβ1–40), which may potentially inhibit the fibril formation. J. Biol. Chem. 277:6273–6279. [DOI] [PubMed] [Google Scholar]
- Kagan, B. L., Y. Hirakura, R. Azimov, R. Azimova, and L. Meng-Chin. 2002. The channel hypothesis of Alzheimer's disease: current status. Peptides. 23:1311–1315. [DOI] [PubMed] [Google Scholar]
- Kang, J., H. G. Lemaire, A. Unterbeck, J. M. Salbaum, C. L. Masters, K. H. Grzeschik, G. Multhaup, K. Beyreuther, and B. Muller-Hill. 1987. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 325:733–736. [DOI] [PubMed] [Google Scholar]
- Lambert, M. P., A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, et al. 1998. Diffusible, nonfibrillar ligands derived from A β1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, T. L., K. J. Barnham, C. C. Curtain, C. L. Masters, and F. Separovic. 2003. Magnetic resonance studies of the β-amyloid peptide. Aust. J. Chem. 56:349–356. [Google Scholar]
- Lin, H., Y. J. Zhu, and R. Lal. 1999. Amyloid β-protein (1–40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry. 38:11189–11196. [DOI] [PubMed] [Google Scholar]
- Lin, H., R. Bhatia, and R. Lal. 2001. Amyloid beta protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15:2433–2444. [DOI] [PubMed] [Google Scholar]
- Longo, M. L., A. J. Waring, L. M. Gordon, and D. A. Hammer. 1998. Area expansion and permeation of phospholipid membrane bilayers by influenza fusion peptides and melittin. Langmuir. 14:2385–2395. [Google Scholar]
- Maget-Dana, R., D. Lelièvre, and A. Brack. 1999. Surface active properties of amphiphilic sequential isopeptides: comparison between α-helical and β-sheet conformations. Biopolymers. 49:415–423. [DOI] [PubMed] [Google Scholar]
- Maget-Dana, R. 1999. The monolayer technique: a potent tool for studying the interfacial properties of antimicrobial and membrane-lytic peptides and their interactions with lipid membranes. Biochim. Biophys. Acta. 1462:109–140. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., and C. Horikiri. 1999. Interactions of amyloid β-peptide (1–40) with ganglioside-containing membranes. Biochemistry. 38:4137–4142. [DOI] [PubMed] [Google Scholar]
- McLaurin, J., T. Franklin, A. Chakrabartty, and P. E. Fraser. 1998. Phosphatidylinositol and inositol involvement in Alzheimer amyloid-β fibril growth and arrest. J. Mol. Biol. 278:183–194. [DOI] [PubMed] [Google Scholar]
- Micelli, S., D. Meleleo, V. Picciarelli, and E. Gallucci. 2004. Effect of sterols on β-amyloid peptide (AβP 1–40) channel formation and their properties in planar lipid membranes. Biophys. J. 86:2231–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham, D., and R. S. Nunn. 1990. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 58:997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillot, T., M. Goethals, B. Vanloo, C. Talussot, R. Brasseur, J. Vandekerckhove, M. Rosseneu, and L. Lins. 1996. Fusogenic properties of the C-terminal domain of the Alzheimer β-amyloid peptide. J. Biol. Chem. 271:28757–28765. [DOI] [PubMed] [Google Scholar]
- Schladitz, C., E. P. Vieira, H. Hermel, and H. Mohwald. 1999. Amyloid-β-sheet formation at the air-water interface. Biophys. J. 77:3305–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig, A. 1987. Local anesthetics and pressure: a comparison of dibucaine binding to lipid monolayers and bilayers. Biochim. Biophys. Acta. 899:196–204. [DOI] [PubMed] [Google Scholar]
- Selkoe, D. J. 1991. The molecular pathology of Alzheimer's disease. Neuron. 4:487–498. [DOI] [PubMed] [Google Scholar]
- Selkoe, D. J. 1993. Physiological production of the β-amyloid protein and the mechanism of Alzheimer's disease. Trends Neurosci. 16:403–409. [DOI] [PubMed] [Google Scholar]
- Tashima, Y., R. Oe, S. Lee, G. Sugihara, E. J. Chambers, M. Takahashi, and T. Yamada. 2004. The effect of cholesterol and monosialoganglioside (GM1) on the release and aggregation of amyloid β-peptide from liposomes prepared from brain membrane-like lipids. J. Biol. Chem. 279:17587–17595. [DOI] [PubMed] [Google Scholar]
- Terzi, E., G. Holzemann, and J. Seelig. 1997. Interaction of Alzheimer β-amyloid peptide (1–40) with lipid membranes. Biochemistry. 36:14845–14852. [DOI] [PubMed] [Google Scholar]
- Tickler, A. K., C. J. Barrow, and J. D. Wade. 2001. Improved preparation of amyloid-β peptides using DBU as Nα-Fmoc deprotection reagent. J. Pept. Sci. 7:488–494. [DOI] [PubMed] [Google Scholar]
- Yankner, B. A., L. K. Duffy, and D. A. Kirschner. 1990. Neurotrophic and neurotoxic effects of amyloid β-protein: reversal by tachykinin neuropeptides. Science. 250:279–282. [DOI] [PubMed] [Google Scholar]