

Abstract
The structural properties of the linker peptide connecting the cellulose-binding module to the catalytic module in bimodular cellulases have been investigated by small-angle x-ray scattering. Since the linker and the cellulose-binding module are relatively small and cannot be readily detected separately, the conformation of the linker was studied by means of an artificial fusion protein, Cel6BA, in which an 88-residue linker connects the large catalytic modules of the cellulases Cel6A and Cel6B from Humicola insolens. Our data showed that Cel6BA is very elongated with a maximum dimension of 178 Å, but could not be described by a single conformation. Modeling of a series of Cel6BA conformers with interdomain separations ranging between 10 Å and 130 Å showed that good Guinier and P(r) profile fits were obtained by a weighted average of the scattering curves of all the models where the linker follows a nonrandom distribution, with a preference for the more compact conformers. These structural properties are likely to be essential for the function of the linker as a molecular spring between the two functional modules. Small-angle x-ray scattering therefore provides a unique tool to quantitatively analyze the conformational disorder typical of proteins described as natively unfolded.
INTRODUCTION
Cellulases, the enzymes that degrade cellulose, are central to the biological recycling of photosynthetically fixed carbon in the biosphere. Because of the recalcitrant nature of their substrate (cellulose is an insoluble crystalline polysaccharide), efficient cellulases have evolved a modular organization consisting of a large catalytic module linked to a smaller cellulose binding module (CBM) (Carrard et al., 2000; Henrissat, 1994). These two modules are usually separated by a long and flexible linker peptide, which is often O-glycosylated in the case of fungal cellulases (Gilkes et al., 1991). Although the structural and functional properties of the individual globular modules are well documented, very little is known about the role of the linker peptide, and its structural properties are a matter of speculation. Previous studies, however, indicate a crucial importance for the linker on the activity of the cellulases, as the shortening or the deletion of the linker dramatically reduces enzymatic activity on crystalline cellulose (Shen et al., 1991; Srisodsuk et al., 1993).
The structural properties of the linker peptide of cellulases require characterization to better understand the mode of action of these enzymes. At present, only limited structural information is available, and most of it is generally inferred from negative results. For example, the few successful crystallographic structures of entire two-module glycoside hydrolases—obtained for enzymes with short linkers—lack electron density for the linker residues, indicating disorder for the residues of the linker (Fujimoto et al., 2000; Pell et al., 2004). Significantly, no crystal structure is available for bimodular glycoside hydrolases with long linkers. However, we have shown recently that small-angle x-ray scattering is a valuable tool to analyze the overall conformation of such cellulases (Receveur et al., 2002). Using this method, we showed that the cellulase linkers are flexible and extended, and we have accordingly proposed a model where cellulases can bind and move on crystalline cellulose with a caterpillar-like motion, thus enhancing their catalytic efficiency (Receveur et al., 2002). However, the degree of flexibility of the linker could not be determined directly, nor was it possible to prove definitely that the linker could adopt a conformation that would enable this motion to take place. This was mainly due to the inability to distinguish between contributions from the linker and from the CBM to the scattering curve, since both contained approximately the same number of amino acids, and both were relatively small compared to the catalytic module.
The saprophytic fungus Humicola insolens produces two cellulases belonging to glycoside hydrolase family 6 (Henrissat and Bairoch, 1993, 1996), namely cellobiohydrolase Cel6A and endoglucanase Cel6B. These two cellulases are bimodular, with a catalytic module and a CBM connected by a glycosylated linker peptide. In Cel6A, the N-terminal CBM (44 residues) is separated from the C-terminal catalytic module (360 residues) by a 52-residue linker (Fig. 1 a). In contrast, the 348-residue catalytic module of Cel6B is N-terminal, followed by a 36-residue linker and a C-terminal CBM (36 residues) (Schulein, 1997). The three-dimensional structure of the catalytic modules of both H. insolens Cel6A (Varrot et al., 1999) and Cel6B (Davies et al., 2000) have been solved. To overcome the insensitivity of the scattering experiment to the small sizes of the linker and of the CBM, we have designed and produced a chimeric double cellulase containing two globular catalytic modules of similar sizes connected by a very long linker. This chimera made use of the different two-module orientations in the H. insolens Cel6A and Cel6B cellulases. The truncation of the CBM from each of the full-length cellulases enabled the remaining catalytic modules in Cel6A and Cel6B to be rejoined by their respective linker peptides (Fig. 1). The resulting chimeric cellulase (called Cel6BA), which has the N-terminal Cel6B and C-terminal Cel6A catalytic modules connected by an 88-residue-long linker, was successfully expressed in Aspergillus orzyae as a soluble and active protein. Here we characterize this chimeric double cellulase by a joint application of small-angle x-ray scattering and molecular modeling. We conclude from this work that the cellulase linker is indeed flexible and disordered, and adopts both compact and extended conformations. We analyze the distribution of these conformations in solution by molecular modeling and show that x-ray scattering is a useful tool for the identification and quantification of disorder in appropriate proteins.
FIGURE 1.

(a) Schematic cartoon (not to scale) of the modular organization of the Cel6B and Cel6A cellulases and of the chimeric variant Cel6BA from H. insolens. (b) Prediction of a long-disorder region (thick black line) in Cel6BA by PONDR, and sequence of the 88-residue linker of Cel6BA predicted as disordered.
MATERIALS AND METHODS
Purification of the chimeric cellulase and the isolated catalytic modules
The chimeric cellulase, Cel6BA, was generated by the overlap extension PCR (OE-PCR) method (Higuchi et al., 1988; Ho et al., 1989) using the entire coding regions for the H. insolens Cel6A and Cel6B genes as DNA templates to amplify individual gene fragments. To construct Cel6BA, an upstream OE-PCR fragment encoding the secretion signal peptide and the catalytic module and linker regions was amplified from the N-terminal portion of the H. insolens Cel6B gene using the oligonucleotide primers, 5′-CGACAACATCACATCAAGCTCTCC* and 5′-TCACCTGGCTGCCAGGGTTACCGCCTCCAGGG. An adjoining downstream OE-PCR fragment encoding only the linker and catalytic module regions was amplified from the C-terminal portion of the H. insolens Cel6A gene using the oligonucleotide primers, 5′-CCCTGGAGGCGGTAACCCTGGCAGCCAGGTGA and 5′-CCCCATCCTTTAACTATAGCGAAATGG*. The upstream and downstream OE-PCR fragments were then reassembled as a full-length chimeric Cel6B-Cel6A gene encoding the Cel6BA protein by an additional PCR step using a pair of flanking 5′- and 3′-end oligonucleotide primers (see asterisk symbols above). The final PCR fragment was cleaved with BamHI and XbaI restriction endonucleases and then cloned into an Escherichia coli-Aspergillus shuttle expression vector that utilizes a fungal α-amylase promoter and glucoamylase terminator (Christensen et al., 1988) for the transcriptional control of the inserted chimeric Cel6B-Cel6A gene. The chimeric Cel6B-Cel6A gene construct was verified by DNA sequencing. For the expression of the Cel6BA protein, the resulting plasmid was co-transformed with an acetamidase selection plasmid (pTOC202) into A. oryzae JaL228 as described previously (Kelly and Hynes, 1985). Established procedures were employed in all DNA manipulations (Sambrook et al., 1989). After fermenter cultivation, the culture supernatant was filtered through three layers of Whatman GF filters (2.7, 1.6, and 1.2 μm, respectively; Whatman, Albertslund, Denmark), and concentrated by ultrafiltration (Filtron equipped with a 10 kDa cutoff filter; Pall Filtron, Northborough, MA). The concentrated culture supernatant was adjusted to pH 8.5 using 1 M Tris-HCl, pH 8.5 and subsequently applied onto a Q-Sepharose FF column (2.6 × 18 cm; Pharmacia, Uppsala, Sweden), pre-equilibrated in 20 mM Tris-HCl, pH 8.5 at 4°C using at flow rate of 300 ml h−1. Cel6BA was detected in the column flowthrough, which was concentrated using an Amicon filtration unit (10-kDa cutoff filter; Amicon, Millipore, Bedford, MA). Cel6BA was subsequently applied onto a Sephacryl S-200 HR column (1.6 × 90 cm; Pharmacia), pre-equilibrated in 20 mM Tris-HCl, 0.2 M NaCl, pH 8.5 at 4°C and eluted at a flow rate of 30 ml h−1. Fractions containing homogenous Cel6BA, as estimated using SDS-PAGE, were pooled and kept at −18°C. The finally purified Cel6BA showed a molecular weight of ∼95 kDa as estimated by SDS-PAGE. The single catalytic modules Cel6A and Cel6B from H. insolens were cloned and expressed in A. oryzae (Rasmussen et al., 1991) and purified as previously described (Fort et al., 2000; Varrot et al., 1999).
Sample preparation and x-ray scattering experiments
The lyophilized proteins were diluted in 50 mM sodium phosphate buffer, pH 8.5 for the chimeric cellulase and in 50 mM sodium phosphate buffer, pH 7.5 for the isolated catalytic modules. Glycerol (10%, v/v) was added to the buffer as a radiation scavenger for x-ray scattering experiments. The proteins were then washed extensively through a microconcentrator equipped with a Filtron polyvinylidene membrane (Pall Filtron) to remove contaminating salts. The filtrate was used as the buffer for the x-ray scattering experiments. The protein concentration of each sample was determined by its absorbance at 280 nm using extinction coefficients calculated from the sequence (Schülein et al., 1993).
Data collection was performed at the European Synchrotron Radiation Facility (Grenoble, France) on beamline ID02. The wavelength λ was 1.0 Å. The sample-to-detector distance was set to 3.0 m for the single catalytic modules, and to 2.0 m for the Cel6BA chimera, and these values resulted in scattering vectors q ranging from 0.01 Å−1 to 0.22 Å−1 and from 0.013 Å−1 to 0.34 Å−1, respectively. The scattering vector is defined as q = 4π/λ sin θ, where 2θ is the scattering angle. The detector was a Thomson x-ray image (Thomson Scientific Instruments, Carlton, Australia) intensified optically, coupled to a European Synchrotron Radiation Facility-developed fast-read, low-noise CCD camera. During acquisitions, 40 successive frames of 0.5 s with 4-s intervals (the dead time) between each frame were recorded for each sample. During the dead time, fresh protein solution was injected into the beam in a 1.5-mm Lindemann-type quartz capillary using a remote-controlled syringe coupled with the data acquisition program. Using this, no protein solution was irradiated longer than 0.5 s. Background scattering was measured after each protein sample-run using the buffer solution. The temperature was set at 20°C. Each protein or buffer frame was inspected to confirm the absence of possible bubble formation or radiation-induced aggregation effects on the scattering pattern. This allowed the individual frames to be averaged. The averaged dataset for the buffer recorded from the immediately subsequent data collection was subtracted from the averaged protein dataset after proper normalization and correction for the detector response. The absolute calibration of scattered intensities was made with a lupolen sample used as a standard on the ID02 beamline.
The radius of gyration Rg was derived from the scattering curve using the Guinier approximation 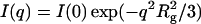 (Guinier and Fournet, 1955), where I(q) is the scattered intensity and I(0) is the forward scattering intensity. In dilute solution, I(0)/c is proportional to the molecular mass M of the scattering object, where c is its concentration, and to the excess scattering of the object relative to the buffer. The distance distribution function P(r) was calculated by the Fourier inversion of the scattering intensity I(q) using GNOM (Svergun, 1992) and GIFT (Bergmann et al., 2000).
(Guinier and Fournet, 1955), where I(q) is the scattered intensity and I(0) is the forward scattering intensity. In dilute solution, I(0)/c is proportional to the molecular mass M of the scattering object, where c is its concentration, and to the excess scattering of the object relative to the buffer. The distance distribution function P(r) was calculated by the Fourier inversion of the scattering intensity I(q) using GNOM (Svergun, 1992) and GIFT (Bergmann et al., 2000).
X-ray scattering curve modeling
Cel6BA models were constructed following the method used for antibody hinge peptides (Perkins et al., 1998; Boehm et al., 1999). Briefly, a 90-residue linker was built in an extended conformation using the BIOPOLYMER package of INSIGHT98 (Molecular Simulations, San Diego, CA). Its conformation was randomized using the DISCOVER3 package of INSIGHT98 (Molecular Simulations). After an initial 300 cycles of energy minimization, the linker was subjected to 1000 fs of dynamics temperature equilibration at 771 K, then a 100,000-fs dynamics simulation was carried out at 771 K. In the latter simulation, a linker conformation was saved every 100 fs, giving 1000 linker conformations. To create the 1000 Cel6BA models, the main-chain atoms of the linker N-terminal residue were superimposed on the main-chain atoms of the C-terminal residue of Cel6B (PDB code 1dys). Likewise, the N-terminal residue of Cel6A (PDB code 1bvw) was superimposed on the C-terminal residue of the linker. The duplicated N- and C-terminal residues of the linker were removed. For the P(r) profile fitting, 124 of these 1000 models were selected to represent C- and N-terminal separations of Cel6A and Cel6B that varied from 6 Å to 129 Å in 1 Å steps.
In addition to the variable linker conformations, the linker is also heterogeneously O-glycosylated. Mass spectrometry measurements showed that the linker contains an average additional mass of 9000 Da arising from glycosylation, and this corresponds to ∼50 sugar residues (M. Schülein, unpublished data). The glycosylation sites were predicted to occur at serine and threonine residues on the Cel6BA linker using the Net-O-Glyc program from Expasy (http://www.cbs.dtu.dk/services/NetOGlyc-3.0/) (Hansen et al., 1998). Thirty potential glycosylation sites were identified on the 88-residue linker. As the exact nature of the sugar residues carried by recombinant Cel6BA is not known, every other of the 30 potential O-linked glycosylation sites was selected for attachment of a NeuNAc·Gal·GalNAc trisaccharide (Boehm et al., 1999), resulting in 16 sites carrying a total of 48 sugar residues. Since no significant difference in the curve fits between the glycosylated and unglycosylated structures was noticed, no further glycosylation analyses were undertaken.
Each Cel6BA model was used to calculate x-ray scattering curves for comparison with the experimental curves. Each set of atomic coordinates for a model was placed within a three-dimensional grid of cubes. A sphere of equal volume to the cube was placed at the center of each cube if a user-specified cutoff for the minimum number of atoms contained within a cube was satisfied. For the Cel6BA model, a cube side-length of 5.44 Å in combination with a cutoff of 4 atoms consistently produced sphere models within 2% of the total dry volume of 109.0 nm3 calculated from its composition. Since the hydration shell surrounding glycoproteins is detected by x-ray scattering, spheres were added to the surface of the dry models using HYPRO (Ashton et al., 1997), based on a hydration of 0.3 g H2O/g glycoprotein and a water molecule volume of 0.0245 nm3. The optimal total of hydrated spheres for the Cel6BA model is 892 (143.6 nm3).
The x-ray scattering curve I (q) was calculated assuming a uniform scattering density for the spheres using the Debye equation as adapted to spheres (Perkins and Weiss, 1983),
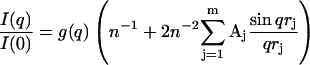 |
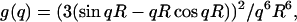 |
where g(q) is the squared form-factor for the sphere of radius r, n is the number of spheres filling the body, Aj is the number of distances rj for that value of j, rj is the distance between the spheres, and m is the number of different distances rj. Other details, including those of calibration studies used to validate this approach, are given elsewhere (Boehm et al., 1999; Perkins, 2001). X-ray curves were calculated from the hydrated sphere models without corrections for wavelength spread or beam divergence, as these are considered to be negligible for synchrotron x-ray data. X-ray scattering models generated in this way were then assessed using a goodness-of-fit R-factor defined by analogy with protein crystallography and based on the experimental curves, in the q-range extending to 0.2 Å−1 (denoted as R; Beavil et al., 1995).
P(r) profiles for the 124 x-ray scattering models were generated using the program GNOM (Svergun, 1992) with the maximal distances derived from the scattering models. To calculate an appropriately-weighted summation of these in a distribution that was more populated at shorter separations, the weighting factor W for each separation, that gave the best fit to the data, was determined from
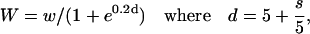 |
in which w = 3 at s = 0 Å, increasing by steps of 0.2 up to w = 14 at s = 55 Å, then down by steps of 0.2 to w = 0 at s = 125 Å, and s is the separation between the C-terminus of the first module and the N-terminus of the second module.
RESULTS
Solution structure of the isolated catalytic modules
Small-angle x-ray scattering experiments were performed on the isolated catalytic modules Cel6A and Cel6B (Fig. 1) to compare their solution structures with their crystal structures. The protein concentration ranged from 4 to 17 mg/ml for the Cel6A module and from 3 to 11 mg/ml for the Cel6B module. No aggregation was observed in both cases. The scattering data are linear in a Guinier plot in the low q-region and are nicely fitted by the Guinier law (data not shown). For both proteins, the radius of gyration Rg inferred from the slope of the fit does not vary with the concentration, indicating the lack of interparticle effects. The Rg of the catalytic modules of the two proteins are equivalent with a value of 20 Å (Table 1). The distance distribution functions have a bell-shaped appearance that is typical of spherical molecules, with similar Dmax values (56 Å and 59 Å for Cel6A and Cel6B, respectively; Table 1). The Rg/Ro ratio (where Ro is the Rg of a spherical protein with the same hydrated volume) is 1.03 for Cel6A and 1.06 for Cel6B. Cel6B is thus slightly less compact than Cel6A. This is consistent with the enclosed active-site tunnel of Cel6A and the open substrate-binding cleft of Cel6B. The comparison of the experimental scattering curves with the crystal structures (PDB codes: 1bvw and 1dys for Cel6A and Cel6B, respectively) using the program CRYSOL (Svergun et al., 1995) showed excellent agreements, and this indicated that the two proteins do not undergo any conformational change and remain globular in solution (Fig. 2).
TABLE 1.
Radius of gyration Rg and maximum dimension Dmax determined by x-ray scattering for the isolated catalytic modules Cel6A and Cel6B (50 mM sodium phosphate buffer, pH 7.5) and for the double cellulase Cel6BA (50 mM sodium phosphate, pH 8.5)
| Protein | Cel6A module | Cel6B module | Cel6BA |
|---|---|---|---|
| Rg (Å) | 20.2 ± 0.2 | 19.9 ± 0.4 | 47.3 ± 0.5 |
| Dmax (Å) | 56 ± 2 | 59 ± 2 | 178 ± 3 |
| Residues | 360 | 348 | 796 + sugars |
| Mw (kDa) | 39.7* | 37.6* | 93–96† |
Calculated from the sequence.
Determined by mass spectrometry.
FIGURE 2.

Fit of the experimental scattering curves with the crystal structures (red line) for the isolated catalytic modules in (a) Cel6A and (b) Cel6B.
Molecular dimensions of the chimeric double cellulase Cel6BA
We measured the solution scattering of the biologically active construct Cel6BA at concentrations varying from 2 to 13 mg/ml. In the Guinier region, the scattering data were linear, indicating that the protein was not aggregated (Fig. 3). The Rg was calculated at each concentration in a q-range extending out to qRg < 1.0. Small repulsive interactions resulted in a decrease of the Rg with increasing concentration. Extrapolation to zero concentration gave an Rg value of 47.3 Å corresponding to one single molecule in solution with no interparticle interaction. The Rg of a globular protein containing the same number of amino acids as Cel6BA would be much smaller (Millett et al., 2002). This suggests that the linker between the two catalytic modules possesses an extended conformation.
FIGURE 3.

Guinier plot of the scattered intensity of Cel6BA in 50 mM sodium phosphate, pH 8.5 buffer. The radius of gyration Rg is inferred from the slope of the straight line fitting the data in the q-range qRg ≤ 1.0. Protein concentration (from top to bottom): 13 mg/ml, 10.1 mg/ml, 6.9 mg/ml, 4.9 mg/ml, and 2.1 mg/ml.
The extended linker conformation was further confirmed by the distance distribution function P(r) of Cel6BA (Fig. 4). The P(r) function is the histogram of all the interatomic distances within the molecule. The experimental P(r) profile exhibits a main peak at an r-value of ∼30 Å and a long tail up to a maximum dimension of 178 Å, indicating that Cel6BA is highly elongated. The main peak is assigned to the intradomain distances within each of the globular catalytic modules, whereas the tail of the curve corresponds to the interdomain distances between the two modules. In the Cel6BA chimera, the proportion of residues in the linker (88 amino acids and ∼48 carbohydrates) compared to those in the catalytic modules (708 amino acids) is small. The difference between the sum of the Dmax values of the catalytic modules (115 Å) and that for the Cel6BA chimera (178 Å) reveals that the length of the linker of the full-length chimera protein can be quite expanded at 63 Å. However, the theoretical P(r) profile for a rigid dumbbell-shaped protein with two globular spherical modules of the same size and separated by an extended linker would exhibit two distinct peaks, the first peak at an r-value corresponding to the radius of each sphere, and the second peak corresponding to the distance between the two spheres (see Fig. 7 a below). The region of low intensity between the two peaks would correspond to the interatomic distances between relatively few linker residues, including its O-glycosylation, and the two spheres. In distinction to this, the experimental P(r) profile showed that the second peak cannot be observed, and that the intermediate distances are much more populated, thus leading to a very broad shoulder from 55 Å to 178 Å. This broad shoulder suggests that the linker adopts all the possible separations between the two catalytic modules, and corresponds to the observation of conformational disorder. Putative disordered regions in proteins can be identified by analysis of the sequence using computing methods such as PONDR (Li et al., 1999). Intrinsically disordered proteins are indeed usually rich in charged or polar residues, and poor in hydrophobic residues (see Fig. 1 b for the sequence of the linker of Cel6BA). PONDR was run on full-length Cel6BA and predicted a long disordered region from residue 343–434 with a prediction score of 0.96. These 92 residues correspond precisely to the linker (Fig. 1 b). This result supports the observation that the linker is disordered, separating the two catalytic modules by a wide range of distances achieved through many different conformations.
FIGURE 4.

Experimental distance distribution profile P(r) of the double cellulase Cel6BA (solid line), superimposed with the distance distribution profiles of the catalytic modules Cel6A (dotted line) and Cel6B (dashed line).
FIGURE 7.

Analysis of P(r) profiles for models of the Cel6A and Cel6B module structure. (a) The P(r) profiles of three models with intermodule separations of 10 Å, 60 Å, and 120 Å were represented by histograms of the distribution of their inter-Cα–Cα distances (red, green, and blue). These are compared with the experimental P(r) profile calculated using GNOM with an assumed maximum length of 180 Å (black dotted line). (b) The modeled P(r) profile (red) was calculated from a weighting scheme based on 124 models with intermodule separations between 6 Å and 129 Å in 1 Å steps. Each model possessed a linker that was energy-minimized (see Fig. 6) in order that each linker adopted a stereochemically reasonable conformation. These models have R-factors as shown in Fig. 6. The weighted summation of the 124 models generated the P(r) profile with a single peak maximum as shown in red. The experimental P(r) curve is shown as the dotted line. The relative weights for the 124 models are shown as an inset, with that at 15 Å set as 1.
Modeling of the x-ray scattering curve for the Cel6BA chimera
The observed scattering curve I(q) (Fig. 5 a, dotted line) is produced by the sum of all the different conformations the protein can adopt in solution. The size of the broad shoulder in the P(r) profile (Fig. 4) indicates thus that there is a distribution of conformations with varying distances between the Cel6A and Cel6B domains, and that the more extended structures are less populated than the more compact structures. Therefore, it is not possible to describe the scattering curve I(q) of the chimeric double cellulase Cel6BA by one single conformation of the molecule. Consequently, we employed molecular modeling of the linker region with different end-to-end separations to establish whether a conformational distribution of linker lengths would account for the experimental data.
FIGURE 5.

Calculated scattering curves for two-module Cel6A and Cel6B structures with and without the 88-residue intermodule linker. (a) The 12 scattering curves correspond to every 10th model generated in which the C-terminal α-carbon atom of the Cel6A module was separated by 10 Å to 120 Å in 10 Å steps from the N-terminal α-carbon atom of the Cel6B module. No linker is present. The minimum at 0.17 Å−1 is indicated by an arrow (see text). The experimental curve is indicated by the dotted line. (b) The 12 scattering curves correspond to the 12 models of a, but now have linkers attached between the Cel6A and Cel6B modules. Although the same general features are observed as in a, the minimum at q of 0.17 Å−1 (arrow) now varies from curve to curve. (c) The effect of simulations with glycosylated linker structures is shown. (d) The goodness-of-fit R-factors calculated from the comparison of the modeled and experimental scattering curves are shown as functions of the separation of the Cel6A and Cel6B modules with and without the linker peptide.
The length of the linker of ∼63 Å in the Cel6BA chimera is much shorter than the 287 Å extension of a theoretical 88-residue β-strand or the 127 Å length of a theoretical 88-residue α-helix. This, together with the fact that the linker adopts several conformations of different lengths, shows that the linker is not an extended regular polypeptide chain, but instead adopts a much more compact structure.
In the following, the assumptions required to perform a multiconformational analysis for Cel6BA were explored. Firstly, models of the fusion protein without the linker peptide were generated by increasing the separation between the Cel6A and Cel6B crystal structures in 6 Å steps up to 129 Å. The resulting scattering curves exhibited a pronounced minimum at 0.17 Å−1 that did not vary with the separation, whereas the I(q) intensities in the q-range below 0.05 Å−1 showed a large dependence on the separation (Fig. 5 a). However, the experimental scattering curve shows no features such as any clear minima (Fig. 5 a). In the next step, 1000 randomized linker conformations were generated by a molecular dynamics simulation using BIOPOLYMER and DISCOVER3 (Materials and Methods). When the Cel6A and Cel6B domains used to generate Fig. 5 a were connected with a linker of appropriate length, the minimum at 0.17 Å−1 decreased in magnitude (Fig. 5 b). The addition of 16 O-linked NeuNAc·Gal·GalNAc trisaccharides at Ser and Thr residues of the linker representing its glycosylation (Materials and Methods) further reduced the minimum at 0.17 Å−1 (Fig. 5 c). It was concluded that better agreement with the observed scattering curve in the reciprocal space may be achieved by taking into account the linker, its disorder, and its glycosylation.
The goodness of fit (R-factor) provides a monitor of the agreement between the modeled and experimental curves: the smaller the R-factor, the better the fit. The R-factors from comparison of the modeled x-ray scattering curves calculated for separations between 1 Å and 129 Å with the experimental scattering curve (Fig. 5 d) varied between 7% and 45%. The best R-factors resulted from models in which the separation was ∼20 Å for the linker-free models, and ∼32 Å for the models with linkers, and not from those with the experimentally determined maximum value of 63 Å estimated above (Fig. 4). The R-factors also showed that changes in the relative orientation of the Cel6A and Cel6B modules s at a fixed linker separation of 32 Å did not significantly affect the curve-fit procedure.
A further 1000 linker-conformations were generated from a second molecular dynamics procedure in which the C-terminal α-carbon atom of Cel6B and the N-terminal α-carbon atom of Cel6A were fixed at a separation of 32 Å. The starting linker model comprised an extended C-shaped unglycosylated loop. As the molecular dynamics simulation proceeded, the linker adopted a quasiglobular structure. The R-factors dropped from 14.7% for the starting model to an acceptable level of 7–8% after 400 models. No further reduction was observed up to 1000 models (Fig. 6). This showed that the best single representation of Cel6BA in solution resulted from a model with a quasiglobular linker conformation. However, the separation of 32 Å and not 63 Å meant that this model was not able to account for the high Dmax given by the distance distribution function P(r) (Fig. 4).
FIGURE 6.

The refinement of a compact linker structure between the Cel6A and Cel6B modules. A total of 1000 linker conformations were generated in the course of an energy minimization by a molecular dynamics procedure. The α-carbon molecular views of the first and last models are shown as insets, together with a 20 Å scale bar.
The final calculations were based on the P(r) profiles (real space) of the models linked with a range of separations (Fig. 5 d). The comparison of the experimental P(r) profile with three of these models with N-terminal–C-terminal separations of 10 Å (short linker), 60 Å (intermediate linker), and 120 Å (long linker) showed large discrepancies between the single peak seen experimentally and the double peak obtained from these three models (Fig. 7 a). Good agreement between the experimental and modeled P(r) profiles requires a summation of an appropriate broad range of separations between the Cel6A and Cel6B modules. One has to apply a weighting scheme such that the weight of a model becomes smaller as the intermodule separation increases. After a procedure in which several weighting schemes were tested we obtained the best results with a function that gave an appropriate distribution of weights (inset to Fig. 7 b) when applied to the 124 models of Fig. 5 d with intermodule separations of 6 Å to 129 Å in 1 Å steps. The R-factor of this I(q) curve fit (Fig. 8) was 7.4%, which is similar to the best of the single-conformation models shown in Fig. 6. Unlike the quasiglobular linker model, however, the resulting weighted P(r) profile shows good agreement with the experimental P(r) profile (Fig. 7 b). The success of this fit showed that the best agreement is obtained with a range of models, i.e., that conformational disorder is present. Typical examples of four models taken from the 124 structures in Fig. 8 show that the linker is conformationally variable in the solution structure of this fusion protein.
FIGURE 8.

Comparison between the experimental (dotted line) and calculated (solid line) scattering curves I(q). The calculated I(q) curve corresponds to the weighted summation of the 124 models used to generate the P(r) profile of Fig. 4. The R-factor is 7.4%. The α-carbon views of four typical molecular structures that were used for the weighted summation are shown below the curve fit.
DISCUSSION
To study the conformational disorder in the cellulase linker we monitored the scattering properties of a double-headed cellulase with two large catalytic modules separated by an 88-amino-acid-long linker (Fig. 1). Contrarily to a normal cellulase where the CBM is too small and could not be discriminated from the linker, the scattering curve of this engineered arrangement is sensitive to the contributions of the two large globular modules at the extremities of the linker. This enabled us to infer structural information on the linker and to determine whether it possesses a static or variable conformation.
We have shown that the intermodule linker peptide in the chimeric double cellulase Cel6BA adopts a wide range of conformations in solution, together with very different end-to-end distances. This is consistent with the prediction of disorder in the linker region by PONDR, the shape of its experimental P(r) profile, and the modeling of Cel6BA as a double-domain structure with variable linker conformations. The modeling was able to restore the major features of the experimental P(r) profile (Fig. 7 b), and gave a reasonable I(q) curve fit in reciprocal space out to a q-value of 0.1 Å−1 (Fig. 8). It has already been reported that the presence of random surface loops in proteins can lead to a marked smoothening of prominent features in the scattering curve (Petoukhov et al., 2002). Here, we have shown that the randomization of a linker peptide joining two large globular modules can likewise lead to the smoothening of such features, as shown by the comparisons between Fig. 5, a and b. Similarly, a proper description of the heterogeneous O-glycosylation would also further improve the quality of the I(q) curve fit, as shown by Fig. 5 c. A good approximation of the experimental x-ray P(r) profile of Cel6BA was obtained by describing the proteins in solution by a series of Cel6BA models with intermodule separations ranging from 6 Å to 129 Å in steps of 1 Å, combined with a distribution of weights that displayed a peak at 15 Å. The weighting scheme might be optimized by the use of, for example, a genetic algorithm to minimize the difference between the observed and calculated P(r) profiles. At the present time, the P(r) profile generated from our weighting scheme is sufficiently accurate to indicate the existence of conformational disorder in the linker, and to describe the resulting distribution of intermodule separations. Moreover, our experiments have been performed in buffer conditions similar to those of other wild-type cellulases, where the protein is the most active and a change in the pH or ionic strength might modulate this distribution.
The distribution of conformations obtained in our modeling study based on the experimental data definitely indicates the existence of conformational disorder in the linker, and that compact linkers are the most frequent. The single Cel6BA model that fits best the I(q) data in reciprocal space is that with an intermodule separation of 32 Å. The distribution of 124 Cel6BA models that best fits the I(q) data shows a maximum at an intermodule separation of 15 Å. The resulting best single model with a 32 Å intermodule separation corresponds to the average conformation weighted by the distribution of conformations (inset to Fig. 7 b). The maximum in the distribution means that the most compact linker conformations are the most stable, but these are able to unwind into longer linkers with a relatively low energy cost. Even though the linker separation would be different in the case of the native Cel6A and Cel6B proteins, the peak of the distribution of linker lengths suggests that the most stable linker conformations position the catalytic module and the CBM at a distance comparable to one cellobiose unit (in crystalline cellulose, the cellobiose repeat unit is 10.4 Å long). After hydrolysis, the flexibility of the linker conformation would allow the catalytic module to readily diffuse away to hydrolyze another glycosidic bond, while leaving the CBM attached to the cellulose surface. Such a mechanism would permit the progressive and efficient hydrolysis of cellulose by the enzyme. The corollary is that any strong restriction to the independence of the movement of the two modules of cellulase would result in a decreased overall efficiency. The particular conformational properties of the linker probably offer the best compromise between the requirement for a tight binding to cellulose by the binding module and the need to reach fresh hydrolysis sites by the catalytic module.
The structural properties of the fungal cellulase linkers are contained in their sequences and O-glycosylation, and are not modified by noncovalent interactions with the appended functional modules. The observed range of linker conformations is most satisfactorily explained by the effect of O-glycosylation. Indeed it is interesting to note that the maximum extension of the linker observed here is larger than that expected for a random coil. A random coil peptide containing 88 residues would have an Rg of ∼26–27 Å and therefore a Dmax of 52–55 Å (Millett et al., 2002), compared to the length of 64 Å observed here. The steric restraint introduced by O-glycosylation probably drives the equilibrium toward more extended conformations. It is interesting to compare the results obtained here with those we reported earlier for cellulase Cel45 from H. insolens (Receveur et al., 2002) . The linkers of Cel6A and Cel6B are, on average, less glycosylated than that of Cel45 (M. Schülein, unpublished), and are much less extended in the chimeric Cel6BA than in Cel45. Comparisons of the Dmax values of the globular modules to those of the intact cellulase gave a length of 50 Å for 36 residues in Cel45 (Receveur et al., 2002), whereas this is only 64 Å for 88 residues in Cel6BA. This suggests that a higher level of O-glycosylation stabilizes conformations with a larger separation between the two globular modules. The role of the linker of bimodular cellulases is likely to ensure an independent action of the two functional modules through the achievement of a wide range of conformations. Adequate linker properties are probably attainable by varying the number of residues in the linker, the extent of glycosylation, the amino-acid composition, and so on, and this could explain why there is nothing apparently conserved in the amino-acid sequence of intermodule linkers.
Even though the work presented here was done on a chimeric double cellulase that does not exist in nature, such a construct is biologically relevant. First, a number of multimodular glycosidases that comprise more than one catalytic module are known (a cellulase from the fungus Neocallimastix patriciarum contains three catalytic modules; Xue et al., 1992). Secondly, a survey of the linker lengths in fungal modular glycoside hydrolases (B. Henrissat, unpublished) shows that many have lengths in the range of 40–120 residues. A few are even longer, such as a bimodular xylanase from the fungus N. patriciarum whose linker is annotated as containing over 480 residues to separate the catalytic module from the CBM (Black et al., 1994). The 88-residue linker studied here is therefore not exceptionally long. Finally, in our previous study on the H. insolens Cel45 cellulase in both its native full-length form and with a CBM-deleted form (Receveur et al., 2002), we have shown that the linker conformation was not affected by the presence or absence of the CBM. We believe therefore that the wide conformational variability found in the linker in the engineered double-headed cellulase is realistic and representative of the linker present in natural cellulases. The recurrence of these linkers in enzymes degrading insoluble polysaccharides is an indication of the clear evolutionary advantage in joining globular functional modules through a flexible, elongated, and unfolded peptide region. The presence of disordered regions in proteins is not restricted to cellulases nor to glycoside hydrolases, and there is a growing interest for proteins whose function requires the lack of folded globular structure (“natively unfolded proteins”) (Dunker and Obradovic, 2001; Uversky, 2002; Wright and Dyson, 1999).
Small-angle x-ray scattering is probably the method of choice to characterize and quantify the structural properties of natively disordered proteins or protein regions. Many types of natively disordered proteins begin to emerge (Tompa, 2002), such as those implicated in molecular recognition processes and which fold upon encounter with physiological partners (Longhi et al., 2003), and the entropic chains that serve as flexible hinges or molecular springs between distinct functional modules such as the linkers described here. In conclusion, the work reported here establishes the first direct experimental measurement of the conformational variability of the linkers of plurimodular fungal glycoside hydrolases. We show that full-length cellulases exist in solution as a set of conformers with very different relative distances between the two functional modules resulting from the ability of their linkers to adopt both compact and extended structures. Such structural properties are typical of those required for the model of action proposed by Receveur et al. (2002), in which cellulases can move on their substrate with a caterpillar-like motion to achieve an efficient hydrolysis.
Acknowledgments
We thank Birte Jacobsen for technical assistance and Johnny Christensen for fermenter scale cultivations.
J.T.E. and S.J.P. are supported by the Wellcome Trust.
Martin Schülein is deceased.
Ingemar von Ossowski and Julian T. Eaton contributed equally to this work.
Ingemar von Ossowski's present address is Biochemistry Division, Dept. of Biological and Environmental Sciences, Faculty of Biosciences, University of Helsinki, Biocenter 2, PO Box 56, FIN-00014 Helsinki, Finland.
Torben P. Frandsen's present address is Symphogen A/S, Elektrovej, Building 375 DK-2800 Lyngby, Denmark.
References
- Ashton, A. W., M. K. Boehm, J. R. Gallimore, M. B. Pepys, and S. J. Perkins. 1997. Pentameric and decameric structures in solution of serum amyloid P component by x-ray and neutron scattering and molecular modelling analyses. J. Mol. Biol. 272:408–422. [DOI] [PubMed] [Google Scholar]
- Beavil, A. J., R. J. Young, B. J. Sutton, and S. J. Perkins. 1995. Bent domain structure of recombinant human IgE-Fc in solution by x-ray and neutron scattering in conjunction with an automated curve-fitting procedure. Biochemistry. 34:14449–14461. [DOI] [PubMed] [Google Scholar]
- Bergmann, A., G. Fritz, and O. Glatter. 2000. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA). J. Appl. Crystallogr. 33:1212–1216. [Google Scholar]
- Boehm, M. K., J. M. Woof, M. A. Kerr, and S. J. Perkins. 1999. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by x-ray and neutron solution scattering and homology modelling. J. Mol. Biol. 286:1421–1447. [DOI] [PubMed] [Google Scholar]
- Black, G. W., G. P. Hazlewood, G. P. Xue, C. G. Orpin, and H. J. Gilbert. 1994. Xylanase B from Neocallimastix patriciarum contains a non-catalytic 455-residue linker sequence comprised of 57 repeats of an octapeptide. Biochem. J. 299:381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrard, G., A. Koivula, H. Soderlund, and P. Beguin. 2000. Cellulose-binding domains promote hydrolysis of different sites on crystalline cellulose. Proc. Natl. Acad. Sci. USA. 97:10342–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, T., H. Wöldike, E. Boel, S. B. Mortensen, K. Hjortshøj, L. Thim, and M. T. Hansen. 1988. High level expression of recombinant genes in Aspergillus oryzae. Biotechnology (NY). 6:1419–1422. [Google Scholar]
- Davies, G. J., A. M. Brzozowski, M. Dauter, A. Varrot, and M. Schulein. 2000. Structure and function of Humicola insolens family 6 cellulases: structure of the endoglucanase, Cel6B, at 1.6 Å resolution. Biochem. J. 348:201–207. [PMC free article] [PubMed] [Google Scholar]
- Dunker, A. K., and Z. Obradovic. 2001. The protein trinity-linking function and disorder. Nat. Biotechnol. 19:805–806. [DOI] [PubMed] [Google Scholar]
- Fort, S., V. Boyer, L. Greffe, G. J. Davies, O. Moroz, L. Christiansen, M. Schulein, S. Cottaz, and H. Driguez. 2000. Highly efficient synthesis of β(1-4)-oligo- and -polysaccharides using a mutant cellulase. J. Am. Chem. Soc. 122:5429–5437. [Google Scholar]
- Fujimoto, Z., A. Kuno, S. Kaneko, S. Yoshida, H. Kobayashi, I. Kusakabe, and H. Mizuno. 2000. Crystal structure of Streptomyces olivaceoviridis E-86 β-xylanase containing xylan-binding domain. J. Mol. Biol. 300:575–585. [DOI] [PubMed] [Google Scholar]
- Gilkes, N. R., B. Henrissat, D. G. Kilburn, R. C. Miller, Jr., and R. A. Warren. 1991. Domains in microbial β-1, 4-glycanases: sequence conservation, function, and enzyme families. Microbiol. Rev. 55:303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinier, A., and F. Fournet. 1955. Small Angle Scattering of X-Rays. Wiley Interscience, New York.
- Hansen, J. E., O. Lund, N. Tolstrup, A. A. Gooley, K. L. Williams, and S. Brunak. 1998. NetOglyc: prediction of mucin type O-glycosylation sites based on sequence context and surface accessibility. Glycoconj. J. 15:115–130. [DOI] [PubMed] [Google Scholar]
- Henrissat, B. 1994. Cellulases and their interaction with cellulose. Cellulose. 1:169–196. [Google Scholar]
- Henrissat, B., and A. Bairoch. 1993. New families in the classification of glycosyl hydrolases based on amino-acid sequence similarities. Biochem. J. 293:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat, B., and A. Bairoch. 1996. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316:695–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi, R., B. Krummel, and R. K. Saiki. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 77:51–59. [DOI] [PubMed] [Google Scholar]
- Kelly, J. M., and M. J. Hynes. 1985. Transformation of Aspergillus niger by the amdS gene of Aspergillus nidulans. EMBO J. 4:475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., P. Romero, M. Rani, A. K. Dunker, and Z. Obradovic. 1999. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 10:30–40. [PubMed] [Google Scholar]
- Longhi, S., V. Receveur-Brechot, D. Karlin, K. Johansson, H. Darbon, D. Bhella, R. Yeo, S. Finet, and B. Canard. 2003. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 278:18638–18648. [DOI] [PubMed] [Google Scholar]
- Millett, I. S., S. Doniach, and K. W. Plaxco. 2002. Toward a taxonomy of the denatured state: small angle scattering studies of unfolded proteins. Adv. Protein Chem. 62:241–262. [DOI] [PubMed] [Google Scholar]
- Pell, G., L. Szabo, S. J. Charnock, H. Xie, T. M. Gloster, G. J. Davies, and H. J. Gilbert. 2004. Structural and biochemical analysis of Cellvibrio japonicus xylanase 10C: how variation in substrate-binding cleft influences the catalytic profile of family GH-10 xylanases. J. Biol. Chem. 279:11777–11788. [DOI] [PubMed] [Google Scholar]
- Perkins, S. J. 2001. X-ray and neutron scattering analyses of hydration shells: a molecular interpretation based on sequence predictions and modelling fits. Biophys. Chem. 93:129–139. [DOI] [PubMed] [Google Scholar]
- Perkins, S. J., A. W. Ashton, M. K. Boehm, and D. Chamberlain. 1998. Molecular structures from low angle x-ray and neutron scattering studies. Int. J. Biol. Macromol. 22:1–16. [DOI] [PubMed] [Google Scholar]
- Perkins, S. J., and H. Weiss. 1983. Low-resolution structural studies of mitochondrial ubiquinol:cytochrome C reductase in detergent solutions by neutron scattering. J. Mol. Biol. 168:847–866. [DOI] [PubMed] [Google Scholar]
- Petoukhov, M. V., N. A. Eady, K. A. Brown, and D. I. Svergun. 2002. Addition of missing loops and domains to protein models by x-ray solution scattering. Biophys. J. 83:3113–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, G., J. M. Mikkelsen, M. Schülein, S. A. Patkar, F. Hagen, C. M. Hjort, and S. Hastrup. 1991. Cellulase preparation comprising endoglucanase enzyme used in detergents for cellulose-containing fabrics or to improve drainage for paper pulp. WO 91/17243.
- Receveur, V., M. Czjzek, M. Schulein, P. Panine, and B. Henrissat. 2002. Dimension, shape, and conformational flexibility of a two-domain fungal cellulase in solution probed by small angle x-ray scattering. J. Biol. Chem. 277:40887–40892. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual, 2nd Ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schulein, M. 1997. Enzymatic properties of cellulases from Humicola insolens. J. Biotechnol. 57:71–81. [DOI] [PubMed] [Google Scholar]
- Schülein, M., D. Tikhomirov, and C. Schou. 1993. Humicola insolens alkaline cellulases. In Trichoderma reesei Cellulases and Other Hydrolases. P. Suominen and T. Reinikainen, editors. Foundation for Biotechnical and Industrial Fermentation Research, Espoo, Finland. 109–116.
- Shen, H., M. Schmuck, I. Pilz, N. R. Gilkes, D. G. Kilburn, R. C. Miller, and R. A. J. Warren. 1991. Deletion of the linker connecting the catalytic and cellulose-binding domains of endoglucanase A (CenA) of Cellulomonas fimi alters its conformation and catalytic activity. J. Biol. Chem. 266:11335–11340. [PubMed] [Google Scholar]
- Srisodsuk, M., T. Reinikainen, M. Penttila, and T. T. Teeri. 1993. Role of the interdomain linker peptide of Trichoderma reesei cellobiohydrolase I in its interaction with crystalline cellulose. J. Biol. Chem. 268:20756–20761. [PubMed] [Google Scholar]
- Svergun, D. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503. [Google Scholar]
- Svergun, D., C. Baraberato, and M. H. Koch. 1995. CRYSOL— a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28:768–773. [Google Scholar]
- Tompa, P. 2002. Intrinsically unstructured proteins. Trends Biochem. Sci. 27:527–533. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N. 2002. What does it mean to be natively unfolded? Eur. J. Biochem. 269:2–12. [DOI] [PubMed] [Google Scholar]
- Varrot, A., S. Hastrup, M. Schulein, and G. J. Davies. 1999. Crystal structure of the catalytic core domain of the family 6 cellobiohydrolase II, Cel6A, from Humicola insolens, at 1.92 Å resolution. Biochem. J. 337:297–304. [PMC free article] [PubMed] [Google Scholar]
- Wright, P. E., and H. J. Dyson. 1999. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293:321–331. [DOI] [PubMed] [Google Scholar]
- Xue, G. P., K. S. Gobius, and C. G. Orpin. 1992. A novel polysaccharide hydrolase cDNA (celD) from Neocallimastix patriciarum encoding three multi-functional catalytic domains with high endoglucanase, cellobiohydrolase and xylanase activities. J. Gen. Microbiol. 138:2397–2403. [DOI] [PubMed] [Google Scholar]