

Abstract
The lateral organization of lipid components within membranes is usually investigated with fluorescence microscopy, which, though highly sensitive, introduces bulky fluorophores that might alter the behavior of the components they label. Secondary ion mass spectroscopy performed with a NanoSIMS 50 instrument also provides high lateral resolution and sensitivity, and many species can be observed in parallel without the use of bulky labels. A tightly focused beam (∼100 nm) of Cs ions is scanned across a sample, and up to five of the resulting small negative secondary ions can be simultaneously analyzed by a high-resolution mass spectrometer. Thin layers of 15N- and 19F-labeled proteins were microcontact-printed on an oxidized silicon substrate and imaged using the NanoSIMS 50, demonstrating the sensitivity and selectivity of this approach. Supported lipid bilayers were assembled on an oxidized silicon substrate, then flash-frozen and freeze-dried to preserve their lateral organization. Lipid bilayers were analyzed with the NanoSIMS 50, where the identity of each specific lipid was determined through detection of its unique secondary ions, including 12C1H−, 12C2H−, 13C−, 12C14N−, and 12C15N−. Steps toward obtaining quantitative composition analysis of lipid membranes that varied spatially in isotopic composition are presented. This approach has the potential to provide a composition-specific analysis of membrane organization that compliments other imaging modalities.
INTRODUCTION
The lateral organization and dynamic rearrangements of lipid and protein components within biological membranes are essential for their functions. The lipid bilayer is the common motif for all membranes; some fraction of the lipid molecules are free to diffuse and mix in the two-dimensional plane of the bilayer, whereas other components are believed to be organized into domains, for example, lipid rafts, that are distinguished by correlated motion and emergent function. Investigation of the associations, organization, and dynamics of membrane components on a length scale that is greater than that of the individual molecules but much less than that which is accessible with light microscopy is a major challenge. Furthermore, since fluid membrane properties, especially those of the lipid components, involve delicate interactions that are highly sensitive to their specific chemical and physical properties, elucidation of the spatial organization of all the components present without the use of large and chemically complex labels, such as those used in fluorescence microscopy, is most desirable. Herein, we demonstrate that secondary ion mass spectrometry (SIMS), implemented using a recently developed instrument from Cameca Instruments (Courbevoie, France), the NanoSIMS 50, can be combined with isotopically labeled membrane components and tools developed for supported bilayer patterning (Groves and Boxer, 2002) to obtain information on the lateral composition of a membrane without altering the chemical composition of the components of interest. Moreover, because the ultimate resolution of the NanoSIMS 50 is on the order of 50 nm, this technique surpasses the lateral resolution of fluorescence microscopy. Through a combination of sample preparation, isotopic substitution strategies, data acquisition, and analysis, this method may ultimately be a powerful technique to probe membrane composition and spatial organization in a manner that complements current approaches.
A comprehensive view of the complex lateral organization of biological membranes requires the application of many methods in parallel. To date, most studies of lipid domains utilize model membrane systems and a multitude of techniques, including atomic force microscopy (AFM) (Lawrence et al., 2003; Rinia et al., 2001; Giocondi et al., 2001; Yuan et al., 2002), differential scanning calorimetry (Gandhavadi et al., 2002; Shaikh et al., 2001), single particle tracking (Dietrich et al., 2001a), electron spin resonance and infrared spectroscopy (Veiga et al., 2001), x-ray diffraction (Gandhavadi et al., 2002), nuclear magnetic resonance (Filippov et al., 2004), and fluorescence microscopy (Dietrich et al., 2001a,b; Hwang et al., 1998; Korlach et al., 1999; Samsonov et al., 2001; Scherfeld et al., 2003; Veatch and Keller, 2003). Fluorescence microscopy is the most powerful approach for achieving the highest sensitivity. However, this requires the addition of fluorescent labels that might interfere with the delicate interactions that occur within membranes (Burns, 2003) and, of course, only the component that is conjugated to the label can be detected. There is an extensive literature on the differential partitioning of fluorescent labels into different phases in monolayers (Dietrich et al., 2001a; Ianoul et al., 2003; Keller, 2002, 2003; Keller et al., 2000; Strottrup et al., 2004), supported bilayers (Dietrich et al., 2001a; Ianoul et al., 2003; Strottrup et al., 2004), and giant unilamellar vesicles (Bacia et al., 2004; Bagatolli, 2003; Bagatolli et al., 2003; Baumgart et al., 2003; Kahya et al., 2003, 2004; Nag et al., 2002; Scherfeld et al., 2003; Strottrup et al., 2004; Veatch and Keller, 2003; Veatch et al., 2004), but the composition of these phases can only be inferred from these measurements. Furthermore, the diffraction limit of optical microscopy restricts the lateral resolution of these measurements. AFM can provide much higher lateral resolution, but assessment of the chemical composition of the observed features is a challenge.
Time-of-flight SIMS (TOF-SIMS) offers an important step toward composition analysis with better spatial resolution than optical microscopy (Bourdos et al., 2000a,b; Harbottle et al., 2003; Ostrowski et al., 2004; Roddy et al., 2002a,b; Ross et al., 2001; Sostarecz et al., 2004). Chemical images of biological samples are constructed from the intensity of the constituent-specific atomic or molecular secondary ions detected at each position. For instance, SIMS images of atomic ions, molecular ions, and significantly higher molecular weight ionized molecular clusters have been employed to determine the subcellular location of biologically relevant ions, atomically or isotopically labeled metabolites, and natural phospholipids (Cannon et al., 2000; Chandra, 2004; Colliver et al., 1997; Fartmann et al., 2004; Lockyer and Vickerman, 2004; Ostrowski et al., 2004; Quong et al., 2004; Stelly et al., 1995). These studies demonstrate the utility of applying SIMS imaging to biological samples, but the approach would greatly benefit from improvements in the sensitivity, resolution, and quantification. At present, the best instruments are considerably less sensitive and have significantly lower lateral resolution than the NanoSIMS 50; however, this is a rapidly evolving area, and technical advances that will expand the application of these instruments can be expected.
The principle of the experimental method utilized in this report is presented in Fig. 1, which shows a schematic diagram of a patterned supported lipid bilayer. For purpose of illustration, a two-component supported lipid bilayer on a patterned substrate is shown, where spatial variations in the chemical composition of the lipids within the membrane in different corralled regions are present (Kam and Boxer, 2000). Because SIMS experiments are performed under ultra-high vacuum (UHV), methods were first developed to rapidly freeze and then remove water from the sample in a manner that preserves the surface organization with minimal perturbations. In our experiments, the Cameca NanoSIMS 50 uses a tightly focused Cs+ ion beam that extensively fragments the components on the surface within the focal area. This beam is rastered over the sample and the resulting small negative ions are extracted and separated by a high-resolution mass spectrometer that has five independent pulse-counting detectors. Relevant negative ions that can be readily distinguished include atomic ions, such as 1H−, D−, 12C−, 13C−, 16O−, 18O−, 19F−, 31P−, and 32S−, and molecular ions, such as 12C1H−, 13C1H−, 12CD−, 13CD−, 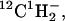 12C14N−, 13C14N−, 12C15N−, and 13C15N−. By careful choice of isotopic composition or unique atomic signatures (e.g., the presence of sulfur, phosphorous, or fluorine), the negative secondary ion signals can provide information on which components lie within the path of the primary ion beam as it is stepped across the surface of the sample. To date, the NanoSIMS 50 has primarily been used to characterize the isotopic compositions of small particles from interstellar sources (Besmehn and Hoppe, 2003; Daulton et al., 2002; Floss et al., 2004; Marhas et al., 2003; Messenger et al., 2003; Nguyen and Zinner, 2004; Hoppe et al., 2004), as well as to determine the structure of hard materials (Cameca_materials). Although limited data is available on organic materials (Audinot et al., 2004; Grignon et al., 2000; Hallegot et al., 2004; Kleinfeld et al., 2004; Peteranderl and Lechene, 2004), nothing as thin as the 5–10 nm thick soft protein and lipid membranes described in this report have been previously characterized with the NanoSIMS 50. These materials present many challenges, and we address a first set of these in the following by imaging protein adlayers and supported lipid membranes that contained simple variations in their lateral composition.
12C14N−, 13C14N−, 12C15N−, and 13C15N−. By careful choice of isotopic composition or unique atomic signatures (e.g., the presence of sulfur, phosphorous, or fluorine), the negative secondary ion signals can provide information on which components lie within the path of the primary ion beam as it is stepped across the surface of the sample. To date, the NanoSIMS 50 has primarily been used to characterize the isotopic compositions of small particles from interstellar sources (Besmehn and Hoppe, 2003; Daulton et al., 2002; Floss et al., 2004; Marhas et al., 2003; Messenger et al., 2003; Nguyen and Zinner, 2004; Hoppe et al., 2004), as well as to determine the structure of hard materials (Cameca_materials). Although limited data is available on organic materials (Audinot et al., 2004; Grignon et al., 2000; Hallegot et al., 2004; Kleinfeld et al., 2004; Peteranderl and Lechene, 2004), nothing as thin as the 5–10 nm thick soft protein and lipid membranes described in this report have been previously characterized with the NanoSIMS 50. These materials present many challenges, and we address a first set of these in the following by imaging protein adlayers and supported lipid membranes that contained simple variations in their lateral composition.
FIGURE 1.

Cross-sectional schematic of a patterned lipid bilayer with different compositions in each corral as prepared under water (not to scale; the SiO2 layer is typically 40 nm thick and barriers are separated by tens of microns). After freeze-drying to remove the bulk water, its composition is sampled by secondary ion mass spectrometry using a Cameca NanoSIMS 50 instrument. The incoming focused Cs-ion beam generates secondary ions; negative ions are collected and analyzed in a high-resolution mass spectrometer (not shown). The identities of different components are encoded by isotopic or atomic labeling.
MATERIALS AND METHODS
Silicon/SiO2 substrates
To dissipate charge on the surface, samples for NanoSIMS 50 measurements require a conducting pathway. This can be achieved by using silicon wafer supports or by overcoating with metals. Although supported bilayers assemble on the native oxide layer of silicon (typically <10 nm thick), we have found that these membranes are not very stable, and thicker oxide layers are preferable. Because charge dissipation decreases with increases in the thickness of the insulating oxide, oxide layers of 2 nm, 24 nm, 38 nm, and 138 nm were initially prepared by dry oxidizing the 〈100〉 surface of p-doped silicon wafers (thickness characterized by ellipsometry) to determine the optimal oxide thickness that balanced stable lipid bilayer formation with maximal charge dissipation. An oxide layer of ∼40 nm was experimentally determined to be a suitable compromise between maximal secondary ion signal intensity and surface stability (data not shown), and this thickness was used for subsequent experiments. For compatibility with the standard NanoSIMS 50 sample holders, the wafer was cut into 5 × 5 mm pieces with a conventional dicing saw. The surfaces were cleaned using procedures developed for supported bilayer assembly (Groves et al., 1998): sonication for 30 min in acetone, piranha solution at 80°C for 15 min, rinse with deionized water, wash in ICN 7X detergent (ICN, Costa Mesa, CA), rinse extensively with deionized water, and anneal at 400°C in air for a minimum of 6 h.
Protein stamping
Proteins were stamped onto the wafer surface by microcontact printing (James et al., 1998) using polydimethylsiloxane (PDMS) stamps (Sylgard 184 silicon elastomer, Dow Corning, Midland, MI). The PDMS was cured on silicon masters that were patterned with photoresist, generating a 1 μm raised grid, in which the bars of the grid were either 2.5 μm or 5 μm wide and spaced 25 μm apart. A droplet of protein solution was deposited on the PDMS stamp and incubated for 2 min. The excess protein solution was absorbed with a tissue and the surface of the stamp was dried under a stream of nitrogen gas. The protein-coated PDMS stamp was pressed against the oxidized silicon substrate with a 12 g weight, which typically left a 5–10 nm thick adlayer of protein on the surface (imaged by AFM, see below). In certain cases, these printed proteins can be used to pattern the assembly of supported lipid bilayers (Kung et al., 2000).
Three proteins were utilized in this work. Fibronectin (FN, C22972H 35644O7362N6412S180) (Sigma-Aldrich, St. Louis, MO) was diluted to a concentration of 100 μg/ml in 10 mM Hepes:150 mM NaCl solution. For some samples, FN was labeled with Cy 3.5 (Amersham Biotech, Piscataway, NJ) for visualization. Acyl carrier protein 2 that was uniformly isotopically labeled with 15N to ∼90% (15N-Acp2, C520H841O152N167 S1) was generously provided by V. Y. Alekseyev in the Khosla lab (Stanford University, Stanford, CA) (Wu et al., 2002). A sample of recombinant human dihydrofolate reductase (DHFR) grown in the presence of 4-fluorophenylalanine (F-DHFR, C841H1275O245N235F∼4.5S7) was generously provided by J. T. Schulte in the Swartz lab (Stanford University). Under these conditions, an average of 4–5 of the six phenylalanines were replaced with 4-fluorophenylalanine (F-Phe).
Supported lipid bilayers
Egg phosphatidylcholine (egg PC, Avanti Polar Lipids, Alabaster, AL), Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (TR-DHPE, Molecular Probes, Eugene, OR) and 1-palmitoyl-D31-2-oleoyl-sn-glycero-3-phosphocholine (D31-POPC, Avanti Polar Lipids) were purchased in chloroform. Egg phosphatidylcholine-15N (15N-egg PC) was prepared from egg phosphatidylethanolamine (Avanti Polar Lipids) and choline chloride-15N (99 atom % 15N, Icon, Summit, NJ) following previously reported procedures (Juneja et al., 1989). Thin layer chromatography analysis (65:25:4 chloroform/methanol/water) showed the purified 15N-egg PC comigrated with an egg PC standard. 1,2-Dioleoyl-sn-glycero-3-phosphatidylcholine (N,N,N-trimethyl-13C3) (DOPC-13C3) was synthesized from 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (Avanti Polar Lipids) and iodomethane-13C (99 atom % 13C, Sigma-Aldrich) according to previous methods (Patel et al., 1979). The purified product comigrated with a DOPC standard using the aforementioned thin layer chromatography solvent system.
Typically, 0.5 mol % TR-DHPE was added to the bilayer samples to aid in imaging the membrane with fluorescence microscopy, which was performed to evaluate the homogeneity of the sample before and after freeze-drying, and for comparison to the images acquired with the NanoSIMS 50. The desired lipid mixtures were dissolved in chloroform, dried under a nitrogen stream, desiccated under vacuum for at least 3 h, and reconstituted with Millipore (Bedford, MA) water (18 MΩ) to a concentration of 5 mg/ml. Small unilamellar vesicles were formed by passing the lipid suspension through a 50 nm polycarbonate membrane 19 times. The extruded vesicles were stored at 4°C and used within 1 week. Supported bilayers were formed by exposing the oxidized silicon substrate to the vesicle suspension, and excess vesicles were removed by rinsing in Millipore water.
Although methods are available to prepare precise variations in lipid composition across a patterned surface (Kam and Boxer, 2000), the small dimensions of the NanoSIMS 50 substrates led to a simplified method for these first experiments. Two 1 μl droplets, each containing an isotopically distinct vesicle population, were deposited on a glass coverslip such that they were separated by ∼2–3 mm. A protein-patterned substrate was then placed over the droplets, causing the two droplets to come into contact with one another and begin to mix. Since membrane formation on the substrate was more rapid than the mixing of the two vesicle suspensions, the ratio of the two lipid components within the resulting bilayer varied from corral to corral. The composition within each corral was allowed to become fully mixed before freezing the samples.
Freeze-drying
Supported bilayers are only stable when bathed by aqueous solutions and delaminate from the surface upon contact with air (Cremer and Boxer, 1999; Holden et al., 2004). To prepare the samples for the NanoSIMS 50 experiments, which are conducted under UHV, the water must be removed without disrupting the lateral organization of the membrane (at least on the scale of the 100 nm area sampled by the Cs+ beam). The freeze-drying method was derived from those employed in freeze-fracture and cryoelectron microscopy experiments (Ryan, 1992), which involves flash-freezing and careful removal of bulk water. After assembly, the supported bilayer samples were manually plunged into liquid ethane (cooling rate ∼15,000°C/s (Ryan, 1992) with a well-separated melting and boiling point, 90 K and 184 K, respectively, that minimizes ice crystallization). Samples were then transferred into a precooled vessel that was submerged in liquid nitrogen to keep the water in a frozen amorphous state during the freeze-drying process, then the ice was removed under reduced pressure (90 μbar) for at least 16 h using an oil-free scroll pump (Varian, Lexington, MA) and a liquid nitrogen trap to minimize surface contamination. During the first 4 h that the samples were subjected to reduced pressure, they were kept cold by immersing the vessel that contained the samples in liquid nitrogen. This process evolved by trial and error, in which the primary criteria for the quality of preservation was gauged by the homogeneity of the lipid membrane within each corral, as determined by imaging the dehydrated sample with fluorescence microscopy and AFM. The most common problem was slow transfer of the sample from the water into the liquid ethane, which adversely affected the bilayer structure. Artifacts of the freeze-drying process were easily identified by fluorescence microscopy (see below).
Secondary ion mass spectrometry
SIMS was performed using the Lawrence Livermore National Laboratory Cameca NanoSIMS 50 in simultaneous secondary ion collection mode with pulse counting on the electron multipliers. A microcesium source was used to generate 16 kV Cs+ primary ions for sample interrogation. The primary beam was tuned to a nominal spot size of ∼100 nm, which determines the lateral resolution of these measurements (100 nm), with ∼2 pA Cs+, and stepped over the sample in a 256 × 256 or 1024 × 1024 pixel raster to generate quantitative secondary ion images. The same region was rerastered (1–7 times), creating a series of images, one for each raster plane, that were compiled together to create the final image, which is presented in this article. The dwell time was 1 ms/pixel and the magnetic field in the mass spectrometer for secondary ions was set to detect 31P− or 32S− on the fixed electron multiplier (EM5) (Cameca_NanoSIMS), and the four movable electron multipliers were positioned to detect 12C−, 12C1H−, 12CD−, 19F−, 12C14N−, 12C15N−, or 31P−. The secondary mass spectrometer was tuned for ∼6800 mass resolving power to resolve isobaric interferences, which arise from mass interferences that occur whenever another ion has the same nominal mass as the analyte ion. The slit at the entrance of the multiplier was typically set at 80 μm; however, when the resolution of 13C1H− (m/e (mass charge ratio) = 14.0106304 amu) (atomic mass unit), 12CD− (m/e = 14.0135524 amu), and 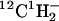 (m/e = 14.0151014 amu) was desired, a slit width of 25 μm was employed. Samples were also simultaneously imaged using the secondary electrons that were detected by a photomultiplier. Samples were first visually located using a charge-coupled device camera. Next, the samples were moved to the analysis position in the NanoSIMS 50 and positioned exactly using real-time secondary electron imaging. A map of the sample was obtained by assembling a series of measurements such that their overlapping regions matched. For the analysis of gradients, the NanoSIMS 50 signals were normalized with respect to 12C− to minimize signal intensity variations that arose during the measurements. Each image was smoothed with a 3-pixel-wide window, which was necessary for compatibility with the software with which isotopic ratios were calculated. As a result of the smoothing, the images acquired for each measurement are outlined with a black frame (e.g., Fig. 5 B).
(m/e = 14.0151014 amu) was desired, a slit width of 25 μm was employed. Samples were also simultaneously imaged using the secondary electrons that were detected by a photomultiplier. Samples were first visually located using a charge-coupled device camera. Next, the samples were moved to the analysis position in the NanoSIMS 50 and positioned exactly using real-time secondary electron imaging. A map of the sample was obtained by assembling a series of measurements such that their overlapping regions matched. For the analysis of gradients, the NanoSIMS 50 signals were normalized with respect to 12C− to minimize signal intensity variations that arose during the measurements. Each image was smoothed with a 3-pixel-wide window, which was necessary for compatibility with the software with which isotopic ratios were calculated. As a result of the smoothing, the images acquired for each measurement are outlined with a black frame (e.g., Fig. 5 B).
FIGURE 5.

Analysis of a bilayer compositional array by fluorescence and secondary ion signals collected by the NanoSIMS 50. A gradient between egg PC (containing 0.5 mol% TR-DHPE) and D31-POPC was formed by depositing one droplet of each of two different vesicle suspensions (egg PC and D31-POPC) onto a FN-patterned surface. (A) Fluorescence image showing the variation in composition, from low fluorescence to high fluorescence. (B) 12CD−/12C− SIMS signal from the same region shows the 12CD−/12C− signal is maximal where the fluorescence is minimal (the total dwell time was 8 ms). (C) Normalized 12CD−/12C− and 12C1H−/12C− signals versus the fluorescence, where each of the eight data points on each line corresponds to one of the eight corrals within the lower row highlighted with arrows in panels A and B. The best fit to a linear relationship is plotted through the points.
Atomic force microscopy
Stamped proteins or freeze-dried lipid bilayers were measured using a Nanoscope III AFM (Digital Instruments, Santa Barbara, CA) to visualize the topography of the sample. The measurements were performed in ambient air in tapping mode (TM) with a 122 μm long Si TM-tip to minimize the interaction between the tip and the surface (Ikai, 1996).
RESULTS AND DISCUSSION
Microcontact-printed proteins
A surface with microcontact-printed proteins is a useful test sample because the composition can be readily varied, the potential complications of freeze-drying are avoided, they are often used to pattern supported lipid bilayers, and ultimately, we will be interested in imaging the organization of membrane-associated proteins. Using the grid pattern described above, a single oxidized silicon substrate was sequentially printed with natural abundance FN, 15N-labeled Acp2, and 19F-DHFR, respectively, such that the grids were rotated and offset from each other. AFM measurements indicated that the adlayer thickness was 5–7 nm for 15N-Acp2 and 19F-DHFR, whereas a height of 25 nm was measured at the thickest area of patterned FN. Using the NanoSIMS 50, the surface was imaged with simultaneous detection of 12C−, 19F−, 12C14N−, 12C15N−, and 32S− secondary ions, and the results are shown in Fig. 2. A schematic of the three patterned proteins is presented in the right column, where each protein grid displayed in the corresponding SIMS image is color-coded. Fig. 2 A shows the 19F− signal: only the 19F-DHFR protein grid was detectable. Fig. 2 B displays the 12C15N− signal, in which only the 15N-Acp2 protein grid was revealed. Note, the NanoSIMS 50 was tuned such that negligible 13C14N− secondary ions contributed to the 12C15N− signal (for 12C15N−–13C14N− Δm/e = 6.3 milli amu). Fig. 2 C shows the ion intensity map of the 12C14N− secondary ion signal, in which two protein grids were clearly imaged. One corresponded to 19F-DHFR because its location was identical to the F− signal shown in panel A, whereas the second protein grid was FN because its position differed from that of 15N-Acp2, as shown in panel B. Closer inspection of the map of the 12C14N− secondary ion signal reveals the third protein grid, 15N-Acp2, which has one-fourth the signal intensity of the other two proteins. The detection of a 12C14N− secondary ion signal from the 15N-Acp2 protein grid reflects the incomplete (90%) isotopic substitution of 15N for 14N. The 12C− and 32S− signals, which are shown in Fig. 2, D and E, were generated by all three protein grids. Defects in the protein patterns are visible in panels A and B; the 15N-Acp2 and 19F-DHFR protein grids were not continuous in the regions where the substrate was already patterned with protein, indicating protein-on-protein deposition typically did not occur. Additionally, protein over stamping also produced defects in the protein grid that was previously deposited on the silicon substrate. As shown in panels C–E, a portion of the FN grid was removed by subsequent contact with the 15N-Acp2 and 19F-DHFR stamps.
FIGURE 2.

SIMS data for a set of differently labeled proteins deposited in a grid pattern by microcontact printing. Fibronectin (FN, natural abundance) was printed first, followed by 15N-labeled-Acp2, and then 19F-Phe-substituted DHFR such that the protein grids were rotated and offset from each other. The left panels present the SIMS data for different species, and a schematic guide is presented to the right. (A) The 19F− image exclusively corresponds to 19F-DHFR. (B) The 12C15N− image corresponds to 15N-Acp2. (C) The 12C14N− image, which shows two separate protein grids, 19F-DHFR and FN. (D) The 32S− image (32S is present in all three proteins). (E) The 12C− image, also present in all three proteins. The dots in the schematic guide correspond to regions with maximal intensities, which are 527, 1068, 6280, 1436, and 3402 counts/pixel s, respectively. All five of the secondary ions were measured at a localized region on each protein, which is denoted by the star. The total dwell time was 4 ms (1 ms each serial image; total of four serial images).
For panels A, B, C, D, and E in Fig. 2, the regions of maximal signal intensity denoted by circles in the schematic drawings are 1, 2, 11.9, 2.7, and 6.5 relative to the 19F− signal, respectively. For comparison of the ion yields in each protein, the signal intensities of all five secondary ions were measured at a localized region on each sample, which is designated with a star. As expected, the ratio of the 12C15N−/12C14N− signal intensities measured for 19F-DHFR (Fig. 2 A) and FN (Fig. 2 C) correspond to the natural abundance ratio of 15N/14N (0.4%). The 19F− yield was 16 times that of the 12C14N− yield for 19F-DHFR, and the relative 32S− yield varied between 4 and 6 times that of 12C14N− for all three different proteins, where the yield was calculated by taking the ratio of the relative SIMS signal intensity to the relative theoretical number of species present in the protein. The high ion yield and uniqueness of the F− ions demonstrates that fluorine is an attractive atomic label for future work, especially because only a relatively small amount of fluorine incorporation is required for detection. Comparison of yields between proteins was not performed because variations in protein thickness can drastically influence their secondary ion signal intensities, especially when the sample is rastered multiple times, since a thicker adlayer continues to produce secondary ions after a thinner adlayer is sputtered away. This complication is absent for lipid membranes since their thickness is roughly constant.
Supported lipid bilayers
Although the thickness of a supported lipid bilayer in an aqueous environment (5 nm) is similar to that of microcontact-printed proteins, the potential amount of high-yielding secondary ions (i.e., 12C14N−) produced by the lipid components is significantly lower than that of the protein. For compatibility with UHV conditions, supported lipid bilayers were flash-frozen and freeze-dried to preserve their lateral organization. Fig. 3 A shows an AFM image of a freeze-dried supported lipid bilayer (egg PC doped with 0.5 mol % TR-DHPE) that was patterned with a FN grid, which prevents the lipid components that were deposited within each corral from mixing with neighboring corrals. The width of the protein grid measured by AFM and SIMS are very similar, being 3.4 ± 0.2 μm and 3.8 ± 0.2 μm, respectively. The protein barrier protruded 3 nm above the lipid membrane within the grid, which was extremely flat with a root mean-square roughness of 0.2 nm over an area of 100 μm2, similar to that of the clean oxidized silicon substrate (0.18 nm over the same area). The presence of the lipid membrane within the corral was confirmed with fluorescence microscopy, which detected the TR-DHPE within the membrane. Although freeze-drying decreased the fluorescence intensity of the TR-DHPE, the fluorescence from the lipid membrane was still 3.3 times greater than that detected for the protein grid after background subtraction. Analysis of the same region using the NanoSIMS 50 demonstrates that supported bilayers can be detected. (To test the sample stability over time, this sample was remeasured after 48 h in UHV, then stored in ambient air for 6 months and reintroduced to the UHV two more times, giving a total time under vacuum of 270 h. The 12C1H−/12C− ratios measured for proteins and lipids after all four different measurements varied by a total of only 4–5%, suggesting that materials did not sublime off the surface under the UHV conditions.) The location of the lipids and proteins was evidenced by contrast in the 12C− (Fig. 3 B), 31P− (Fig. 3 C), and, when appropriate, 12CD− (vide infra, see Figs. 4 A and 5 B) and 12C15N− (Figs. 4 B and 6 B) signals. The 12C, 31P−, and 12C14N− signal intensities from the supported bilayers were 61%, 1150%, and 9%, respectively, compared to that obtained from the protein grid.
FIGURE 3.

Freeze-dried egg PC supported lipid membrane patterned with a fibronectin grid and imaged by (A) AFM, (B) 12C−, and (C) 31P− signals using the NanoSIMS 50 (12C− maximum intensity is 6.5 times that of 31P−). The AFM image demonstrates that the bilayer region is flat and that the protein grid protrudes above the membrane by several nanometers. Fluorescence is readily seen in the regions covered by the egg PC doped with 0.5 mol % Texas Red-DHPE membrane, but not those covered by the protein (not shown, cf. Figs. 5–7). Since the relatively thicker protein grid contains more carbon than the bilayer, the protein grids appear brighter in panel B, whereas the FN grid does not contain phosphorous and appears dark in panel C. The total dwell time was 5 ms.
FIGURE 4.

SIMS data of isotopically labeled lipids patterned with a fibronectin grid. (A) 12CD− signal of D31-POPC. (B) 12C15N− signal of 15N-egg PC with 0.5 mol % TR-DHPE, where the protein stamp was wider than the one used in A. The contrast of both images is substantially higher than the natural abundance of D and 15N, clearly demonstrating that lipid bilayers can be detected, imaged, and their compositions analyzed. The total dwell times were 8 ms and 2 ms, respectively.
FIGURE 6.

Isotopically labeled bilayer compositional array was analyzed using fluorescence and the secondary ion signals collected by the NanoSIMS 50. A gradient between 15N-egg PC (containing 0.5 mol % TR-DHPE) and DOPC-13C3 (terminal methyls in choline headgroup labeled with carbon-13) was formed by contacting a FN-patterned surface with two droplets of vesicles, each consisting of one of the two isotopically labeled lipids (15N-egg PC or DOPC-13C3). (A) Fluorescence image showing the variation in composition from little fluorescence to maximum fluorescence. (B) 12C15N−/12C− SIMS signal from the same region indicates the substitution of a single nitrogen-15 atom into each lipid is adequate for detection with the NanoSIMS 50 (the total dwell time was 2 ms). (C) Normalized 13C−/12C− and 12C15N−/12C− signals as a function of the fluorescence taken along the lower row highlighted with arrows in panels A and B. The normalized 12C15N− signal also increased with the fluorescence. Note that the data range of SIMS signals differs from Fig. 5 C due to lower intensities of 12C15N− and 13C−. The best fit to a linear relationship is plotted through the points.
Although the silicon substrates were carefully cleaned, contamination of the substrate by residual material from microcontact printing and contaminants from the air and buffers was difficult to avoid. Therefore, whether the differences in the signal intensities detected for the protein grid and the lipid membrane-covered regions could be attributed to surface contamination was investigated. The background signals collected from the lipid- and protein-free regions of the oxidized silicon substrate were 17%, 74%, and 7% for 12C−, P−, and 12C14N−, respectively, relative to a 10 nm-thick microcontact-printed FN protein grid. Using these values as an estimate of the magnitude of the background signal in the lipid membrane samples, the signal intensities observed for the lipid membrane relative to those for FN were appreciably greater than the background, confirming that the lipid membrane did, indeed, produce the secondary ions collected in that region. Because each lipid has only a single phosphorous atom (two per ∼60 Å2), the detection of P− demonstrates both the extraordinary sensitivity of the NanoSIMS 50 and its ability to image thin films of biological materials with high contrast, where contrast refers to the difference between the light and dark regions of the image produced by the NanoSIMS 50, which is dictated by the signal intensities at these regions. Although the presence of an element that is unique to the phospholipids, phosphorous, simplifies differentiating the membrane from the protein adlayer, the NanoSIMS 50 can also distinguish isotopically substituted lipids from other components. Fig. 4 A shows the 12CD− secondary ion signal of a D31-POPC lipid membrane patterned with a FN grid. The 12CD− secondary ion signal was 6.1 times greater than that of the protein adlayer, which contains deuterium at its natural abundance level (0.02%). Likewise, the presence of an 15N-egg PC membrane doped with 0.5 mol % TR-DHPE was evidenced by a 12C15N− signal that was 5.0 times that of FN, which contains 15N at natural abundance (0.4%). The lower than expected excess of 12CD− and 12C15N− suggests that the lipid membrane also contained a significant amount of isotopically unmodified lipid; however, as mentioned in the previous section, the differences in the thickness of the protein and lipid layers makes comparison of their ion yields unreliable.
Bilayer composition variations
Although the results presented above demonstrate that biological materials can be imaged with high contrast and high lateral resolution by exploiting differences in the masses of their secondary ions, ultimately, quantitative information on the composition of a mixture is most desirable. As a first step in this direction, we investigated whether the NanoSIMS 50 could detect variations in membrane composition by forming a lipid bilayer that spatially varied in isotopic composition on a substrate patterned with protein grids, which serve as barriers to lateral diffusion. As described above, variations in the composition within the corralled regions were created by allowing two drops of different vesicle solutions to mix on a patterned surface during membrane deposition. Fig. 5 shows results for a freeze-dried sample with a compositional gradient between partially deuterated lipids (D31-POPC) and isotopically unmodified egg PC (doped with 0.5 mol % TR-DHPE for visualization). Fig. 5 A shows a fluorescence image of this surface, where the fluorescence intensity was digitized at a magnification of 400. The unlabeled microcontact-printed FN barriers appeared dark, but the fluorescence intensity, and, therefore, the composition of the lipid membrane, varied from left to right. Subsequent AFM measurements of this sample indicated that the regions covered by the lipid membrane were topographically homogeneous (data not shown), similar to that shown in Fig. 3 A. The normalized 12CD− (12CD−/12C−) signal acquired by the NanoSIMS 50 at the same region is shown in Fig. 5 B. As expected, the natural isotopic abundance FN barriers appear homogeneously dark in contrast to the lipid membrane. The normalized 12CD− (12CD−/12C−) signals generated by each corralled membrane patch varied in intensity in parallel with the fluorescence intensity, suggesting that the quantity of deuterium-enriched lipid within each corral differed. The rectangular region of low secondary ion signal intensity, represented by a dark silhouette located approximately six corrals from the left side of Fig. 5 B, resulted from exposing this area to the Cs+ beam for 3 min before acquiring the entire image. Further investigation with AFM revealed this region corresponded to a crater with a depth of 0.2 nm, indicating that the damage produced by extended exposure of the sample to the Cs+ ion beam was limited to the soft adlayer and did not significantly affect the silicon substrate.
To verify that the secondary ion signal intensities were quantitatively dependent on the lipid components within the sample, we examined the relationship between the intensities of component-specific secondary ions and the fluorescence signal. Both the normalized 12CD− (12CD−/12C−) and 12C1H− (12C1H−/12C−) signals are plotted as a function of the fluorescence intensity in Fig. 5 C, where each data point represents the average fluorescence and secondary ion signals collected from one of the eight corrals depicted in the lower row of Fig. 5, A and B (highlighted with arrows). Because the natural abundance egg PC contained the fluorescent lipid and the deuterium-enriched lipid was not doped with a fluorescent lipid, a rise in the normalized 12C1H− signal should accompany an increase in fluorescence, whereas the 12CD− signal should increase when the fluorescence falls if the secondary ion signal is quantitative. Indeed, both plots exhibited these trends, and the normalized 12CD− and 12C1H− signals were also linearly correlated to the fluorescence intensity (R2 = 0.97 and 0.95, respectively), suggesting the possibility for quantitative analysis of lipid mixtures using the NanoSIMS 50. Exploration of the quantitative aspects of this technique is under way and will be presented in a detailed report.
Choice of isotopic labels
The commercial availability of many deuterium-enriched compounds, primarily due to their widespread application for NMR experiments, renders deuterium an obvious choice as an isotopic label for a component of interest. However, to investigate the spatial organization of multiple lipids within a mixture, each unique component of interest must be individually labeled with a specific stable isotope that is exclusive to that component. Besides deuterium, there are three more potential isotopic labels for lipids: 13C, 18O, and 15N. 15N can be selectively substituted into the headgroup of a phospholipid, but this generates only a single unique nuclide per lipid molecule. Although there is not a simple method to replace the six oxygen atoms within a phospholipid with 18O, an 18O label can be easily incorporated into cholesterol (Hudgins et al., 1988). Preliminary results indicate that the background signal from the natural abundance of 18O (0.2%) in the oxidized silicon substrate overwhelms the 18O− secondary ion signal collected from the sample, obscuring visualization of the 18O-cholesterol (substrates prepared with 18O-depleted oxygen may curtail this background signal). Therefore, 15N- or 13C-enriched lipids were employed in the following experiments. To determine the efficacy of 15N- and 13C-labeled lipid detection by the NanoSIMS 50, a single substrate that supported an array of compositionally distinct lipid bilayers was employed, in which the composition of the membrane assembled within each corral exhibited a smooth transition from 15N-egg PC doped with 0.5 mol % TR-DHPE at one side of the substrate to 13C-enriched lipids (DOPC-13C3, N,N,N-trimethyl-13C3 choline, 99 atom % 13C) at the other. After freeze-drying, the compositional gradient was clearly evidenced by a variation in the fluorescence signal intensity of the membrane, which increased from left to right in Fig. 6 A, where the fluorescence intensity was digitized at a magnification of 100. In addition, small regions of bright fluorescence were distributed across the sample; these regions were disregarded in the experimental analysis that follows but can be exploited as discussed in the next section. The normalized 12C15N− (12C15N−/12C−) signal collected from the region shown in Fig. 6 A using the NanoSIMS 50 corroborates the presence of a compositional variation in the sample. Similar to the fluorescence, the 12C15N− (12C15N−/12C−) signal intensity increased from left to right in Fig. 6 B, which was expected since the 15N-egg PC solution also contained the fluorescent lipid. In contrast, a change in the composition of the membrane within this region was not visible from the image of the normalized 13C− signal (13C−/12C−) produced by DOPC-13C3 (not shown), indicating the ∼7% 13C-enrichment (relative to 1.1% 13C natural abundance) was insufficient for unambiguous visualization of the compositional gradient with the NanoSIMS 50. (We have not yet explored the use of other combinations of isotopically labeled CN−, such as 13C15N− or 13C14N− as potential component-specific secondary ions. However, 13C14N− and 12C15N− cannot be simultaneously detected due to constraints on the physical placement of the detectors.) Fig. 6 C graphically illustrates the relationship between the normalized secondary ion and fluorescence signal intensities acquired from the 15 corrals displayed in the indicated row in Fig. 6, A and B, where each point depicts the average signal intensities collected from a small region (100–180 μm2) within a single corral that was devoid of abnormally bright fluorescent spots. Again, increases in the fluorescence were paralleled by a rise in the 12C15N− signal with a linear correlation (R2 = 0.99) between the two signals. Although the gradual change in membrane composition was not evident from the SIMS image of the normalized 13C− signal, its presence was suggested in the quantitative analysis and plot of the 13C− versus fluorescence signal intensities. A decrease in the normalized 13C− signal was linearly correlated (R2 = 0.45) with an elevation in fluorescence intensity, as predicted a priori since the 13C-enriched lipid was not doped with a fluorescent label. The correlation factor R2 increases to 0.9 if only the last nine corrals are considered, where a significant variation in the fluorescence intensity (from 216 to 536) also indicates that the membrane composition changes appreciably. Nevertheless, the maximum normalized 13C− signal was only 1.3 times greater than its minimum value, so a more highly enriched 13C-labeled lipid would be necessary for future experiments.
Detection and analysis of microdomains
As mentioned above, fluorescence imaging revealed localized regions of elevated fluorescence scattered across some of the corrals in the 15N- and 13C-enriched lipid gradient sample. The origin of this effect is unknown, but we find that these bright spots are more frequently observed when TR-DHPE is employed for visualization with fluorescence microscopy, and protein barriers are used to pattern the membrane, compared to the use of 12-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-labeled lipids and chrome grids for these respective roles. Although these bright spots were unintended defects within the membrane, we can exploit their presence to test whether the NanoSIMS 50 can image structures within a membrane (i.e., microdomains). The elevated fluorescence signal that typified these defects suggested they contained a TR-DHPE concentration that was higher than the rest of the membrane, but lower than that which leads to self-quenching. Because TR-DHPE is the only component that contains sulfur, the 32S− signal was used to image TR-DHPE, as shown in Fig. 7. The fluorescence signal (green) is shown in Fig. 7 A and the 32S− SIMS signal is presented in Fig. 7 B (red). Both images are overlaid in Fig. 7 C, where an increased 32S− signal (Fig. 7, B and C) and a depressed 12C15N− signal (not shown) was detected at a majority of the highly fluorescent regions, confirming they contained an elevated abundance of TR-DHPE. Though the microdomains examined in this experiment differ in both complexity and chemical composition from those of biological relevance, these results demonstrate that the NanoSIMS 50 has the sensitivity and resolution to detect composition variations and identify their constituents.
FIGURE 7.

Membrane defects induced by freeze-drying were analyzed with the NanoSIMS 50 to demonstrate the lateral resolution of this technique. Highly fluorescent inhomogeneities were observed in a freeze-dried two-component lipid membrane (egg PC-0.5 mol % TR-DHPE and D31-POPC), providing an opportunity to compare their location in the fluorescence (in green in A and C) and 32S-SIMS (in red in B and C) images. TR-DHPE is the only membrane component that contains sulfur. As shown in the overlaid image C, the TR-DHPE specific signal, 32S−, was elevated at a majority of the bright spots, demonstrating the NanoSIMS 50 is capable of imaging microdomains. The dwell time of NanoSIMS 50 was 2 ms.
CONCLUSION
By combining isotopic labeling strategies with SIMS, we have demonstrated that thin biological films can be detected and imaged by the NanoSIMS 50 without the necessity of bulky chemical labels that may change the properties of the labeled component. Isotopically labeled proteins can be imaged and detected with high sensitivity and selectivity. The high relative yield of F− secondary ions and relative ease of introducing F-Phe into proteins makes this an attractive atomic label for proteins. A lipid membrane can be distinguished from a protein adlayer through detection of the P− secondary ions, which exclusively originate from the single phosphorous atom within each lipid during sample interrogation with the NanoSIMS 50. Likewise, isotopically substituted lipid membranes can be differentiated from protein adlayers and even each other with the proper selection of unique isotopic labels. Our experiments indicate that quantitative information about the composition of a lipid mixture might be extracted from the intensity of the signal of interest, and small heterogeneous regions within a single membrane can be detected, and components that reside there identified, with the NanoSIMS 50. We expect that rapid progress in multifaceted isotopic labeling strategies, membrane patterning techniques, and quantitative methods will follow and prove to be a useful addition to the tools available for characterizing complex biological membranes.
Acknowledgments
We thank Dr. Eun Sun Park and Paul Abbyad for help in the early stages of this work, Donguen Huh for the preparation of SiO2 surfaces, Viktor Alekseyev in Chaitan Khosla's laboratory for a gift of 15N-Acp2, and Jennifer T. Schulte in Jim Swartz's laboratory for a gift of F-Phe DHFR. Dr. Anders Meiborn (Stanford Earth Sciences) provided early help and guidance in the application of the NanoSIMS 50.
C.G.M. is supported by a postdoctoral fellowship from the Swiss National Science Foundation; M.L.K is supported by a National Institutes of Health National Research Service Award fellowship. This work is supported by grants from the National Science Foundation Biophysics program and National Institutes of Health GM06930 (to S.G.B.) and the work at the Lawrence Livermore National Laboratory was performed under the auspices of the U.S. Department of Energy. We are grateful to the Stanford Nanofabrication Facility for fabrication and the National Science Foundation Materials Research Science and Engineering Centers Center on Polymer Interfaces and Macromolecular Assemblies for analysis (ellipsometry and AFM).
References
- Audinot, J. N., S. Schneider, M. Yegles, P. Hallegot, R. Wennig, and H. N. Migeon. 2004. Imaging of arsenic traces in human hair by nano-SIMS 50. Appl. Sur. Sci. 231–232:490–496. [Google Scholar]
- Bacia, K., D. Scherfeld, N. Kahya, and P. Schwille. 2004. Fluorescence correlation spectroscopy relates rafts in model and native membranes. Biophys. J. 87:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli, L. A. 2003. Direct observation of lipid domains in free standing bilayers: from simple to complex lipid mixtures. Chem. Phys. Lipids. 122:137–145. [DOI] [PubMed] [Google Scholar]
- Bagatolli, L. A., S. A. Sanchez, T. Hazlett, and E. Gratton. 2003. Giant vesicles, Laurdan, and two-photon fluorescence microscopy: evidence of lipid lateral separation in bilayers. Methods Enzymol. 360:481–500. [DOI] [PubMed] [Google Scholar]
- Baumgart, T., S. T. Hess, and W. W. Webb. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425:821–824. [DOI] [PubMed] [Google Scholar]
- Besmehn, A., and P. Hoppe. 2003. A NanoSIMS study of Si- and Ca-Ti-isotopic compositions of presolar silicon carbide grains from supernovae. Geochim. Cosmochim. Acta. 67:4693–4703. [Google Scholar]
- Bourdos, N., F. Kollmer, A. Benninghoven, M. Ross, M. Sieber, and H.-J. Galla. 2000a. Analysis of lung surfactant model systems with time-of-flight secondary ion mass spectrometry. Biophys. J. 79:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdos, N., F. Kollmer, A. Benninghoven, M. Sieber, and H.-J. Galla. 2000b. Imaging of domain structures in a one-component lipid monolayer by time-of-flight secondary ion mass spectrometry. Langmuir. 16:1481–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, A. R. 2003. Domain structure in model membrane bilayers investigated by simultaneous atomic force microscopy and fluorescence imaging. Langmuir. 19:8358–8363. [Google Scholar]
- Cameca_materials. http://www.cameca.fr/html/nanosims_material.html.
- Cameca_NanoSIMS. http://www.cameca.fr/html/product_nanosims.html.
- Cannon, D. M., Jr., M. L. Pacholski, N. Winograd, and A. G. Ewing. 2000. Molecule specific imaging of freeze-fractured, frozen-hydrated model membrane systems using mass spectrometry. J. Am. Chem. Soc. 122:603–610. [Google Scholar]
- Chandra, S. 2004. Subcellular SIMS imaging of isotopically labeled amino acids in cryogenically prepared cells. Appl. Sur. Sci. 231–232:462–466. [Google Scholar]
- Colliver, T. L., C. L. Brummel, M. L. Pachloski, F. D. Swanek, A. G. Ewing, and N. Winograd. 1997. Atomic and molecular imaging at the single-cell level with TOF-SIMS. Anal. Chem. 69:225–231. [DOI] [PubMed] [Google Scholar]
- Cremer, P. S., and S. G. Boxer. 1999. Formation and spreading of lipid bilayers on planar glass supports. J. Phys. Chem. B. 103:2554–2559. [Google Scholar]
- Daulton, T. L., T. J. Bernatowicz, R. S. Lewis, S. Messenger, F. J. Stadermann, and S. Amari. 2002. Polytype distribution in circumstellar silicon carbide. Science. 296:1852–1855. [DOI] [PubMed] [Google Scholar]
- Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001a. Lipid raft reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, C., Z. N. Volovyk, M. Levi, and N. L. Thompson. 2001b. Partitioning of Thy-1, GM1, and cross-linked phospholipids analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl. Acad. Sci. USA. 98:10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fartmann, M., C. Kriegeskotte, S. Dambach, A. Wittig, W. Sauerwein, and H. F. Arlinghaus. 2004. Quantitative imaging of atomic and molecular species in cancer cell cultures with TOF-SIMS and Laser SNMS. Appl. Sur. Sci. 231–232:428–431. [Google Scholar]
- Filippov, A., G. Orädd, and G. Lindblom. 2004. Lipid lateral diffusion in ordered and disordered phases in raft mixtures. Biophys. J. 86:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floss, C., F. J. Stadermann, J. Bradley, Z. R. Dai, S. Bajt, and G. Graham. 2004. Carbon and nitrogen isotopic anomalies in an anhydrous interplanetary dust particle. Science. 303:1355–1358. [DOI] [PubMed] [Google Scholar]
- Gandhavadi, M., D. Allende, A. Vidal, S. A. Simon, and T. J. McIntosh. 2002. Structure, composition, and peptide binding properties of detergent soluble bilayers and detergent resistant rafts. Biophys. J. 82:1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocondi, M.-C., V. Vié, E. Lesniewska, P.-E. Milhiet, M. Zinke-Allmang, and C. L. Grimellec. 2001. Phase topology and growth of single domains in lipid bilayers. Langmuir. 17:1653–1659. [Google Scholar]
- Grignon, N., J. J. Vidmar, F. Hillion, and B. Jaillard. 2000. Physiological application of the nanoSIMS 50 ion microscope: localization at subcellular level of 15N labeling in Arabidopsis thaliana. Proc. 12th Int. Conf. Secondary Ion Mass Spectroscopy. A. Benninghoven, P. Bertrand, H. N. Migeon, and H. W. Werner, editors. Elsevier Science, Brussels. 903–906.
- Groves, J. T., and S. G. Boxer. 2002. Micropattern formation in supported lipid membranes. Acc. Chem. Res. 35:149–157. [DOI] [PubMed] [Google Scholar]
- Groves, J. T., N. Ulman, P. S. Cremer, and S. G. Boxer. 1998. Substrate-membrane interactions: mechanisms for imposing patterns on a fluid bilayer membrane. Langmuir. 14:3347–3350. [Google Scholar]
- Hallegot, P., R. Peteranderl, and C. Lechene. 2004. In-situ imaging mass spectrometry analysis of melanin granules in the human hair shaft. J. Invest. Dermatol. 122:381–386. [DOI] [PubMed] [Google Scholar]
- Harbottle, R. R., K. Nag, N. S. McIntyre, F. Possmayer, and N. O. Petersen. 2003. Molecular organization revealed by time-of-flight secondary ion mass spectrometry of a clinically used extracted pulmonary surfactant. Langmuir. 19:3698–3704. [Google Scholar]
- Holden, M. A., S.-Y. Jung, T. Yang, E. T. Castellana, and P. S. Cremer. 2004. Creating fluid and air-stable solid supported lipid bilayers. J. Am. Chem. Soc. 126:6512–6513. [DOI] [PubMed] [Google Scholar]
- Hoppe, P., U. Ott, and G. Lugmair. 2004. NanoSIMS, the new tool of choice: 26 Al, 44 Ti, 49 V, 53 Mn, 60 Fe, and more. N. Astron. Rev. 48:171–176. [Google Scholar]
- Hudgins, L. C., T. S. Parker, D. J. McNamara, F. A. Bencsath, and F. H. Field. 1988. Limits for the use of (18O)cholesterol and (18O)sitosterol in studies of cholesterol metabolism in humans. Biomed. Environ. Mass Spectrom. 17:463–470. [DOI] [PubMed] [Google Scholar]
- Hwang, J., L. A. Gheber, L. Margolis, and M. Edidin. 1998. Domains in cell plasma membranes investigated by near-field scanning optical microscopy. Biophys. J. 74:2184–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianoul, A., P. Burgos, Z. Lu, R. S. Taylor, and L. J. Johnston. 2003. Phase separation in supported phospholipid bilayers visualized by near-field scanning optical microscopy in aqueous solution. Langmuir. 19:9246–9254. [Google Scholar]
- Ikai, A. 1996. STM and AFM of bio/organic molecules and structures. Surf. Sci. Rep. 26:261–332. [Google Scholar]
- James, C. D., R. C. Davis, L. Kam, H. G. Craighead, M. Isaacson, J. N. Turner, and W. Shain. 1998. Patterned protein layers on solid substrates by thin stamp microcontact printing. Langmuir. 14:741–744. [Google Scholar]
- Juneja, L. R., T. Yamane, and S. Shimizu. 1989. Enzymatic method of increasing phosphatidylcholine content of lecithin. J. Am. Oil Chem. Soc. 66:714–717. [Google Scholar]
- Kahya, N., D. Scherfeld, K. Bacia, B. Poolman, and P. Schwille. 2003. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 278:28109–28115. [DOI] [PubMed] [Google Scholar]
- Kahya, N., D. Scherfeld, K. Bacia, and P. Schwille. 2004. Lipid domain formation and dynamics in giant unilamellar vesicles explored by fluorescence correlation spectroscopy. Struct. Biol. 147:77–89. [DOI] [PubMed] [Google Scholar]
- Kam, L., and S. G. Boxer. 2000. Formation of supported lipid bilayer composition arrays by controlled mixing and surface capture. J. Am. Chem. Soc. 122:12901–12902. [Google Scholar]
- Keller, L. P. 2002. Coexisting liquid phases in lipid monolayers and bilayers. J. Phys.-Condens. Mat. 14:4763–4766. [Google Scholar]
- Keller, L. P. 2003. Miscibility transitions and lateral compressibility in liquid phases of lipid monolayers. Langmuir. 19:1451–1456. [Google Scholar]
- Keller, L. P., T. G. Anderson, and H. M. McConnell. 2000. Miscibility critical pressures in monolayers of ternary lipid mixtures. Biophys. J. 79:2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfeld, A. M., J. P. Kampf, and C. Lechene. 2004. Transport of 13C-oleate in adipocytes measured using multi imaging mass spectrometry J. Am. Soc. Mass Spectrom. 15:1572–1580. [DOI] [PubMed] [Google Scholar]
- Korlach, J., P. Schwille, W. W. Webb, and G. W. Feigenson. 1999. Characterization of lipid bilayer phases by confocal and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 96:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung, L. A., L. Kam, J. S. Hovis, and S. G. Boxer. 2000. Patterning hybrid surfaces of proteins and supported lipid bilayers. Langmuir. 16:6773–6776. [Google Scholar]
- Lawrence, J. C., D. E. Saslowsky, J. M. Edwardson, and R. M. Henderson. 2003. Real-time analysis of the effects of cholesterol on lipid raft behavior using atomic force microscopy. Biophys. J. 84:1827–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockyer, N. P., and J. C. Vickerman. 2004. Progress in cellular analysis using TOF-SIMS. Appl. Sur. Sci. 212–232:377–384. [Google Scholar]
- Marhas, K. K., P. Hoppe, and U. Ott. 2003. A NanoSIMS study of C-, Si-, and Ba-isotopic compositions of presolar silicon carbide grains from the Murchison meteorite. Meteorit. Planet. Sci. 38:A58. (Abstr.) [Google Scholar]
- Messenger, S., L. P. Keller, F. J. Stadermann, R. M. Walker, and E. Zinner. 2003. Samples of stars beyond the solar system: silicate grains in interplanetary dust. Science. 300:105–108. [DOI] [PubMed] [Google Scholar]
- Nag, K., J.-S. Pao, R. R. Harbottle, F. Possmayer, N. O. Petersen, and L. A. Bagatolli. 2002. Segregation of saturated chain lipids in pulmonary surfactant films and bilayers. Biophys. J. 82:2041–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, A. N., and E. Zinner. 2004. Discovery of ancient silicate stardust in a meteorite. Science. 303:1496–1499. [DOI] [PubMed] [Google Scholar]
- Ostrowski, S. G., C. T. Van Bell, N. Winograd, and A. G. Ewing. 2004. Mass spectrometric imaging of highly curved membranes during tetrahymena mating. Science. 305:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, K. M., J. D. Morrisett, and J. T. Sparrow. 1979. The conversion of phosphatidylethanolamine into phosphatidylcholine labeled in the choline group using methyl iodide, 18-crown-6 and potassium carbonate. Lipids. 14:596–597. [Google Scholar]
- Peteranderl, R., and C. Lechene. 2004. Measurement of carbon and nitrogen stable isotope ratios in cultured cells J. Am. Soc. Mass Spectrom. 15:478–485. [DOI] [PubMed] [Google Scholar]
- Quong, J. N., M. G. Knize, K. S. Kulp, and K. J. Wu. 2004. Molecule-specific imaging analysis of carcinogens in breast cancer cells using time-of-flight secondary ions mass spectrometry. Appl. Sur. Sci. 231–232:424–427. [Google Scholar]
- Rinia, H. A., M. M. Snel, J. P. van der Eerden, and B. de Kruijff. 2001. Visualizing detergent resistant domains in model membranes with atomic force microscopy. FEBS Lett. 501:92–96. [DOI] [PubMed] [Google Scholar]
- Roddy, T. P., D. M. Cannon, C. A. Meserole, N. Winograd, and A. G. Ewing. 2002a. Imaging of freeze-fractured cells with in situ florescence and time-of-flight secondary ion mass spectrometry. Anal. Chem. 74:4011–4019. [DOI] [PubMed] [Google Scholar]
- Roddy, T. P., D. M. Cannon, S. G. Ostrowski, N. Winograd, and A. G. Ewing. 2002b. Identification of cellular sections with imaging mass spectrometry following freeze fracture. Anal. Chem. 74:4020–4026. [DOI] [PubMed] [Google Scholar]
- Ross, M., C. Steinem, H.-J. Galla, and A. Janshoff. 2001. Visualization of chemical and physical properties of calcium-induced domains in DPPC/DPPS Langmuir-Blodgett layers. Langmuir. 17:2437–2445. [Google Scholar]
- Ryan, K. P. 1992. Cryofixation of tissues for electron microscopy: a review of plunge cooling methods. Scanning Microsc. 6:715–743. [Google Scholar]
- Samsonov, A. V., I. Mihalyov, and F. S. Cohen. 2001. Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes. Biophys. J. 81:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherfeld, D., N. Kahya, and P. Schwille. 2003. Lipid dynamics and domain formation in model membranes composed of ternary mixtures of unsaturated and saturated phosphatidylcholines and cholesterol. Biophys. J. 85:3758–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh, S. R., A. C. Dumaual, L. J. Jenski, and W. Stillwell. 2001. Lipid phase separation in phospholipids bilayers and monolayers modeling the plasma membrane. Biochim. Biophys. Acta. 1512:317–328. [DOI] [PubMed] [Google Scholar]
- Sostarecz, A. G., C. M. McQuaw, A. G. Ewing, and N. Winograd. 2004. Phosphatidylethanolamine-induced cholesterol domains chemically identified with mass spectrometric imaging. J. Am. Chem. Soc. 126:13882–13883. [DOI] [PubMed] [Google Scholar]
- Stelly, N., S. Halpern, G. Nicolas, P. Fragu, and A. Adoutte. 1995. Direct visualization of a vast cortical calcium compartment in Paramecium by secondary ion mass spectrometry (SIMS) microscopy: possible involvement in exocytosis. J. Cell Sci. 108:1895–1909. [DOI] [PubMed] [Google Scholar]
- Strottrup, B., S. L. Veatch, and L. P. Keller. 2004. Nonequilibrium behavior in supported lipid membranes containing cholesterol. Biophys. J. 86:2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch, S. L., and S. L. Keller. 2003. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85:3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch, S. L., I. V. Polozov, K. Gawrisch, and L. P. Keller. 2004. Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophys. J. 86:2910–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga, M. P., L. R. Arrondo, F. M. Goni, A. Alonso, and D. Marsh. 2001. Interaction of cholesterol with sphingomyelin in mixed membranes containing phosphatidylcholines, studied by spin-label ESR and IR spectroscopies. A possible stabilization of gel-phase sphingolipid domains by cholesterol. Biochemistry. 40:2614–2622. [DOI] [PubMed] [Google Scholar]
- Wu, N., D. E. Cane, and C. Khosla. 2002. Quantitative analysis of the relative contributions of donor acyl carrier proteins, acceptor ketosynthases, and linker regions to intermodular transfer of intermediates in hybrid polyketide synthases. Biochemistry. 41:5056–5066. [DOI] [PubMed] [Google Scholar]
- Yuan, C., J. Furlong, P. Burgos, and L. J. Johnston. 2002. The size of lipid rafts: an atomic force microscopy study of ganglioside GM1 domains in sphingomyelin/DOPC/cholesterol. Biophys. J. 82:2526–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]