

Abstract
In the presence of high concentrations of inert macromolecules, the self-association of proteins is strongly enhanced through an entropic, excluded-volume effect variously called macromolecular crowding or depletion attraction. Despite the predicted large magnitude of this universal effect and its far-reaching biological implications, few experimental studies of macromolecular crowding have been reported. Here, we introduce a powerful new technique, fast field-cycling magnetic relaxation dispersion, for investigating crowding effects on protein self-association equilibria. By recording the solvent proton spin relaxation rate over a wide range of magnetic field strengths, we determine the populations of coexisting monomers and decamers of bovine pancreatic trypsin inhibitor in the presence of dextran up to a macromolecular volume fraction of 27%. Already at a dextran volume fraction of 14%, we find a 30-fold increase of the decamer population and 5105-fold increase of the association constant. The analysis of these results, in terms of a statistical-mechanical model that incorporates polymer flexibility as well as the excluded volume of the protein, shows that the dramatic enhancement of bovine pancreatic trypsin inhibitor self-association can be quantitatively rationalized in terms of hard repulsive interactions.
INTRODUCTION
The importance of excluded-volume interactions for the solution behavior of proteins was first recognized in studies of the effect of polymers on protein partitioning (Ogston and Phelps, 1960) and solubility (Laurent, 1963; Atha and Ingham, 1981), which laid the foundations for the current widespread use of nonadsorbing polymers, like polyethylene glycol, for protein separation (Albertsson, 1986) and crystallization (McPherson, 1985). More recently, the realization that macromolecules occupy 20–30% of the intracellular volume has provided a biological motivation for studying the effects of nominally inert background macromolecules on protein association equilibria and rate processes (Minton, 1981, 1998, 2000; Ellis, 2001). Now loosely referred to as “macromolecular crowding”, such entropic, excluded-volume effects are thought to play important roles in protein folding (van den Berg et al., 1999; Qu and Bolen, 2002; Sasahara et al., 2003), protein self-association into functional native oligomers (Lindner and Ralston, 1995; Rivas et al., 1999, 2001; Zorrilla et al., 2004a) or amyloid aggregates (Hatters et al., 2002), intracellular compartmentation (Walter and Brooks, 1995; Hancock, 2004), and cell volume regulation (Garner and Burg, 1994; Al-Habori, 2001).
Addition of inert macromolecules to a protein solution shifts equilibria toward the more compact state because this minimizes the volume excluded to the inert macromolecules. Under typical physiological conditions, macromolecular crowding is therefore a powerful driving force for protein self-association, which may increase the association constant by several orders of magnitude as compared to dilute in vitro conditions. Whereas theoretical predictions of macromolecular crowding effects abound, there are remarkably few experimental studies of this universal phenomenon. The principal experimental challenge is to resolve and quantify populations of protein oligomers in dynamic equilibrium. This task is further complicated by the background of abundant macromolecular crowding agent. One of the few techniques that has been applied to this problem is analytical ultracentrifugation, where tracer sedimentation equilibrium data yield an apparent buoyant molar mass, which, after certain approximations, provides the weight-average molar mass of the self-associating protein (Rivas et al., 1999). Because this technique does not resolve the different oligomers, the interpretation becomes somewhat model dependent.
In this work, we demonstrate that the macromolecular crowding effect can be studied in a direct way by an NMR technique known as magnetic relaxation dispersion (MRD). The MRD technique has recently been used to characterize quantitatively the self-association of bovine pancreatic trypsin inhibitor (BPTI) (Gottschalk et al., 2003a), bovine β-lactoglobulin (Gottschalk et al., 2003b), and hen lysozyme (Gottschalk and Halle, 2003), in all cases without crowding agent. By recording the solvent proton spin relaxation rate over a wide range of magnetic field strengths, it is possible to resolve (in the frequency domain) protein oligomers with different rotational diffusion coefficients and to determine the populations of coexisting oligomers. Here, we use the MRD technique to study the BPTI monomer-decamer equilibrium (Hamiaux et al., 2000; Gottschalk et al., 2003a) in the presence of dextran as a crowding agent. We find that BPTI self-association is strongly enhanced by the polymer, with a 30-fold increase of the decamer population and 5105-fold increase of the association constant at a dextran volume fraction of merely 14%.
Dextran is commonly used as a crowding agent because it is uncharged and does not adsorb to protein surfaces (Laurent, 1963; Lindner and Ralston, 1995; Rivas et al., 1999; van den Berg et al., 1999; Qu and Bolen, 2002; Hatters et al., 2002; Sasahara et al., 2003). On the other hand, the conformational flexibility of the dextran polymer chain complicates the statistical-mechanical analysis of the crowding effect. In contrast to previous work, we take the flexibility into account explicitly in the theoretical analysis (Lue, 1998). We find that the MRD data can be quantitatively accounted for by a hard-repulsion (excluded-volume) interaction and a Kuhn length for dextran consistent with light-scattering results. Our theoretical analysis also incorporates, in a self-consistent manner, the crowding effect of the protein itself.
MATERIALS AND METHODS
Materials and sample preparation
Bovine pancreatic trypsin inhibitor was supplied by Bayer HealthCare AG (Trasylol, lot no. 9104, 97% purity by high-performance liquid chromatography). After exhaustive dialysis to remove residual salt the protein was lyophilized. Dextran, manufactured by sucrose fermentation with Leuconostoc mesenteroides bacteria (strain B-512), limited hydrolysis, ethanol fractionation, and spray drying, was obtained from Sigma (product no. D9260). According to the manufacturer, the mean molecular mass was 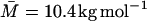 (determined by size exclusion chromatography), which corresponds to a mean degree of polymerization of
(determined by size exclusion chromatography), which corresponds to a mean degree of polymerization of 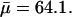 The residual water content of the dextran preparation was taken to be 5%.
The residual water content of the dextran preparation was taken to be 5%.
Samples for MRD measurements were prepared by dissolving, at room temperature, dextran and/or BPTI in millipore water and adjusting pH to 4.5 by microliter additions of 3 M HCl. No buffers were used. The BPTI concentration was determined spectrophotometrically (GBC UV-VIS 920) at 280 nm (before addition of dextran), using an extinction coefficient of 0.837 mL mg−1 cm−1 (Gottschalk et al., 2003a). Volume fractions were calculated with partial specific volumes of 0.611 mL g−1 for dextran (Granath, 1958), 0.720 mL g−1 for BPTI (Filfil et al., 2004), and 1.000 mL g−1 for water. The mixed BPTI/dextran samples were prepared by adding known amounts of dextran powder to a BPTI solution of known concentration. The relevant concentration variables for all samples are summarized in Table 1: mass (w) and volume (φ) fractions, molarities (C), and the number of water molecules per glucose unit  or per BPTI molecule
or per BPTI molecule  To assess the degree of polymer chain overlap, we include in Table 1 the mean spacing, d, between dextran molecules. For a uniformly expanded face-centered cubic spatial distribution,
To assess the degree of polymer chain overlap, we include in Table 1 the mean spacing, d, between dextran molecules. For a uniformly expanded face-centered cubic spatial distribution,  with
with  the dextran volume fraction and VM = 10,540 Å3 the anhydrous volume of a dextran molecule.
the dextran volume fraction and VM = 10,540 Å3 the anhydrous volume of a dextran molecule.
TABLE 1.
Concentrations of dextran and BPTI in MRD samples
| Sample | wM* | φM† | φBPTI† | CBPTI (mM) | d (Å) | 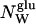 |
 |
|---|---|---|---|---|---|---|---|
| D1 | 0.090 | 0.057 | – | – | 64 | 91.5 | – |
| D2 | 0.164 | 0.107 | – | – | 52 | 46.0 | – |
| D3 | 0.226 | 0.151 | – | – | 46 | 30.8 | – |
| D4 | 0.279 | 0.191 | – | – | 43 | 23.2 | – |
| D5 | 0.325 | 0.227 | – | – | 40 | 18.7 | – |
| P0 | 0 | 0 | 0.072 | 15.4 | – | – | 3345 |
| P1 | – | 0.053 | 0.068 | 14.5 | – | 91.5 | 3417 |
| P2 | – | 0.100 | 0.064 | 13.7 | – | 46.0 | 3490 |
| P3 | – | 0.142 | 0.061 | 13.0 | – | 30.8 | 3562 |
| P4 | – | 0.180 | 0.058 | 12.4 | – | 23.2 | 3634 |
| P5 | – | 0.214 | 0.055 | 11.8 | – | 18.7 | 3707 |
Mass fraction dextran.
Volume fraction dextran (M) or BPTI.
Relaxation dispersion measurements
The longitudinal relaxation rate of the water 1H resonance was measured over more than four frequency decades, from 10 kHz to 200 MHz. To cover this frequency range, we used three types of NMR spectrometer: 1), a Stelar Spinmaster (Stelar, Mede, Italy) fast field-cycling (FC) spectrometer (from 10 kHz to 10 MHz); 2), a field-variable iron-core magnet (Drusch, Hanstedt, Germany or GMW, San Carlos, CA) equipped with a Tecmag (Houston, TX) Discovery or Apollo console (16–78 MHz); and 3), Bruker (Billerica, MA) Avance DMX 100 and 200 spectrometers with conventional cryomagnets (100 and 200 MHz). The temperature was maintained at 27.0 ± 0.1°C using a Stelar variable temperature control unit (below 100 MHz) or a Bruker Eurotherm regulator (at 100 and 200 MHz). Temperatures were checked with a thermocouple referenced to an ice-water bath. No attempt was made to purge oxygen from the solutions, because the small paramagnetic relaxation contribution form dissolved O2 (Teng et al., 2001) is expected to cancel out when taking the difference of the relaxation rates measured with and without dextran.
In the non-FC experiments (with variable detection field), the longitudinal relaxation rate, R1, was measured with the 180° − τ − 90° inversion recovery sequence, an eight-step phase cycle, and 20 randomly ordered delay times. The total 1H magnetization recovers biexponentially, because it includes not only water and rapidly exchanging macromolecular protons but also a minor contribution from nonexchanging macromolecular protons. The latter contribution, which can increase the apparent R1, was eliminated by integrating the water peak over a range where the spectral overlap was negligible (for non-FC experiments), or by using an acquisition delay sufficiently long that the protein magnetization had decayed before the signal was recorded (FC experiments). In this way, single-exponential recovery curves were obtained, from which R1 was determined by a three-parameter fit. The accuracy of R1 is estimated to ±1% (one standard deviation).
The FC technique overcomes the sensitivity problem of conventional fixed-field experiments in weak magnetic fields (Noack, 1986; Kimmich and Anoardo, 2004). The polarization and detection fields (in 1H frequency units) were set, respectively, to 20 and 9 MHz (D series of samples) or to 10 and 8 MHz (P series). A field slew rate of 15 MHz ms−1 (D series) or 4 MHz ms−1 (P series) and a switching time of 3 ms (D series) or 10 ms (P series) were used. Relaxation measurements were performed with two different field cycles (Kimmich and Anoardo, 2004): the prepolarized cycle below 10 MHz (D series) or 4 MHz (P series) and the nonpolarized cycle above 4 MHz (P series). In either case, 20 (D series) or 15 (P series) different relaxation delays (evolution times) were used. The magnitude of the quadrature-detected signal after a 90° pulse was recorded with a four-step phase cycle. For the P series, the signal was averaged over 32 transients. The relaxation curves were invariably single exponential. The accuracy (mean ± SD of 1) of R1 determined by the FC technique is estimated to 1% (D series) or 1–2% (P series).
Analysis of relaxation dispersion data
The measured 1H relaxation rate is due to thermal fluctuations of intramolecular and intermolecular magnetic dipole-dipole couplings experienced by water protons and labile macromolecular protons in fast or intermediate exchange (residence time < 10 ms, typically) with the water protons (Venu et al., 1997; Halle et al., 1999). The relaxation dispersion, i.e., the frequency dependence of R1, is produced by protons in long-lived (residence time 10−9 − 10−2 s) association with a macromolecule. Such protons belong either to water molecules trapped within the macromolecule or to solvent-exposed hydroxyl, carboxyl, ammonium, or other groups in the macromolecule that engage in rapid proton exchange with water.
Under fast-exchange conditions, the relaxation rate measured on a dextran solution can be expressed as a population-weighted average:
 |
(1) |
where 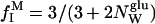 is the fraction of the observed protons that reside in dextran hydroxyl groups (three per glucose unit) and
is the fraction of the observed protons that reside in dextran hydroxyl groups (three per glucose unit) and  is the fraction of the observed protons that reside in water molecules in contact with (and thus significantly dynamically perturbed by) dextran. (The number,
is the fraction of the observed protons that reside in water molecules in contact with (and thus significantly dynamically perturbed by) dextran. (The number, 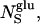 of such water molecules is ∼6 per glucose unit (Uedaira et al., 1989).) Further,
of such water molecules is ∼6 per glucose unit (Uedaira et al., 1989).) Further,  is the relaxation rate of oxygen-free bulk water (0.27 s−1 at 27 C; Hindman et al., 1973),
is the relaxation rate of oxygen-free bulk water (0.27 s−1 at 27 C; Hindman et al., 1973), 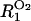 is the paramagnetic relaxation enhancement from dissolved oxygen (0.1 s−1 for water in equilibrium with air; Teng et al., 2001), and
is the paramagnetic relaxation enhancement from dissolved oxygen (0.1 s−1 for water in equilibrium with air; Teng et al., 2001), and 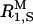 and
and 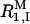 are, respectively, the intrinsic relaxation rates of protons in water molecules hydrating dextran and in the hydroxyl groups of dextran.
are, respectively, the intrinsic relaxation rates of protons in water molecules hydrating dextran and in the hydroxyl groups of dextran.
For a solution containing dextran as well as BPTI, the relaxation rate contains additional contributions from internal water molecules (four per BPTI molecule; Venu et al., 1997) and labile BPTI protons (at pH 4.5, mainly the eight hydroxyl groups), with proton fraction 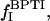 and from water molecules in the hydration layer of BPTI, with proton fraction
and from water molecules in the hydration layer of BPTI, with proton fraction 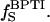 Thus,
Thus,
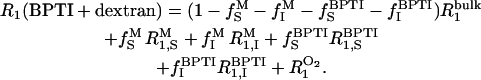 |
(2) |
At sufficiently low dextran concentrations, we can assume that the dextran contribution is unaffected by the protein. Also the small oxygen contribution should be the same as without protein. These contributions then cancel out in the difference of the relaxation rates measured on the (BPTI + dextran) sample, henceforth denoted Pn, and on the dextran sample, henceforth denoted Dn, with the same water/glucose mol ratio (see Table 1). Thus,
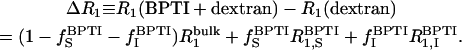 |
(3) |
The frequency dependence of the difference relaxation rate, 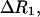 is produced by the last term in Eq. 3. If the residence times of all protons contributing to this term are long compared to the rotational correlation time,
is produced by the last term in Eq. 3. If the residence times of all protons contributing to this term are long compared to the rotational correlation time, 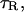 of the protein, as is the case for BPTI (Denisov et al., 1995, 1996; Venu et al., 1997; Denisov and Halle, 2002), then the difference dispersion profile,
of the protein, as is the case for BPTI (Denisov et al., 1995, 1996; Venu et al., 1997; Denisov and Halle, 2002), then the difference dispersion profile,  from a solution containing BPTI monomers (A) and decamers (B) is described by the following relations (Venu et al., 1997; Halle et al., 1999):
from a solution containing BPTI monomers (A) and decamers (B) is described by the following relations (Venu et al., 1997; Halle et al., 1999):
 |
(4) |
 |
(5) |
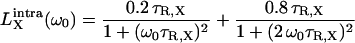 |
(6) |
 |
(7) |
Here, □X is the mean-square fluctuation amplitude and τR,X is the rank-two rotational correlation time associated with BPTI oligomer X (X = A for monomer and X = B for decamer). Furthermore, ξX = bX,inter / bX is the relative contribution from intermolecular dipole-dipole couplings to the overall fluctuation amplitude, bX = bX,intra + bX,inter. The functions 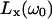 will be referred to as Lorentzians, even though they are, in fact, linear combinations of two Lorentzian (reduced) spectral density functions differing by a factor 2 in frequency. Apart from an overall scaling by a factor 0.9, the functions
will be referred to as Lorentzians, even though they are, in fact, linear combinations of two Lorentzian (reduced) spectral density functions differing by a factor 2 in frequency. Apart from an overall scaling by a factor 0.9, the functions 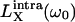 and
and 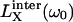 differ very little (Venu et al., 1997). The value of ξX therefore has no significant effect on the oligomer fractions that we deduce from the data. We set ξX = 0.33, as previously found for the four internal water molecules in BPTI (Venu et al., 1997). The quantity α in Eq. 4 represents all frequency-independent contributions to ΔR1, including the secular (zero-frequency) intermolecular contribution (Venu et al., 1997).
differ very little (Venu et al., 1997). The value of ξX therefore has no significant effect on the oligomer fractions that we deduce from the data. We set ξX = 0.33, as previously found for the four internal water molecules in BPTI (Venu et al., 1997). The quantity α in Eq. 4 represents all frequency-independent contributions to ΔR1, including the secular (zero-frequency) intermolecular contribution (Venu et al., 1997).
The experimental difference dispersion data were subjected to nonlinear Marquardt-Levenberg χ2 minimization (Press et al., 1992) with the model function given by Eqs. 4–7 and with the products bX τR,X constrained to be nonnegative. At the two highest dextran concentrations (samples P4 and P5), it was necessary to include a third (larger) oligomeric species, as found previously at high salt concentrations (Gottschalk et al., 2003a). All five difference dispersions (samples P1–P5) plus the dispersion from the dextran-free BPTI solution (sample P0) were fitted jointly with the correlation times τR,A and τR,B constrained to have the same values for all samples. The amplitude parameters bX obtained from the fit can be expressed as (Gottschalk et al., 2003a):
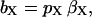 |
(8) |
where pX is the fraction of BPTI molecules that belongs to oligomer species X or, equivalently, the weight fraction of that species. The intrinsic mean-square fluctuation amplitude βX is proportional to the number of protons (per BPTI monomer) with residence times long enough (>τR,X) to sample the rotational diffusion of the oligomer but short enough (<(βX τR,X)−1) to act as a relaxation sink for the observed water 1H magnetization (Halle et al., 1999). A previous MRD study of salt-induced BPTI decamer formation established that, to a good approximation, βA = βB (Gottschalk et al., 2003a). This is, indeed, expected because: 1), the four internal water molecules in the BPTI monomer are conserved in the decamer; 2), the two small, predominantly nonpolar, cavities formed at the intermolecular contacts in the decamer appear to be empty; 3), the central channel in the decamer is too wide to provide the geometric constraints necessary for long-lived hydration; and 4), most of the hydroxyl and carboxyl protons that contribute to the dispersion at pH 4.5 are fully exposed in the decamer (Wlodawer et al., 1987; Lubkowski and Wlodawer, 1999; Hamiaux et al., 1999, 2000; Gottschalk et al., 2003a). The oligomer fractions, which must sum to unity, can then be obtained as
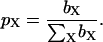 |
(9) |
Quoted uncertainties in the fitted parameter values correspond to one standard deviation and were obtained by the Monte Carlo method (Press et al., 1992) using 1000 synthetic data sets.
Modeling of the excluded-volume effect
The model used to describe the effect of dextran on the BPTI monomer-decamer equilibrium is outlined in the following Theory section. Here we provide further details about calculations, fits, and choice of geometric parameters.
The decamer fraction, pB, was calculated self-consistently with the aid of Eqs. 11, 18, and 20–22. For given values of the experimental variables, φBPTI (or CBPTI) and φM, and of the parameters, K0, RA, RB, RM, LM, and λM, we use the following iterative scheme:
Set lnΓ = 0 initially (ideal solution).
Substitute K0, CBPTI, and Γ into Eq. 11 and solve for pB.
Calculate ΔbBPTI and ΔbM from Eqs. 18, 21, and 22, using pB and geometric parameters.
Calculate lnΓ from Eq. 20.
Repeat from step 2 until lnΓ and pB converge to desired accuracy.
In the analysis of the experimentally determined decamer fraction, pB, at different dextran volume fractions, φM, we implement this self-consistent scheme in a nonlinear least-squares fit, with K0 and λM as adjustable parameters. In the calculation of pB, we take into account the slight variation of the BPTI volume fraction φBPTI (and concentration CBPTI) resulting from dilution by added dextran (see Table 1).
The values of the geometric parameters needed to calculate the crowding effect on pB were assigned as follows. The radii RX (X = A, B, or M) were obtained by adding a distance δ/2 to the bare radius, 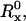 deduced from the macromolecular volume:
deduced from the macromolecular volume:
 |
(10) |
The parameter-δ models solvent-mediated short-range repulsion between the macromolecules. We use a default value of δ = 3.0 Å, corresponding to one water layer. This appears to be consistent with thermodynamic data on the preferential solvation of proteins in glucose/water mixtures (Shimizu and Smith, 2004) and with the linearity of the water 17O magnetic relaxation rate up to very high glucose concentrations (at least 20% by volume) (Uedaira et al., 1989).
The radius of a sphere with the same volume as the BPTI monomer, with molar mass 6.50 kg mol−1 and partial specific volume 0.720 mL g−1 (Filfil et al., 2004), is  The BPTI decamer is compact, with a high degree of shape complementarity at the monomer interfaces (Lubkowski and Wlodawer, 1999; Hamiaux et al., 1999, 2000). However, it is pierced by a 10–15 Å wide central channel, with a volume comparable to that of a BPTI monomer. We therefore set VB = 12 VA, corresponding to
The BPTI decamer is compact, with a high degree of shape complementarity at the monomer interfaces (Lubkowski and Wlodawer, 1999; Hamiaux et al., 1999, 2000). However, it is pierced by a 10–15 Å wide central channel, with a volume comparable to that of a BPTI monomer. We therefore set VB = 12 VA, corresponding to  The α-(16)-d-linked glucose monomer of dextran is modeled as a cylinder of length lM = 4.4 Å (Marszalek et al., 1998). The volume,
The α-(16)-d-linked glucose monomer of dextran is modeled as a cylinder of length lM = 4.4 Å (Marszalek et al., 1998). The volume, 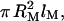 is obtained from the molar mass of the glucose residue, 162.1 g mol−1, and the partial specific volume of dextran, 0.611 mL g−1 (Granath, 1958), yielding
is obtained from the molar mass of the glucose residue, 162.1 g mol−1, and the partial specific volume of dextran, 0.611 mL g−1 (Granath, 1958), yielding 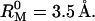 The contour length of the dextran molecule is
The contour length of the dextran molecule is 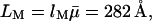 with
with 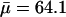 the average degree of polymerization (see above). Finally, the Kuhn length λM is regarded as an adjustable parameter. As expected for the flexible α-(16)-d-glycosidic linkage in dextran, we find that λM is an order of magnitude shorter than LM, as assumed in the derivation of Eq. 17.
the average degree of polymerization (see above). Finally, the Kuhn length λM is regarded as an adjustable parameter. As expected for the flexible α-(16)-d-glycosidic linkage in dextran, we find that λM is an order of magnitude shorter than LM, as assumed in the derivation of Eq. 17.
THEORY
Analytical treatments of solution nonideality resulting from excluded volume can be based either on the virial expansion (McMillan and Mayer, 1945; Zimm, 1946; Kihara, 1953) or on scaled particle theory (Reiss et al., 1959; Boublík, 1974), whereas numerical treatments make use of simulation techniques or density functional theory (Kinjo and Takada, 2002). Here, we adopt the virial expansion approach, which (before truncation) is formally exact and which, furthermore, allows the effect of polymer flexibility to be included in a simple way.
The stoichiometric association constant for the monomer-decamer equilibrium, 10 A B, can be expressed as
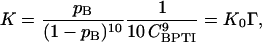 |
(11) |
where CBPTI is the total BPTI concentration (see Table 1) and K0 is the dimensionless “ideal” association constant. The excluded-volume effect on the monomer-decamer equilibrium is described by the crowding factor Γ, which is related to the monomer and decamer activity coefficients through (Minton, 1998)
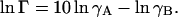 |
(12) |
According to McMillan-Mayer solution theory, the activity coefficients may be expressed in terms of a virial expansion of the form (Hill, 1986)
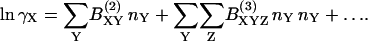 |
(13) |
In our case, the summation indices run over three species: BPTI monomer (A) and decamer (B) and dextran (M). We assume that the number densities nA, nB, and nM are sufficiently low that we can truncate the expansion after the first term, involving the second virial coefficient BXY (we henceforth omit the superscript). Furthermore, we assume that the interactions among the three species are short ranged and can be approximated by hard repulsions. The second virial coefficient, BXY, is then simply the covolume, VXY, i.e., the volume excluded by molecule X to the center of molecule Y (or vice versa), averaged over all relative orientations of the two molecules. We thus obtain from Eqs. 12 and 13:
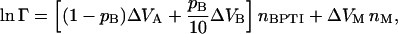 |
(14) |
where
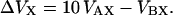 |
(15) |
To calculate the covolumes VXY, we model the BPTI monomer and decamer as spheres of radii RA and RB and the dextran polymer as a flexible cylinder of radius RM, contour length LM, and statistical segment length (or Kuhn length) λM. The sphere-sphere covolumes VAA, VAB, and VBB are simply
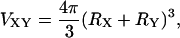 |
(16) |
whereas the sphere-polymer covolumes VAM and VBM are given by (Lue, 1998)
 |
(17) |
where X = A or B and
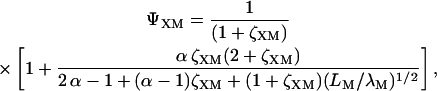 |
(18) |
with 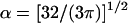 and
and
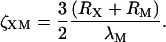 |
(19) |
Equation 17 is a rational fraction approximation that interpolates between the more well-known rod limit (Ogston, 1970) ζXM ≪ 1, where ΨXM = 1, and the random-coil limit (Jansons and Phillips, 1990) ζXM ≫ 1, where 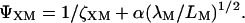 This approximate formula compares favorably with Monte Carlo calculations of the covolume (Lue, 1998). Although Eq. 17 is valid for any ratio of the sphere radius, RX, to the radius of gyration, RG, of the polymer, it assumes that the polymer is sufficiently long that LM ≫ λM (as is the case for our dextran). The original derivation of Eq. 17 (Lue, 1998) and its random-coil limit (Jansons and Phillips, 1990) pertain to a polymer of vanishing thickness (RM = 0). In Eqs. 17–19, we have incorporated the finite thickness of the polymer chain simply by replacing RX with the distance of closest approach, RX + RM (Ogston, 1958).
This approximate formula compares favorably with Monte Carlo calculations of the covolume (Lue, 1998). Although Eq. 17 is valid for any ratio of the sphere radius, RX, to the radius of gyration, RG, of the polymer, it assumes that the polymer is sufficiently long that LM ≫ λM (as is the case for our dextran). The original derivation of Eq. 17 (Lue, 1998) and its random-coil limit (Jansons and Phillips, 1990) pertain to a polymer of vanishing thickness (RM = 0). In Eqs. 17–19, we have incorporated the finite thickness of the polymer chain simply by replacing RX with the distance of closest approach, RX + RM (Ogston, 1958).
Combination of Eqs. 14–17 now yields
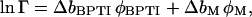 |
(20) |
where φBPTI and φM are the BPTI and dextran volume fractions (see Table 1) and
 |
(21) |
 |
(22) |
The first term in Eq. 20 represents the nonideality contribution to the BPTI monomer-decamer equilibrium from the excluded volume of the protein itself. This contribution is often neglected, but it is significant at the relatively high protein concentration used here. Because the protein contribution to lnΓ depends on the decamer fraction pB, the latter must be calculated self-consistently (see Materials and Methods).
RESULTS AND DISCUSSION
Magnetic relaxation dispersion from dextran solutions
Fig. 1 shows water 1H MRD profiles recorded from five dextran solutions at pH 4.5, 27.0 C, and dextran volume fractions φM in the range 0.057–0.227 (samples D1–D5 in Table 1). Because dextran is highly flexible and unstructured, we do not expect any long-lived water molecules in these samples. Apart from a minor contribution from paramagnetic O2 (Teng et al., 2001), the observed dispersions can therefore be attributed to the three labile hydroxyl protons per glucose residue in dextran. On the basis of previously reported 1H CPMG T2 dispersions from dextran (Hills et al., 1991) and glucose (Hills, 1991) solutions, we estimate that the mean hydroxyl proton residence time is τH ≈ 0.5 ms under our conditions. Because τH is two orders of magnitude shorter than the zero-frequency intrinsic 1H relaxation time of the hydroxyl protons, estimated as 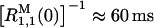 from Eq. 1 and the data in Fig. 1, we are in the fast-exchange regime, as assumed in Eq. 1. This conclusion is corroborated by complete 1H MRD profiles of sample D2 recorded at pH 3.0 and 6.0 (data not shown).
from Eq. 1 and the data in Fig. 1, we are in the fast-exchange regime, as assumed in Eq. 1. This conclusion is corroborated by complete 1H MRD profiles of sample D2 recorded at pH 3.0 and 6.0 (data not shown).
FIGURE 1.

1H relaxation dispersion profiles from aqueous solutions of dextran at pH 4.5 and 27°C. The dextran concentrations for samples D1–D5 are given in Table 1. The curves were obtained by three-Lorentzian fits as described in the text.
The spectral density function for the hydroxyl protons in dextran is expected to have a complicated form (Tylianakis et al., 1999; Dejean de la Batie et al., 1988), including contributions from various dipole-dipole couplings (glucose CH protons at 2–3 Å as well as H-bonded water molecules) and various motions (bond librations on 10-ps timescale, localized and cooperative conformational motions in the dextran chain on 1–10-ns timescale, and local-global reorientation modes on timescale 10–100 ns). Furthermore, because the high-frequency plateau in the MRD profile is not well defined (see Fig. 1) and because the zero-frequency contribution from the intermolecular (OH–CH) part of 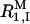 is unknown, it is not possible to determine
is unknown, it is not possible to determine 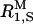 (see Eq. 1) and thereby extract information about dextran hydration dynamics. However, our aim here is not to characterize polymer or water dynamics in dextran solutions. We merely wish to subtract the dextran contribution from the MRD profiles measured on mixed BPTI/dextran solutions. For this purpose, each of the dextran dispersions in Fig. 1 was represented by a three-Lorentzian spectral density function, as in Eqs. 4–6 with ξX = 0 and one additional Lorentzian. With the aid of the six parameters obtained from each fit (three correlation times τk, three associated amplitudes bk, and a frequency-independent contribution α), the dextran contribution to R1 can be obtained at any frequency in the range from 10 kHz to 200 MHz.
(see Eq. 1) and thereby extract information about dextran hydration dynamics. However, our aim here is not to characterize polymer or water dynamics in dextran solutions. We merely wish to subtract the dextran contribution from the MRD profiles measured on mixed BPTI/dextran solutions. For this purpose, each of the dextran dispersions in Fig. 1 was represented by a three-Lorentzian spectral density function, as in Eqs. 4–6 with ξX = 0 and one additional Lorentzian. With the aid of the six parameters obtained from each fit (three correlation times τk, three associated amplitudes bk, and a frequency-independent contribution α), the dextran contribution to R1 can be obtained at any frequency in the range from 10 kHz to 200 MHz.
If the dextran solutions are sufficiently dilute, the correlation times τk should be independent of dextran concentration, whereas the amplitude parameters βk should be proportional to the fraction  of the observed protons that reside in dextran hydroxyl groups. If the small O2 contribution is neglected, it then follows from Eq. 1 (because
of the observed protons that reside in dextran hydroxyl groups. If the small O2 contribution is neglected, it then follows from Eq. 1 (because  is proportional to
is proportional to 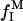 ) that the excess relaxation rate
) that the excess relaxation rate 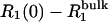 should be proportional to
should be proportional to  As seen from Fig. 2, this is the case for samples D1 and D2, whereas the two most concentrated dextran solutions, D4 and D5, show substantial deviations from linearity.
As seen from Fig. 2, this is the case for samples D1 and D2, whereas the two most concentrated dextran solutions, D4 and D5, show substantial deviations from linearity.
FIGURE 2.

Dependence of the zero-frequency excess 1H relaxation rate, 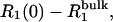 in dextran solutions D1–D5 on the dextran hydroxyl proton fraction,
in dextran solutions D1–D5 on the dextran hydroxyl proton fraction, 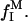 The bulk water relaxation rate at 27°C is
The bulk water relaxation rate at 27°C is 
In dilute aqueous solution, dextran behaves as an unstructured, flexible random coil (Nordmeier, 1993; Ioan et al., 2000). The size of the coil may be characterized by the (root-mean square) radius of gyration, RG. The mean center-to-center separation of adjacent dextran molecules, denoted by d, is given for our samples in Table 1. The dextran solution is said to be dilute if d > 2RG, so that adjacent polymer coils do not interpenetrate extensively. If d is smaller, the coils overlap and the solution is said to be semidilute. The crossover from dilute to semidilute occurs at the overlap concentration, φ*, where the polymers, regarded as spheres of radius RG, are close packed (on a face-centered cubic lattice). Thus,
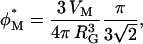 |
(23) |
where the first factor is the dextran volume fraction within a sphere of radius RG and the second factor is the volume fraction of such spheres at close packing. The radius of gyration has been determined by static light scattering for dextran preparations of higher mean molecular mass than used here, e.g., RG = 86 Å for 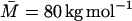 (corresponding to a contour length, LM = 2170 Å) (Nordmeier, 1993) and RG = 120 Å for
(corresponding to a contour length, LM = 2170 Å) (Nordmeier, 1993) and RG = 120 Å for 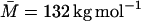 (LM = 3580 Å) (Ioan et al., 2000). For the freely jointed chain model (Flory, 1969),
(LM = 3580 Å) (Ioan et al., 2000). For the freely jointed chain model (Flory, 1969),
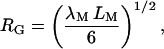 |
(24) |
where λM is the Kuhn length. The radii of gyration measured by light scattering thus correspond to λM = 20 and 24 Å. Shorter values of the Kuhn length for dextran have been deduced from hydrodynamic data, λM = 13 Å (Pavlov et al., 1999; and references cited therein), and from single molecule force measurements, λM = 8 Å (Rief et al., 1998) and λM = 4.4 Å (Marszalek et al., 1998). The smallest of these values, equal to the residue length bM (see above), seems unphysically short; there are surely some steric constraints between adjacent glucose residues. For the following estimates of RG, we use the value λM = 15 Å.
For the 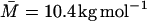 (LM = 282 Å) dextran preparation used for our MRD experiments, Eq. 24 with λM = 15 Å yields RG = 26.5 Å. Inserted into Eq. 23, this yields
(LM = 282 Å) dextran preparation used for our MRD experiments, Eq. 24 with λM = 15 Å yields RG = 26.5 Å. Inserted into Eq. 23, this yields  Comparing with the φM and d values in Table 1, we conclude that samples D1 and D2 are dilute, whereas samples D3–D5 are semidilute. This conclusion is consistent with the experimental findings reported in Fig. 2, where deviations from linearity, signaling overlap of dextran coils, are evident for samples D3–D5. Our analysis is also consistent with previously reported (Tylianakis et al., 1999) 13C relaxation data from solutions of 35 kg mol−1 dextran (RG = 49 Å according to Eq. 24 with λM = 15 Å), which showed no concentration dependence up to 10% (w/v) dextran (corresponding to d = 94 Å).
Comparing with the φM and d values in Table 1, we conclude that samples D1 and D2 are dilute, whereas samples D3–D5 are semidilute. This conclusion is consistent with the experimental findings reported in Fig. 2, where deviations from linearity, signaling overlap of dextran coils, are evident for samples D3–D5. Our analysis is also consistent with previously reported (Tylianakis et al., 1999) 13C relaxation data from solutions of 35 kg mol−1 dextran (RG = 49 Å according to Eq. 24 with λM = 15 Å), which showed no concentration dependence up to 10% (w/v) dextran (corresponding to d = 94 Å).
Magnetic relaxation dispersion from BPTI/dextran solutions
1H MRD profiles were recorded from six BPTI solutions at pH 4.5 with varying amounts of dextran added. The dextran volume fractions φM was in the range 0–0.214 (samples P0–P5 in Table 1). In the dextran-free protein solution, only a small fraction of the BPTI molecules are expected to form decamers (Gottschalk et al., 2003a). On addition of dextran, the magnitude of the dispersion step increases markedly. The origin of this increase is twofold. First, there is a direct contribution to the dispersion from the rapidly exchanging hydroxyl protons of dextran (see Fig. 1). Second, there is an indirect effect of macromolecular crowding, whereby the configurational entropy of the dextran molecules shifts the BPTI self-association equilibrium toward the decamer, which produces a much larger dispersion than 10 monomers. To isolate the crowding effect, we subtract the relaxation rate from the corresponding protein-free dextran sample, forming the difference ΔR1 = R1(BPTI+dextran) − R1(dextran). This correction will remove the dextran contribution to R1(BPTI+dextran) provided that two conditions are met. First, the water/glucose mol ratio must be the same in the (BPTI+dextran) sample and in the dextran sample. This is the case here (see Table 1). Second, the dextran relaxation contribution must be unaffected by the protein. This is likely to be the case if the dextran concentration is so low that the polymer coils do not overlap (dilute regime) and if the protein does not interact specifically with dextran. As discussed above, we expect these conditions to be met for samples P1 and P2, but not for samples P4 and P5 (sample P3 is a border-line case).
If the direct dextran contribution has been completely removed, the difference dispersion ΔR1 only reflects labile protons and internal water molecules in BPTI (see Eq. 3). For samples P0–P3, the data are well described by a two-Lorentzian dispersion law as in Eqs. 4–7. For samples P4 and P5, a third Lorentzian component was required. Fig. 3 shows the dispersion from sample P0 and the difference dispersions from samples P1–P5. To exhibit the crowding effect more clearly, we display the normalized rate,
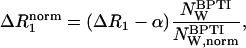 |
(25) |
with  This normalization corrects for the variation of the water/BPTI mol ratio,
This normalization corrects for the variation of the water/BPTI mol ratio, 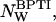 with increasing dextran concentration (see Table 1).
with increasing dextran concentration (see Table 1).
FIGURE 3.

1H difference relaxation dispersion profiles from aqueous solutions of BPTI and dextran at pH 4.5 and 27°C. BPTI and dextran concentrations for samples P0–P5 are given in Table 1. The curves were obtained by two-Lorentzian (samples P0–P3) or three-Lorentzian (samples p4 and p5) fits as described in the text. The data have been normalized to a water/BPTI mol ratio of 
The six dispersions in Fig. 3 were fitted jointly with two (samples P0–P3) or three (samples P4 and P5) Lorentzians and with the two shortest correlation times in common for all samples. In all, 24 parameters were thus fitted to 226 data points. These fits are displayed in Fig. 3. The two common correlation times came out as τ1 = 4.3 ± 0.1 ns and τ2 = 27.0 ± 0.5 ns, close to the values, τ1 = 3.3 ns and τ2 = 26.3 ns, expected for the BPTI monomer and decamer in dilute H2O solution at 27°C and pH 4.5 (Gottschalk et al., 2003a). The slight slowing down of protein tumbling is attributed to dextran (see below), which should have a larger effect on the somewhat elongated monomer (rotational diffusion anisotropy 1.28) than for the nearly spherical decamer (rotational diffusion anisotropy 1.08) (Gottschalk et al., 2003a). The third correlation time obtained from the fit was τ3 = 89 ± 5 ns and 147 ± 5 ns for samples P4 and P5, respectively. This is in the same range as observed in previous work (at high salt concentration), where it was attributed to loose clusters of a few decamers (Gottschalk et al., 2003a).
A macromolecular crowding agent, such as dextran, can affect protein rotational diffusion by indirect hydrodynamic interactions as well as by direct potential-derived interactions. Hydrodynamic theory (Cichocki et al., 1999) and Stokesian dynamics simulations (Phillips et al., 1988) yield τR(φ) = τR(0)/[1 − 0.6310 φ − 0.726 φ2 − 0.52 φ3] for a suspension of equal-sized hard spheres at volume fraction φ. For φ = 0.25, this becomes τR(φ) = 1.27τR(0). This modest (27%) retardation is essentially due to configurations where two spheres are very close to contact. In fact, a dominant contribution comes from separations comparable to or less than the size of a water molecule. At such short separations, the validity of the hydrodynamic continuum description may be questioned. If such contact configurations are not present, e.g., because of the energetic cost of removing the first hydration layer from the macromolecules, then the hydrodynamic retardation effect is negligibly small even at high volume fractions (of spherical particles) (Watzlawek and Nägele, 1997). In the presence of soft repulsive forces, the hydrodynamic retardation effect is greatly diminished (Watzlawek and Nägele, 1997). On the other hand, the dextran coils are not compact and therefore permeate space more uniformly than compact spheres. At the higher dextran concentrations, some polysaccharide segments must therefore be near BPTI molecules and may thus account for the observed (modest) rotational retardation.
Fluorescence anisotropy measurements of protein rotational diffusion in the presence of macromolecular crowding agents have shown stronger retardation effects than expected from hydrodynamic interactions alone, e.g., τR(φ)/τR(0) ≈ 5 for green fluorescent protein in the presence of dextran at φ = 0.25 (Swaminathan et al., 1997) and τR(φ)/τR(0) ≈ 1.4 (monomer) or 2.5 (flexible dimer) for apomyoglobin in the presence of ribonuclease A at φ = 0.18 (Zorrilla et al., 2004b). These larger retardation factors may be attributed to attractive interactions between the reference protein and the crowding agent. The smaller retardation factor of ∼1.3 for the BPTI monomer and even less for the BPTI decamer (see above) obtained from our MRD data indicate that the BPTI-dextran interaction is essentially repulsive. In conclusion, whereas a large rotational retardation, as observed in some other systems (Swaminathan et al., 1997; Zorrilla et al., 2004b), is incompatible with these MRD data, a modest retardation has little effect on the data analysis. Fits with the two correlation times τ1 and τ2 frozen to their dilute-solution values thus gave essentially the same oligomer populations as obtained when τ1 and τ2 were freely adjustable parameters.
The third correlation time was τ3 = 89 ± 5 ns and 147 ± 5 ns for samples 4 and 5, respectively. This is in the same range as observed in previous work (at high salt concentration), where it was attributed to loose clusters of a few decamers (Gottschalk et al., 2003a).
The oligomer fractions, pX, derived from the corresponding dispersion amplitudes, bX, with the aid of Eq. 9, are given in Table 2. The BPTI decamer fraction is only 1.4% in the absence of dextran, but it increases greatly as dextran is added, reaching 40% for sample P3 at a dextran volume fraction of only 0.14 (see Table 1). For samples P4 and P5, the monomer fraction appears to increase with increasing dextran concentration. We believe that this anomalous behavior is related to the unexpected large increase of the amplitude sum ΣX bX = B in going from sample P3 to sample P4 (see Table 2). If the number of long-lived water molecules and labile protons that contribute to the dispersion does not change on self-association of BPTI, then B should be independent of the oligomer fractions. A previous study of salt-induced BPTI decamer formation indicates that this is the case, with B = (1.1–1.3)108 s−2 (Gottschalk et al., 2003a). The B values of samples P0–P2 are essentially within this range, whereas sample P3 deviates somewhat and samples P4 and P5 have about threefold larger B values.
TABLE 2.
BPTI oligomer fractions derived from MRD data
| Sample | 100 pA | 100 pB | 100 pC | Σ bX (108 s−2) |
|---|---|---|---|---|
| P0 | 98.6 ± 0.3 | 1.4 ± 0.3 | – | 0.93 ± 0.02 |
| P1 | 89.0 ± 0.5 | 11.0 ± 0.5 | – | 1.05 ± 0.02 |
| P2 | 73.2 ± 0.9 | 26.8 ± 0.9 | – | 1.29 ± 0.03 |
| P3 | 60.5 ± 1.3 | 39.5 ± 1.3 | – | 1.60 ± 0.05 |
| P4 | 69.7 ± 1.5 | 21.9 ± 1.4 | 8.5 ± 0.8 | 3.02 ± 0.09 |
| P5 | 74.1 ± 0.9 | 19.2 ± 0.8 | 6.8 ± 0.3 | 3.85 ± 0.09 |
Two different scenarios might explain the anomalous behavior of samples P4 and P5. One possibility is that the structure and dynamics of dextran, in the semidilute regime, are significantly perturbed by the protein. In that case, the additivity assumption (see Materials and Methods) would break down so that the direct dextran contribution to R1(BPTI + dextran) would not be completely removed by subtracting R1(dextran) (see Eq. 3). This explanation is consistent with the strong deviations from linearity in Fig. 2 for samples D4 and D5, suggesting a slowing down of chain dynamics on confinement of the polymer coils. Addition of a relatively large amount (φBPTI ≈ 0.06) of charged protein molecules (along with counterions) to these semidilute dextran solutions might amplify this effect. Another possibility is that dextran affects the structure and dynamics of BPTI in other ways than shifting the monomer-decamer equilibrium. The solubility of BPTI decreases with increasing dextran concentration (Laurent, 1963) and it is therefore conceivable that a fraction of the protein in samples P4 and P5 exists in the form of microaggregates. Either of these two scenarios may be supported by the observation that samples P4 and P5 had a markedly higher viscosity than samples P0–P3 and by the finding that dextran forms a gel at high KCl concentrations (Naji et al., 2003). Further experimental work would be needed to resolve this issue. In the following analysis of the crowding effect, we will only consider the results obtained with samples P0–P3.
Quantitative analysis of macromolecular crowding
We shall now attempt to rationalize the observed dependence of the BPTI decamer fraction, pB, on the dextran volume fraction, φM, in terms of statistical-mechanical theory. As described in the Theory section, we use the second-virial approximation and a hard-particle interaction model (only short-range repulsion) to express the nonideality factor Γ = K/K0 (see Eq. 11) in terms of the covolumes VXY, with X = A or B and Y = A, B, or M. To calculate the covolume, we model the BPTI monomer and decamer as spheres of radius RA and RB and the dextran as a freely jointed chain polymer with radius RM, contour length 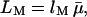 and Kuhn length λM. To model the additional repulsion due to desolvation, we take the distance of closest approach between the centers of two molecular species (A, B, or M) to be the sum of their anhydrous radii
and Kuhn length λM. To model the additional repulsion due to desolvation, we take the distance of closest approach between the centers of two molecular species (A, B, or M) to be the sum of their anhydrous radii  (deduced from the known molecular dimensions) and a solvent layer of thickness δ (see Materials and Methods).
(deduced from the known molecular dimensions) and a solvent layer of thickness δ (see Materials and Methods).
Our theoretical analysis differs from most earlier treatments of crowding effects on self-association equilibria in two respects. First, we incorporate the nonideality arising from the excluded volume of the protein itself. Because this effect depends on the oligomer fractions, the calculation must be done self-consistently (see Materials and Methods). For sample P1, where the volume fractions of BPTI and dextran are similar (see Table 1), BPTI and dextran make comparable contributions to the nonideality (see below). The second novel aspect of our analysis is that we model the dextran polymer as a flexible cylinder, rather than as a rigid rod (Ogston, 1970) or a random coil (Jansons and Phillips, 1990). The more general description (Lue, 1998) used here interpolates between these limits. Indeed, the parameter values, ζAM = 1.2 and ζBM = 2.2, resulting from our analysis imply that the investigated system is neither in the rod limit (ζXM ≪ 1) nor in the random-coil limit (ζXM ≫ 1).
Fig. 4 shows the result of fitting the model to the experimentally determined decamer fractions for samples P0–P3. The two adjustable parameters obtained from this fit are λM = 24 ± 3 Å and K0 = (1.0 ± 0.2)1012. As seen from Fig. 4, the model can account quantitatively for the observed crowding effect and the resulting Kuhn length is similar to the values that have been deduced from light-scattering studies of dextran solutions (Nordmeier, 1993; Ioan et al., 2000). As noted above, single-molecule force measurements (Rief et al., 1998; Marszalek et al., 1998), on the other hand, have suggested a much shorter (in our view, unphysically short) Kuhn length for dextran. The fit in Fig. 4 was performed with δ = 3.0 Å, corresponding to a monolayer of nondisplacable water for BPTI-BPTI and BPTI-dextran pairs at the distance of closest approach. Equally good fits were obtained with any δ-value in the range 0–4 Å. Fig. 5 shows how the values of the two adjustable parameters, λM and K0, depend on the fixed δ-value.
FIGURE 4.

The fraction, pB, of decamer-forming BPTI molecules as a function of dextran volume fraction, φM. The points were derived from the MRD data (samples P0–P3) and the curve resulted from a fit to the model described in the text (with δ = 3.0 Å). The dashed curve was calculated with the same model parameters, but without the nonideality contribution from BPTI.
FIGURE 5.

Variation of (A) the Kuhn length, λM, and (B) the natural logarithm of the ideal association constant, K0, with the thickness, δ, of the undisplacable water layer. The shaded regions correspond to one standard deviation in the fitted parameters.
Fig. 6 shows how the nonideality factor, Γ, varies with the dextran volume fraction. The quantity −RT ln Γ may be regarded as the crowding (excluded volume) contribution to the standard free energy of decamer formation for a standard-state BPTI concentration of CBPTI = 1 M. We may thus write
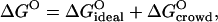 |
(26) |
where  (with K0 obtained from δ = 3 Å fit). The crowding contribution at φM = 0.14 (sample P3) is
(with K0 obtained from δ = 3 Å fit). The crowding contribution at φM = 0.14 (sample P3) is  (at the standard-state BPTI concentration of CBPTI = 1 M), corresponding to a 5.5105-fold increase of the association constant, K. For this sample, the protein itself contributes −1.4 kcal mol−1 to
(at the standard-state BPTI concentration of CBPTI = 1 M), corresponding to a 5.5105-fold increase of the association constant, K. For this sample, the protein itself contributes −1.4 kcal mol−1 to 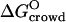 corresponding to a 10-fold increase of K. In view of Eq. 20, we may write
corresponding to a 10-fold increase of K. In view of Eq. 20, we may write
 |
(27) |
FIGURE 6.

Dependence of the natural logarithm of the nonideality factor, Γ, on the dextran volume fraction, φM. The solid curve was calculated from the model with the parameter values resulting from the fit in Fig. 4. The dashed curve was calculated with the same parameter values, but without the nonideality contribution from BPTI.
Within the adopted hard-particle model (where partial specific volumes are independent of temperature) and neglecting the weak temperature dependence of ΔbBPTI (via pB) and of ΔbM (via λM), this is a purely entropic contribution, i.e.,  related to the excess osmotic pressure.
related to the excess osmotic pressure.
It does not appear to be widely recognized among biochemists and cell biologists that the physical phenomenon underlying the macromolecular crowding effect on protein self-association has received considerable attention by colloid chemists (Kulkarni et al., 2000 and references cited therein). Fifty years ago, Asakura and Oosawa predicted that addition of an inert macromolecular species to a dilute solution of colloidal particles induces an effective attraction between the colloidal particles (Asakura and Oosawa, 1954, 1958). This so-called depletion attraction arises from an imbalance in the local osmotic pressure associated with configurations where the separation between two colloidal particles is too small to accommodate a macromolecule. In an alternative (and equivalent) view, the effective attraction between the colloidal particles is attributed to the increase in accessible volume, and hence configurational (translational) entropy, of the macromolecules as the two colloidal particles approach to contact. The depletion attraction is the origin of the effects described in this work.
CONCLUSIONS
Previous contributions from this laboratory have established field-cycling 1H MRD as a quantitative method for quantifying coexisting populations of protein oligomers in solution (Gottschalk et al., 2003a,b; Gottschalk and Halle, 2003). Here, we have demonstrated that the MRD technique also can be used to study protein self-association in the presence of a second macromolecular species. The ability of the MRD method to resolve the different oligomeric species on the basis of their rotational correlation times, with little or no influence of long-range (electrostatic or hydrodynamic) interactions is of particular importance in such studies. Provided that the protein and crowding agent contribute independently to the measured relaxation rate, the protein contribution can be isolated by a straight-forward difference experiment. The MRD method can therefore be used also when the crowding agent is another protein. On the other hand, by choosing a crowding agent, such as polyethylenene glycol, without labile protons or trapped water molecules, it is not necessary to perform a difference experiment because the dispersion is then produced exclusively by the self-associating protein.
The results reported here for the effect of dextran on the self-association of BPTI to decameric aggregates have been analyzed with the aid of a statistical-mechanical model that explicitly incorporates polymer flexibility as well as the excluded volume of the protein. This analysis shows that the observed dramatic enhancement of BPTI self-association can be quantitatively rationalized in terms of hard repulsions, with no need to invoke other interactions. Our results thus confirm the prediction that macromolecular crowding at physiologically relevant volume fractions is a potent modulator of self-association equilibria (Minton, 1981, 1998, 2000; Ellis, 2001) with potentially far-reaching biological implications (Garner and Burg, 1994; Walter and Brooks, 1995; Al-Habori, 2001; Hancock, 2004).
Acknowledgments
We thank Fabian Vaca Chavez, Michael Gottschalk, and Hans Lilja for experimental assistance, and Bayer Healthcare AG (Wuppertal, Germany) for a generous supply of BPTI.
This work was supported by the Swedish Research Council and the Wenner-Gren Foundations.
Karim Snoussi's present address is Biophysics Laboratories, School of Biological Sciences, St. Michael's Bldg., University of Portsmouth, White Swan Rd., Portsmouth PO1 2DT, UK.
References
- Albertsson, P.-Å. 1986. Partition of Cell Particles and Macromolecules. Wiley-Interscience, New York.
- Al-Habori, M. 2001. Macromolecular crowding and its role as intracellular signaling of cell volume regulation. Int. J. Biochem. Cell Biol. 33:844–864. [DOI] [PubMed] [Google Scholar]
- Asakura, S., and F. Oosawa. 1954. On interaction between two bodies immersed in a solution of macromolecules. J. Chem. Phys. 22:1255–1256. [Google Scholar]
- Asakura, S., and F. Oosawa. 1958. Interaction between particles suspended in solutions of macromolecules. J. Polym. Sci. [B]. 33:183–192. [Google Scholar]
- Atha, D. A., and K. C. Ingham. 1981. Mechanism of precipitation of proteins by polyethylene glycols. J. Biol. Chem. 256:12108–12117. [PubMed] [Google Scholar]
- Boublík, T. 1974. Statistical thermodynamics of convex molecule fluids. Mol. Phys. 27:1415–1427. [Google Scholar]
- Cichocki, B., M. L. Ekiel-Jezewska, and E. Wajnryb. 1999. Lubrication corrections for three-particle contribution to short-time self-diffusion coefficients in colloidal dispersions. J. Chem. Phys. 111:3265–3273. [Google Scholar]
- Dejean de la Batie, R., F. Lauprêtre, and L. Monnerie. 1988. Carbon-13 NMR investigation of local dynamics in bulk polymers at temperatures well above the glass transition temperature. 1. Poly(vinyl methyl ether). Macromolecules. 21:2045–2052. [Google Scholar]
- Denisov, V. P., and B. Halle. 2002. Hydrogen exchange rates in proteins from water 1H transverse magnetic relaxation. J. Am. Chem. Soc. 124:10264–10265. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., B. Halle, J. Peters, and H. D. Hörlein. 1995. Residence times of the buried water molecules in bovine pancreatic trypsin inhibitor and its G36S mutant. Biochemistry. 34:9046–9051. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., J. Peters, H. D. Hörlein, and B. Halle. 1996. Using buried water molecules to explore the energy landscape of proteins. Nat. Struct. Biol. 3:505–509. [DOI] [PubMed] [Google Scholar]
- Ellis, R. J. 2001. Macromolecular crowding: an important but neglected aspect of the intramolecular environment. Curr. Opin. Struct. Biol. 11:114–119. [DOI] [PubMed] [Google Scholar]
- Filfil, R., A. Ratavosi, and T. V. Chalikian. 2004. Binding of bovine pancreatic trypsin inhibitor to trypsinogen: spectroscopic and volumetric studies. Biochemistry. 43:1315–1322. [DOI] [PubMed] [Google Scholar]
- Flory, P. J. 1969. Statistical Mechanics of Chain Molecules. Interscience, New York.
- Garner, M. M., and M. B. Burg. 1994. Macromolecular crowding and confinement in cells exposed to hypertonicity. Am. J. Physiol. 266:C877–C892. [DOI] [PubMed] [Google Scholar]
- Gottschalk, M., and B. Halle. 2003. Self-association of lysozyme as seen by magnetic relaxation dispersion. J. Phys. Chem. B. 107:7914–7922. [Google Scholar]
- Gottschalk, M., H. Nilsson, H. Roos, and B. Halle. 2003b. Protein self-association in solution: the bovine β-lactoglobulin dimer and octamer. Protein Sci. 12:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk, M., K. Venu, and B. Halle. 2003a. Protein self-association in solution: the bovine pancreatic trypsin inhibitor decamer. Biophys. J. 84:3941–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granath, K. A. 1958. Solution properties of branched dextrans. J. Colloid Sci. 13:308–328. [Google Scholar]
- Halle, B., V. P. Denisov, and K. Venu. 1999. Multinuclear relaxation dispersion studies of protein hydration. In Biological Magnetic Resonance, Vol. 17. N. R. Krishna and L. J. Berliner, editors. Kluwer Academic/Plenum, New York. 419–484.
- Hamiaux, C., J. Pérez, T. Prangé, S. Veesler, M. Riès-Kautt, and P. Vachette. 2000. The BPTI decamer observed in acidic pH crystal forms pre-exists as a stable species in solution. J. Mol. Biol. 297:697–712. [DOI] [PubMed] [Google Scholar]
- Hamiaux, C., T. Prangé, M. Riès-Kautt, A. Ducruix, S. Lafont, J. P. Astier, and S. Veesler. 1999. The decameric structure of bovine pancreatic trypsin inhibitor (BPTI) crystallized from thiocyanate at 2.7 Å resolution. Acta Crystallogr. D. 55:103–113. [DOI] [PubMed] [Google Scholar]
- Hancock, R. 2004. A role for macromolecular crowding effects in the assembly and function of compartments in the nucleus. J. Struct. Biol. 146:281–290. [DOI] [PubMed] [Google Scholar]
- Hatters, D. M., A. P. Minton, and G. J. Howlett. 2002. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C–II. J. Biol. Chem. 277:7824–7830. [DOI] [PubMed] [Google Scholar]
- Hill, T. L. 1986. An Introduction to Statistical Thermodynamics. Dover, New York.
- Hills, B. P. 1991. Multinuclear NMR studies of water in solutions of simple carbohydrates. I. Proton and deuterium relaxation. Mol. Phys. 72:1099–1121. [Google Scholar]
- Hills, B. P., C. Cano, and P. S. Belton. 1991. Proton NMR relaxation studies of aqueous polysaccharide systems. Macromolecules. 24:2944–2950. [Google Scholar]
- Hindman, J. C., A. Svirmickas, and M. Wood. 1973. Relaxation processes in water. A study of the proton spin-lattice relaxation time. J. Chem. Phys. 59:1517–1522. [Google Scholar]
- Ioan, C. E., T. Aberle, and W. Burchard. 2000. Structure properties of dextran. 2. Dilute solution. Macromolecules. 33:5730–5739. [Google Scholar]
- Jansons, K. M., and C. G. Phillips. 1990. On the application of geometric probability theory to polymer networks and suspensions, I. J. Colloid Interface Sci. 137:75–91. [Google Scholar]
- Kihara, T. 1953. Virial coefficients and models of molecules in gases. Rev. Mod. Phys. 25:831–852. [Google Scholar]
- Kimmich, R., and E. Anoardo. 2004. Field-cycling NMR relaxometry. Prog. Nucl. Magn. Reson. Spectrosc. 44:257–320. [DOI] [PubMed] [Google Scholar]
- Kinjo, A. R., and S. Takada. 2002. Effects of macromolecular crowding on protein folding and aggregation studied by density functional theory: statics. Phys. Rev. E. 66:031911. [DOI] [PubMed] [Google Scholar]
- Kulkarni, A. M., A. P. Chatterjee, K. S. Schweizer, and C. F. Zukoski. 2000. Effects of polyethylene glycol on protein interactions. J. Chem. Phys. 113:9863–9873. [Google Scholar]
- Laurent, T. C. 1963. The interaction between polysaccharides and other macromolecules. Biochem. J. 89:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner, R., and G. Ralston. 1995. Effects of dextran on the self-association of human spectrin. Biophys. Chem. 57:15–25. [DOI] [PubMed] [Google Scholar]
- Lubkowski, J., and A. Wlodawer. 1999. Decamers observed in the crystals of bovine pancreatic trypsin inhibitor. Acta Crystallogr. D. 55:335–337. [DOI] [PubMed] [Google Scholar]
- Lue, L. 1998. An analytic formula for the excluded volume between a sphere and a freely jointed chain. J. Colloid Interface Sci. 202:558–561. [Google Scholar]
- Marszalek, P. E., A. F. Oberhauser, Y.-P. Pang, and J. M. Fernandez. 1998. Polysaccharide elasticity governed by chair-boat transitions of the glucopyranose ring. Nature. 396:661–664. [DOI] [PubMed] [Google Scholar]
- McMillan, W. G., and J. E. Mayer. 1945. The statistical thermodynamics of multicomponent systems. J. Chem. Phys. 13:276–305. [Google Scholar]
- McPherson, A. 1985. Use of polyethylene glycol in the crystallization of macromolecules. Methods Enzymol. 114:120–125. [DOI] [PubMed] [Google Scholar]
- Minton, A. P. 1981. Excluded volume as a determinant of macromolecular structure and reactivity. Biopolymers. 20:2093–2120. [Google Scholar]
- Minton, A. P. 1998. Macromolecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 295:127–149. [DOI] [PubMed] [Google Scholar]
- Minton, A. P. 2000. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 10:34–39. [DOI] [PubMed] [Google Scholar]
- Naji, L., J. Schiller, J. Kaufmann, F. Stallmach, J. Kärger, and K. Arnold. 2003. The gel-forming behaviour of dextran in the presence of KCl: a quantitative 13C and pulsed field gradient (PFG) NMR study. Biophys. Chem. 104:131–140. [DOI] [PubMed] [Google Scholar]
- Noack, F. 1986. NMR field-cycling spectroscopy: principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 18:171–276. [Google Scholar]
- Nordmeier, E. 1993. Static and dynamic light-scattering solution behavior of pullulan and dextran in comparison. J. Phys. Chem. 97:5770–5785. [Google Scholar]
- Ogston, A. G. 1958. The spaces in a uniform random suspension of fibres. Trans. Faraday Soc. 54:1754–1757. [Google Scholar]
- Ogston, A. G. 1970. On the interaction of solute molecules with porous networks. J. Phys. Chem. 74:668–669. [Google Scholar]
- Ogston, A. G., and C. F. Phelps. 1960. The partition of solutes between buffer solutions and solutions containing hyaluronic acid. Biochem. J. 78:827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov, G. M., A. E. Grishchenko, E. I. Rjumtsev, and N. P. Yevlampieva. 1999. Optical properties of dextran in solution and in films. Carbohydr. Polym. 38:267–271. [Google Scholar]
- Phillips, R. J., J. F. Brady, and G. Bossis. 1988. Hydrodynamic transport properties of hard-sphere dispersions. 1. Suspensions of freely mobile particles. Phys. Fluids. 31:3462–3472. [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling, and B. P. Flannery. 1992. Numerical Recipes in C, 2nd Ed. Cambridge University Press, Cambridge, UK.
- Qu, Y., and D. W. Bolen. 2002. Efficacy of macromolecular crowding in forcing proteins to fold. Biophys. Chem. 101–102:155–165. [DOI] [PubMed] [Google Scholar]
- Reiss, H., H. L. Frisch, and J. L. Lebowitz. 1959. Statistical mechanics of rigid spheres. J. Chem. Phys. 31:369–380. [Google Scholar]
- Rief, M., J. M. Fernandez, and H. E. Gaub. 1998. Elastically coupled two-level systems as a model for biopolymer extensibility. Phys. Rev. Lett. 81:4764–4767. [Google Scholar]
- Rivas, G., J. A. Fernandez, and A. P. Minton. 1999. Direct observation of the self-association of dilute proteins in the presence of inert macromolecules at high concentration via tracer sedimentation equilibrium: theory, experiment, and biological significance. Biochemistry. 38:9379–9388. [DOI] [PubMed] [Google Scholar]
- Rivas, G., J. A. Fernandez, and A. P. Minton. 2001. Direct observation of the enhancement of noncooperative protein self-assembly by macromolecular crowding: indefinite linear self-association of bacterial cell division protein FtsZ. Proc. Natl. Acad. Sci. USA. 98:3150–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahara, K., P. McPhie, and A. P. Minton. 2003. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 326:1227–1237. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., and D. J. Smith. 2004. Preferential hydration and the exclusion of cosolvents from protein surfaces. J. Chem. Phys. 121:1148–1154. [DOI] [PubMed] [Google Scholar]
- Swaminathan, R., C. P. Hoang, and A. S. Verkman. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 72:1900–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, C.-L., H. Hong, S. Kiihne, and R. G. Bryant. 2001. Molecular oxygen spin-lattice relaxation in solutions measured by proton magnetic relaxation dispersion. J. Magn. Reson. 148:31–34. [DOI] [PubMed] [Google Scholar]
- Tylianakis, M., A. Spyros, P. Dais, F. R. Taravel, and A. Perico. 1999. NMR study of the rotational dynamics of linear homopolysaccharides in dilute solutions as a function of linkage position and stereochemistry. Carbohydr. Res. 315:16–34. [Google Scholar]
- Uedaira, H., M. Ikura, and H. Uedaira. 1989. Natural-abundance oxygen-17 magnetic relaxation in aqueous solutions of carbohydrates. Bull. Chem. Soc. Jpn. 62:1–4. [Google Scholar]
- van den Berg, B., R. J. Ellis, and C. M. Dobson. 1999. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18:6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venu, K., V. P. Denisov, and B. Halle. 1997. Water 1H magnetic relaxation dispersion in protein solutions. A quantitative assessment of internal hydration, proton exchange, and cross-relaxation. J. Am. Chem. Soc. 119:3122–3134. [Google Scholar]
- Walter, H., and D. E. Brooks. 1995. Phase separation in cytoplasm, due to macromolecular crowding, is the basis for microcompartmentation. FEBS Lett. 361:135–139. [DOI] [PubMed] [Google Scholar]
- Watzlawek, M., and G. Nägele. 1997. Self-diffusion coefficients of charged particles: prediction of nonlinear volume fraction dependence. Phys. Rev. E. 56:1258–1261. [Google Scholar]
- Wlodawer, A., J. Deisenhofer, and R. Huber. 1987. Comparison of two highly refined structures of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 193:145–156. [DOI] [PubMed] [Google Scholar]
- Zimm, B. H. 1946. Application of the methods of molecular distribution to solutions of large molecules. J. Chem. Phys. 14:164–179. [Google Scholar]
- Zorrilla, S., G. Rivas, A. U. Acuña, and M. P. Lillo. 2004a. Protein self-association in crowded protein solutions: a time-resolved fluorescence polarization study. Protein Sci. 13:2960–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorrilla, S., G. Rivas, and M. P. Lillo. 2004b. Fluorescence anisotropy as a probe to study tracer proteins in crowded solutions. J. Mol. Recognit. 17:408–416. [DOI] [PubMed] [Google Scholar]