

Abstract
We study the folding mechanism of an analog of the C-peptide of ribonuclease A in explicit water by a replica-exchange multicanonical molecular dynamics simulation based on all-atom models. The multicanonical weight factor was determined by the combined use of the multicanonical replica-exchange method and the replica-exchange multicanonical algorithm. Using statistically reliable data thus obtained, we have examined the free-energy landscape of the peptide system. The global-minimum free-energy state in the landscape at room temperature has an α-helix structure with a distortion near the N-terminus. The state also has a salt bridge between Glu−-2 and Arg+-10 and an aromatic-aromatic interaction between Phe-8 and His+-12, both of which have been observed in x-ray and other experimental measurements. Principal component analysis clearly shows the different roles of these side-chain interactions in the peptide folding. The side-chain interaction between Phe-8 and His+-12 greatly enhances the stability of helical structure toward the C-terminal end, whereas the salt bridge between Glu−-2 and Arg+-10 mainly works as a restraint to prevent the α-helix structure from extending to the N-terminus. The free-energy landscape of C-peptide reveals a funnel-like shape where all of these interactions consistently exist only in the global-minimum state. This is the major reason why the native structure of the short helical peptide shows significant stability at low temperatures.
INTRODUCTION
A protein native structure is encoded in its amino acid sequence information and the structure is essential for a protein to work as a molecular machine (Anfinsen, 1973). Understanding the mechanism of protein folding and stability is therefore one of the central problems in molecular biology. Recent advances in experimental studies such as stopped-flow methods, hydrogen-exchange NMR, solution x-ray scattering, and protein engineering techniques allow us to characterize the folding intermediates and the denatured states of proteins. Owing to the information on such nonnative states, the understanding of the folding kinetics or thermodynamics has been greatly advanced in the last decade (Baldwin, 1995; Kuwajima and Arai, 1999).
Another progress in the protein-folding study has appeared in theoretical studies. Computational studies on protein folding comprise various approaches from simple lattice models to all-atom models with explicit solvent. Lattice models for proteins are useful to study the essence of the mechanism of protein folding and have often been employed (Chikenji et al., 1999; Dill and Chan, 1997; Go, 1983; Kolinski and Skolnick, 1994; Onuchic et al., 1997; Pande et al., 1998). Go and co-workers first studied the protein-folding mechanism by carrying out Monte Carlo (MC) simulations of simple lattice models and proposed the consistency principle of protein folding (Go, 1983). They claimed that there exists consistency among various interactions such as long-range and short-range interactions in the native structure of foldable proteins. Wolynes and co-workers formulated the principle of minimal frustration by taking advantage of analogy with spin glass problem (Onuchic et al., 1997). In their theory, the native structure is “minimally frustrated” compared with misfolded states. From this point of view, it is possible to describe protein folding in terms of free-energy landscapes (Dill and Chan, 1997; Dobson et al., 1998; Onuchic et al., 1997; Pande et al., 1998). Rapid transitions occur in a smooth energy landscape, whereas a rough energy landscape causes kinetic traps that slow down protein folding.
An all-atom model for a protein with explicit representation of solvent gives us the most detailed information about proteins. However, as mentioned above, insights on the free-energy landscape of proteins were first elucidated in lattice-model simulations or more theoretical studies. It is because there exist a large number of local-minimum states in the potential energy function and statistically reliable free-energy landscape could not be obtained by all-atom simulations until recently. Brooks and co-workers have overcome the problem by using the umbrella sampling techniques (Torrie and Valleau, 1977) combined with all-atom molecular dynamics (MD) simulations and shown the correlation between the shapes of free-energy landscape and the “topology” of protein native structures (Shea and Brooks, 2001). Their approach is very useful and gives us a lot of new insights into the protein-folding problem. However, it cannot be applied to a protein whose structure is unknown, because the method requires the native protein structure for constructing useful umbrella potentials before carrying out the simulations. Thus, to study the protein-folding mechanism of any proteins by using all-atom models, more powerful and more efficient algorithm for conformational sampling is necessary.
The generalized-ensemble algorithm that has been first developed in the studies of spin systems is now becoming a popular method in biological simulations (for recent reviews, see Mitsutake et al., 2001; Sugita and Okamoto, 2002). The generalized-ensemble algorithm has two advantages compared with the conventional simulation methods. First, the simulation can avoid getting trapped in states of local-minimum-energy states and sample a wide conformational space by realizing a random walk in the potential energy space. Secondly, just from one simulation run, thermodynamic quantities at any temperature can be obtained by using the single-histogram or multiple-histogram reweighting techniques (Ferrenberg and Swendsen, 1988, 1989; Kumar et al., 1992). In the last decade, various new generalized-ensemble algorithms such as the multicanonical algorithm (MUCA) (Berg and Neuhaus, 1991, 1992), simulated tempering method (Lyubartsev et al., 1992; Marinari and Parisi, 1992), the method based on Tsallis statistics (Hansmann and Okamoto, 1997), the replica-exchange method (REM) (Hukushima and Nemoto, 1996), and so on have been proposed. Generalized-ensemble algorithms are particularly suitable for studying the protein-folding problem, where we have to explore a wide range of conformational space: not only the native folded state but also the intermediate and denatured states. For instance, the free-energy landscape of a short peptide in gas phase was studied by one of the above generalized-ensemble algorithms (Hansmann et al., 1997, 1999). However, we are interested in a protein in aqueous solution, and for this purpose a more powerful generalized-ensemble algorithm is desirable. MUCA and REM have been originally developed in combination with MC simulations, but MD in the Cartesian coordinate space can be easily performed for the system containing a protein with explicit solvent. Thereby, MD versions of the multicanonical algorithm (Hansmann et al., 1996; Nakajima et al., 1997) and the replica-exchange method (Sugita and Okamoto, 1999) (the latter MD algorithm is referred to as REMD) have been developed and are becoming popular methods for protein simulations (Bartels and Karplus, 1998; Cheung et al., 2002; Feig et al., 2003; Garcia and Sanbonmatsu, 2001; Jang et al., 2002; Kamiya et al., 2002; Pitera and Swope, 2003; Rhee and Pande, 2003; Zhou et al., 2001). We then developed powerful generalized-ensemble algorithms that combine the merits of MUCA and REM; they are referred to as replica-exchange multicanonical algorithm (REMUCA) and multicanonical replica-exchange method (MUCAREM) (Mitsutake et al., 2003a,b; Sugita and Okamoto, 2000). We also generalized REM into multidimensions, which gives an efficient method for free-energy calculations (Sugita et al., 2000).
In this article, we study the mechanism of folding and stability of an analog of the C-peptide of ribonuclease A (Shoemaker et al., 1990, 1987) by carrying out a REMUCA simulation. The C-peptide analogs have been observed to have a partially distorted α-helix structure by CD (Shoemaker et al., 1990, 1987) and NMR experiments (Bruschweiler et al., 1995; Osterhout et al., 1989). Two side-chain interactions are found to be important for the folding and stability of the peptide conformation. One is the salt bridge between Glu−-2 and Arg+-10 and the other is the aromatic-aromatic interaction between Phe-8 and His+-12 (Osterhout et al., 1989; Shoemaker et al., 1990). The effects of the latter interaction, namely the (i, i + 4) Phe-His+ interaction, on helix stability has also been studied in an alanine-based peptide by CD experiments (Armstrong et al., 1993). In theoretical studies, the peptide has been studied by simulated annealing in gas phase (Okamoto et al., 1991), by multicanonical algorithm in implicit solvent (Hansmann and Okamoto, 1999; Schaefer et al., 1998), and by replica-exchange method in implicit solvent (La Penna et al., 2003; Ohkubo and Brooks, 2003). Here, we study the molecular mechanism of folding and stability for this peptide in explicit solvent by a generalized-ensemble simulation starting from a completely random initial conformation. By performing a 15-ns multicanonical MD simulation based on REMUCA, we successfully obtained an α-helix structure with the above two side-chain interactions as the global-minimum free-energy state. The results are further analyzed by using the principal component analysis (PCA) (Amadei et al., 1993; Garcia, 1992; Kitao et al., 1991) combined with the histogram reweighting techniques (Ferrenberg and Swendsen, 1988, 1989). PCA has been developed to study the dynamics of native proteins and it has been shown that a few principal components (PCs) are sufficient to represent the global motion of a native protein (for a recent review, see Kitao and Go, 1999). Recently, PCA has also been used to study the mechanism of protein folding and stability by combining with the generalized-ensemble algorithms such as multicanonical algorithm (Higo et al., 2001; Kamiya et al., 2002) and replica-exchange method (Garcia and Sanbonmatsu, 2001; Zhou et al., 2001). By describing the free-energy landscape along the first two PCs, we study the detailed molecular mechanism of folding and stability of C-peptide.
MATERIALS AND METHODS
Generalized-ensemble algorithms
We first briefly review the generalized-ensemble algorithms employed in this work (Mitsutake et al., 2003a,b; Sugita and Okamoto, 2000). The multicanonical ensemble is based on the following non-Boltzmann weight factor 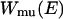 so that the probability distribution of potential energy E,
so that the probability distribution of potential energy E, 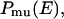 is flat and a one-dimensional random walk in potential energy space is realized:
is flat and a one-dimensional random walk in potential energy space is realized:
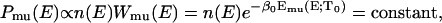 |
(1) |
where 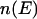 is the density of states and
is the density of states and 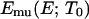 is the so-called multicanonical potential energy function at an arbitrary reference temperature
is the so-called multicanonical potential energy function at an arbitrary reference temperature 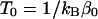 (
(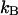 is Boltzmann's constant). Hence, the multicanonical weight factor
is Boltzmann's constant). Hence, the multicanonical weight factor 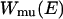 is inversely proportional to the density of states
is inversely proportional to the density of states 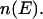 Multicanonical molecular dynamics algorithm is based on solving a “modified” Newton equation at temperature
Multicanonical molecular dynamics algorithm is based on solving a “modified” Newton equation at temperature 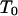 (Hansmann et al., 1996; Nakajima et al., 1997). Namely, the gradient of the multicanonical potential energy
(Hansmann et al., 1996; Nakajima et al., 1997). Namely, the gradient of the multicanonical potential energy  rather than the regular potential energy E is used for the force term. After a multicanonical production run, the canonical-ensemble average of a physical quantity A at any temperature
rather than the regular potential energy E is used for the force term. After a multicanonical production run, the canonical-ensemble average of a physical quantity A at any temperature 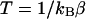 is obtained from
is obtained from
 |
(2) |
where the density of states can be determined from the single-histogram reweighting techniques (Ferrenberg and Swendsen, 1988):
 |
(3) |
Here, 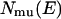 is the potential energy histogram that was obtained from the multicanonical production run.
is the potential energy histogram that was obtained from the multicanonical production run.
In REM, on the other hand, a random walk is realized by simulating 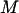 noninteracting copies (or, replicas) of the original system at different temperatures,
noninteracting copies (or, replicas) of the original system at different temperatures, 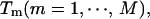 and by trying to exchange neighboring temperatures with the Metropolis criterion. The canonical ensemble at any temperature can be reproduced by the density of states that can be determined from the multiple-histogram reweighting techniques (Ferrenberg and Swendsen, 1989; Kumar et al., 1992) as follows:
and by trying to exchange neighboring temperatures with the Metropolis criterion. The canonical ensemble at any temperature can be reproduced by the density of states that can be determined from the multiple-histogram reweighting techniques (Ferrenberg and Swendsen, 1989; Kumar et al., 1992) as follows:
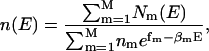 |
(4) |
and
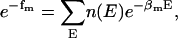 |
(5) |
where nm, 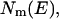 and
and 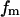 are the total number of snapshots (samples), the potential-energy histogram, and dimensionless Helmholtz free energy, respectively, at temperature
are the total number of snapshots (samples), the potential-energy histogram, and dimensionless Helmholtz free energy, respectively, at temperature 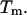 Eqs. 4 and 5 are solved self-consistently to obtain
Eqs. 4 and 5 are solved self-consistently to obtain  and
and 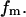
In REMUCA the multicanonical weight factor  or equivalently, the density of states
or equivalently, the density of states  is first determined from the results of a short REM simulation by the multiple-histogram reweighting techniques of Eqs. 4 and 5. A multicanonical production run is then performed with this weight factor. In MUCAREM a replica-exchange production run with replicas corresponding to multicanonical ensembles with different energy ranges is performed. The details of REMUCA and MUCAREM have been given in the original articles (Mitsutake et al., 2003a,b; Sugita and Okamoto, 2000) and the recent review articles (Mitsutake et al., 2001; Sugita and Okamoto, 2002).
is first determined from the results of a short REM simulation by the multiple-histogram reweighting techniques of Eqs. 4 and 5. A multicanonical production run is then performed with this weight factor. In MUCAREM a replica-exchange production run with replicas corresponding to multicanonical ensembles with different energy ranges is performed. The details of REMUCA and MUCAREM have been given in the original articles (Mitsutake et al., 2003a,b; Sugita and Okamoto, 2000) and the recent review articles (Mitsutake et al., 2001; Sugita and Okamoto, 2002).
Computational details
In the model of simulations, the N-terminus and the C-terminus of the C-peptide analog were blocked with the acetyl group and the N-methyl group, respectively. The number of amino acids is 13 and the amino acid sequence is: Ace-Ala-Glu−-Thr-Ala-Ala-Ala-Lys+-Phe-Leu-Arg+-Ala-His+-Ala-Nme (Shoemaker et al., 1990, 1987). The initial configuration of our simulation was first generated by a high-temperature molecular dynamics simulation (at T = 1000 K) in gas phase, starting from a fully extended conformation. We randomly selected one of the structures that do not have any secondary structures such as α-helix and β-sheet. The peptide was then solvated in a sphere of radius 22 Å, in which 1387 water molecules were included (see Fig. 1). A harmonic restraint was applied to prevent the water molecules from going out of the sphere. The dielectric constant was set equal to 1.0. TIP3P model (Jorgensen et al., 1983) was used for water molecules and the force-field parameters for protein were taken from the all-atom version of AMBER parm99 (Cornell et al., 1995; Wang et al., 2000). We have recently compared the secondary-structure contents of C-peptide in explicit water for six force-field parameters, namely, AMBER parm94, AMBER parm96, AMBER parm99, CHARMM22, OPLS-AA/L, and GROMOS96, and we found that the results with AMBER parm99 were the most consistent with the experimental implications (Yoda et al., 2004a,b). This is why we used AMBER parm99 in this study.
FIGURE 1.

The initial configuration of C-peptide in explicit water, which was used in all of the 32 replicas of the first REMD simulation (REMD1 in Table 1). The red solid circles stand for the oxygen atoms of water molecules. The number of water molecules is 1387, and they are placed in a sphere of radius 22 Å. As for the peptide, besides the backbone structure (blue), side chains of only Glu−-2, Phe-8, Arg+-10, and His+-12 are shown (yellow). The figure was created with Molscript (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997).
In this article, we employed MD simulations based on REMUCA algorithm. As a thermostat of MD simulations, we have adopted the constraint method in which the total kinetic energy is constant (Evans and Moriss, 1983; Hoover et al., 1982). The peptide was treated as a flexible molecule, whereas water molecules were treated as rigid bodies. Newton equations in the Cartesian coordinates for the peptide and translational motion of water molecules were integrated by using the leap-frog algorithm. The Euler equations for the quarternion parameters for rotational motions of water molecules were integrated by using the leap-frog method of Fincham's algorithm (Fincham, 1992). The unit time step, Δt, was set to 0.5 fs. The long-range interactions were calculated by using the cell multipole method (Ding et al., 1992) to avoid the artifact of the truncation of the electrostatic interactions. The modified version (Kitao et al., 1998; Sugita and Kitao, 1998) of the software PRESTO version 2 (Morikami et al., 1992) was used.
RESULTS AND DISCUSSION
Random walks in potential energy space and in conformational space
In Table 1 the essential parameters of the simulations performed in this article are summarized. We first performed a REMD simulation with 32 replicas for 100 ps per replica (REMD1 in Table 1). During this REMD simulation, replica exchange was tried every 200 MD steps. Using the obtained potential-energy histogram of each replica as input data to the multiple-histogram analysis in Eqs. 4 and 5, we obtained the first estimate of the multicanonical weight factor, or the density of states. We divided this multicanonical weight factor into four multicanonical weight factors that cover different energy regions (Mitsutake et al., 2003a,b; Sugita and Okamoto, 2000) and assigned these multicanonical weight factors into four replicas (the weight factors cover the potential energy ranges from −13,791.5 to −11,900.5 kcal/mol, from −12,962.5 to −10,796.5 kcal/mol, from −11,900.5 to −9524.5 kcal/mol, and from −10,796.5 to −8293.5 kcal/mol). We then carried out a MUCAREM simulation with four replicas for 1 ns per replica (MUCAREM1 in Table 1), in which replica exchange was tried every 1000 MD steps. We again used the potential-energy histogram of each replica as the input data to the multiple-histogram analysis and finally obtained the multicanonical weight factor with high precision. As a production run, we carried out a 15-ns multicanonical MD simulation with one replica (REMUCA1 in Table 1) and the results of this production run were analyzed in detail.
TABLE 1.
Summary of parameters in REMD, MUCAREM, and REMUCA simulations
| Number of replicas, M | Temperature, Tm (K) (m = 1, …, M) | MD steps per replica | |
|---|---|---|---|
| REMD1* | 32 | 250, 258, 267, 276, 286, 295, 305, 315, 326, 337, 348, 360, 372, 385, 398, 411, 425, 440, 455, 470, 486, 502, 519, 537, 555, 574, 593, 613, 634, 655, 677, 700 | 2.0 × 105 |
| MUCAREM1 | 4 | 360, 440, 555, 700 | 2.0 × 106 |
| REMUCA1 | 1 | 700 | 3.0 × 107 |
REMD1 stands for the replica-exchange molecular dynamics simulation, MUCAREM1 stands for the multicanonical replica-exchange molecular dynamics simulation, and REMUCA1 stands for the final multicanonical molecular dynamics simulation (the production run) of REMUCA. The results of REMD1 were used to determine the multicanonical weight factors for MUCAREM1, and those of MUCAREM1 were used to determine the multicanonical weight factor for REMUCA1. See Mitsutake et al. (2003a,b) and Sugita and Okamoto (2000) for details of these generalized-ensemble algorithms.
In Fig. 2 we show the probability distributions of potential energy that were obtained from the above three generalized-ensemble simulations, namely, REMD1, MUCAREM1, and REMUCA1. We see in Fig. 2 a that there are enough overlaps between all pairs of neighboring canonical distributions, suggesting that there were sufficient numbers of replica exchange in REMD1. We see in Fig. 2 b that there are good overlaps between all pairs of neighboring multicanonical distributions, implying that MUCAREM1 also performed properly. Finally, the multicanonical distribution in Fig. 2 c is completely flat between ∼−13,000 kcal/mol and ∼−8000 kcal/mol. The results suggest that a free random walk was realized in this energy range. The canonical distributions at 290, 300, 500, and 700 K reweighted by the histogram analyses are also plotted in Fig. 2 c. These canonical distributions lie within the energy range covered by the REMUCA1 simulation (two extreme distributions lie at the edges). This implies that any thermodynamic quantities at temperatures between 290 and 700 K can be evaluated accurately from the trajectory of REMUCA1 by the reweighting techniques.
FIGURE 2.

Probability distributions of potential energy of the C-peptide system obtained from (a) REMD1, (b) MUCAREM1, and (c) REMUCA1. See Table 1 for the parameters of the simulations. Dashed curves in panel c are the reweighted canonical distributions at 290, 300, 500, and 700 K (from left to right).
In Fig. 3 a we show the time series of potential energy from REMUCA1. We indeed observe a random walk covering as much as 5000 kcal/mol of energy range. We show in Fig. 3 b the average potential energy as a function of temperature, which was obtained from the trajectory of REMUCA1 by the reweighting techniques. The average potential energy monotonically increases as the temperature increases. We have also calculated the average helicity as a function of temperature by using the definition in DSSP (Kabsch and Sander, 1983). The helicity monotonically decreases as the temperature increases. The values at 300, 400, and 600 K are 23, 19, and 15%, respectively, which are consistent with the experimental values of ∼50% at 276 K (Shoemaker et al., 1990, 1987) and ∼30% at 300 K (Yoda et al., 2004b).
FIGURE 3.

Time series of potential energy of the C-peptide system from the REMUCA production run (REMUCA1 in Table 1) (a) and the average potential energy as a function of temperature (b). The latter was obtained from the trajectory of REMUCA1 by the single-histogram reweighting techniques.
Note that the accuracy of these average quantities depends on the “quality” of the random walk, and the measure for this quality can be given by the number of tunneling events (Berg and Neuhaus, 1992; Mitsutake et al., 2003b). One tunneling event is defined by a trajectory that goes from EH to EL and back, where EH and EL are the values near the highest energy and the lowest energy, respectively, which the random walk can reach. If EH is sufficiently high, the trajectory gets completely uncorrelated when it reaches EH. On the other hand, when the trajectory reaches near EL, it tends to get trapped in local-minimum states. We thus consider that the more tunneling events we observe during a fixed number of MD steps, the more efficient the method is as a generalized-ensemble algorithm (or, the average quantities obtained by the reweighting techniques are more reliable). Here, we took EH = −8250 kcal/mol and EL = −12,850 kcal/mol. The random walk in REMUCA1 yielded as many as 55 tunneling events in 15 ns. The corresponding numbers of tunneling events for REMD1 and for MUCAREM1 were 0 in 3.2 ns and 5 in 4 ns, respectively. Hence, REMUCA is the most efficient and reliable among the three generalized-ensemble algorithms.
Given a random walk in potential energy space, the question is how wide a conformational space this multicanonical molecular dynamics production run (REMUCA1) sampled. Because there is no unique parameter that can be used as a measure of sampling efficiency in the conformational space, we checked time series of four quantities: the radius of gyration of Cα atoms (Rg), the root-mean-square deviation of Cα atoms from the corresponding x-ray structure (Tilton et al., 1992) (RMSD), the salt-bridge distance (D_Salt) that is defined to be the minimal distance between the side-chain carboxyl oxygens of Glu−-2 and the side-chain amide hydrogens of Arg+-10, and the distance between the center of geometry of the side chains of Phe-8 and His+-12 (D_FH). The first two parameters are selected as a measure of the backbone motion, whereas the last two are representative side-chain interactions. The results are shown in Fig. 4. We observe good random walks for all these quantities. It means that both the backbone atoms and the side-chain atoms moved greatly during 15 ns without getting trapped in any local-minimum states.
FIGURE 4.

Time series of conformational parameters of C-peptide from the REMUCA production run (REMUCA1 in Table 1). (a) Radius of gyration with respect to Cα atoms (Rg), (b) RMSD of Cα atoms from the corresponding x-ray structure (RMSD), (c) the salt-bridge distance between Glu−-2 and Arg+-10 (D_Salt), and (d) the distance between side chains of Phe-8 and His+-12 (D_FH). See Table 2 footnote for the definitions.
These good random walks in the potential-energy space and conformational space achieved multiple events of structural transitions from an unfolded conformation to the native conformation during the multicanonical molecular dynamics (REMUCA) simulation. It means that our generalized-ensemble simulation converged very well. However, if generalized-ensemble simulations for protein folding had been started from the native conformation or the multiple conformations that are close to the native conformation, the convergence of the simulations should be checked carefully. The most reliable test for the convergence would be to perform two independent simulation runs starting from the following two different initial structures: the native conformation and an unfolded conformation (Young and Pande, 2003) and to compare the averaged quantities obtained with the two simulation runs.
We also checked the correlation coefficients of these four parameters during REMUCA1 to study the mutual relationship. We list the results in Table 2. We find a positive correlation between RMSD and D_Salt, whereas a negative correlation is found between RMSD and Rg. The latter is due to the fact that in C-peptide the backbone collapse is not the major folding event and that too much collapsed conformations are those of misfolded states, because helical conformations will be destroyed. In Table 2 we also list correlation coefficients of helicity with the above parameters. We separately considered the residues 1–3 and 4–13. Helix(1–3) and Helix(4–13) stand for the number of helical residues in residues 1–3 and in residues 4–13, respectively. We find negative correlations between Helix(4–13) and RMSD as well as D_FH. This implies that as the conformation approaches the native structure and as the side chains of Phe-8 and His+-12 get closer, the helicity in residues 4–13 is enhanced. However, these observations are based not on the canonical ensemble but on the artificial multicanonical ensemble that contains all the information of physical quantities at a wide range of temperatures. In the next subsection we further study the folding mechanism of C-peptide in detail by investigating the free-energy landscape in canonical ensemble reweighted from the multicanonical ensemble.
TABLE 2.
Correlation coefficients, 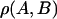 , between structural quantities
, between structural quantities
| Rg* | RMSD | D_Salt | D_FH | Helix(1–3) | Helix(4–13) | |
|---|---|---|---|---|---|---|
| Rg | – | – | – | – | – | – |
| RMSD | −0.447 | – | – | – | – | – |
| D_Salt | 0.123 | 0.440 | – | – | – | – |
| D_FH | −0.106 | 0.188 | −0.093 | – | – | – |
| Helix(1–3) | 0.136 | −0.148 | −0.07 | 0.00 | – | – |
| Helix(4–13) | −0.07 | −0.343 | −0.05 | −0.284 | 0.06 | – |
Correlation coefficient, 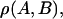 is defined by the following equation:
is defined by the following equation: 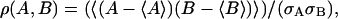 where 〈A〉 stands for the average of a physical quantity
where 〈A〉 stands for the average of a physical quantity 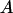 over the trajectory of REMUCA1 in Table 1 and
over the trajectory of REMUCA1 in Table 1 and 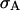 is the corresponding standard deviation. The radius of gyration, Rg, is with respect to only Cα atoms. The root-mean-square deviation, RMSD, from the corresponding x-ray backbone structure is also with respect to Cα atoms. The salt-bridge distance, D_Salt, is defined to be the minimal distance between the side-chain carboxyl oxygens of Glu−-2 and the side-chain amide hydrogens of Arg+-10. The side-chain distance, D_FH, is defined to be the distance between the center of geometry of the side chains of Phe-8 and His+-12. Helix(1–3) and Helix(4–13) stand for the number of helical residues in residues 1–3 and in residues 4–13, respectively.
is the corresponding standard deviation. The radius of gyration, Rg, is with respect to only Cα atoms. The root-mean-square deviation, RMSD, from the corresponding x-ray backbone structure is also with respect to Cα atoms. The salt-bridge distance, D_Salt, is defined to be the minimal distance between the side-chain carboxyl oxygens of Glu−-2 and the side-chain amide hydrogens of Arg+-10. The side-chain distance, D_FH, is defined to be the distance between the center of geometry of the side chains of Phe-8 and His+-12. Helix(1–3) and Helix(4–13) stand for the number of helical residues in residues 1–3 and in residues 4–13, respectively.
Free-energy landscape along the principal component axes
To study the free-energy landscape of a protein, principal component analysis (PCA) is known to be a useful tool. Here, we also applied PCA to characterize the free-energy landscape of C-peptide in solution (namely, the free-energy landscape is given as a potential of mean force with principal component axes as reaction coordinates). We first calculated the average structure of C-peptide by using 30,000 conformations that were stored every 1000 MD steps during REMUCA1. We then prepared a variance-covariance matrix and the obtained principal component axes by diagonalizing the matrix. Finally, we projected each peptide conformation with the statistical weight calculated by the histogram reweighting techniques to draw the free-energy landscape at each temperature. Before drawing the free-energy landscape, we checked the contribution of each principal component to the total root-mean-square fluctuations. The ratios of the contribution of the first five principal components are 22.8, 17.0, 9.1, 5.7, and 4.6%. The first two PCs contribute ∼40% of root-mean-square fluctuations. It means that even in large number of partially unfolded conformations in the folding pathway, a few reaction coordinates that characterize the folding pathway can be clearly defined.
Although PCs are not direct conformational parameters, it is meaningful to check the relation between the first PCs and other conformational parameters. We again calculated the correlation coefficients of time series of PCs and the four parameters (Rg, RMSD, D_Salt, and D_FH). The correlation coefficients between PC1 and Rg, RMSD, D_Salt, and D_FH are 0.21, 0.14, 0.60, and −0.44, respectively. Thus, PC1 is found to be closely related to the side-chain motion of C-peptide (i.e., D_Salt and D_FH). On the other hand, PC2 is correlated well with the backbone motion of C-peptide, because the correlation coefficients between PC2 and Rg, RMSD, D_Salt, and D_FH are 0.63, −0.65, −0.10, and −0.33, respectively. Hence, the first two PCs are shown to represent both backbone and side-chain motions of C-peptide. We believe that these two PCs are sufficient to characterize the free-energy landscape of C-peptide in this study.
In Fig. 5 the potentials of mean force along the first two principal component axes at 300, 350, 400, and 500 K are shown. The potential of mean force at temperature T, 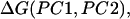 is defined by the following equation:
is defined by the following equation:
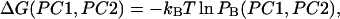 |
(6) |
where PC1 and PC2 are the first two principal component values and 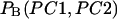 is the “reweighted” canonical probability distribution at temperature T. At high temperatures, we observe smooth free-energy landscapes where the generalized-ensemble simulation can sample a large number of conformational states. On the other hand, at 300 K, the free-energy landscape is more rugged and there exist three distinct minima in the landscape (LM1, LM2, and LM3 in Fig. 5 a), which correspond to three local-minimum free-energy states. Because the probabilities staying in each minima at 300 K are ∼20.1% for LM1 (−0.5 ≤ PC1 ≤ 0.2, 0.2 ≤ PC2 ≤ 1.0), 12.5% for LM2 (1.8 ≤ PC1 ≤ 2.1, −1.5 ≤ PC2 ≤ −0.8), and 5.1% for LM3 (−1.6 ≤ PC1 ≤ −1.4, −0.3 ≤ PC1 ≤ 0.2), respectively, LM1 is, hereafter, referred to as the global-minimum free-energy state (GM).
is the “reweighted” canonical probability distribution at temperature T. At high temperatures, we observe smooth free-energy landscapes where the generalized-ensemble simulation can sample a large number of conformational states. On the other hand, at 300 K, the free-energy landscape is more rugged and there exist three distinct minima in the landscape (LM1, LM2, and LM3 in Fig. 5 a), which correspond to three local-minimum free-energy states. Because the probabilities staying in each minima at 300 K are ∼20.1% for LM1 (−0.5 ≤ PC1 ≤ 0.2, 0.2 ≤ PC2 ≤ 1.0), 12.5% for LM2 (1.8 ≤ PC1 ≤ 2.1, −1.5 ≤ PC2 ≤ −0.8), and 5.1% for LM3 (−1.6 ≤ PC1 ≤ −1.4, −0.3 ≤ PC1 ≤ 0.2), respectively, LM1 is, hereafter, referred to as the global-minimum free-energy state (GM).
FIGURE 5.

Potential of mean force (kcal/mol) of the C-peptide system along the first two principal components at (a) 300, (b) 350, (c) 400, and (d) 500 K. The free energy was calculated from the results of REMUCA production run (REMUCA1 in Table 1) by the single-histogram reweighting techniques and normalized so that the global-minimum state has the value zero. LM1 (GM), LM2, and LM3 represent three distinct minimum free-energy states.
To characterize these three minima, we considered only the low-energy conformations that have the total potential energy <−12,500 kcal/mol (including contributions from water molecules), which is the highest value that can be reached by conformations in the canonical ensemble at T = 300 K (see Fig. 2 c) (it is actually the value in which lnP(E) = −20). These conformations contribute mainly to the average values or the probabilities in free-energy landscape at 300 K (conformations with potential energy E > −12,500 kcal/mol hardly contribute to the properties at 300 K). In Fig. 6 we projected these peptide conformations onto the first two principal axes. The conformations that have the salt bridge between Glu−-2 and Arg+-10 are plotted in green, and those with the side-chain interaction between Phe-8 and His+-12 in red. We also plotted the conformations that have more than six helical residues in the DSSP definition (Kabsch and Sander, 1983) (blue). Here, the criterion for the salt-bridge formation is that D_Salt < 3.0 Å where the border between a clear peak of the salt bridge and a broad distribution is located (data not shown). In Fig. 6 the global-minimum free-energy state has more than six helical residues with the salt bridge between Glu−-2 and Arg+-10 and the aromatic-aromatic interactions between Phe-8 and His+-12. The structures in LM2 have a contact between Phe-8 and His+-12, but the salt bridge is not formed. On the other hand, the structures in LM3 have the salt bridge between Glu−-2 and Arg+-10, but it does not have a contact between Phe-8 and His+-12. To visualize these observations in the free-energy landscape of C-peptide at 300 K, we show representative conformations at these minima in Fig. 7. Each of them corresponds to one of the free-energy minimum structures at the lowest points in the basins and have the principal component values listed in Table 3. Other characteristics of these conformations are also listed in the table. Thus, only the structures at GM satisfy all of the interactions that have been observed by x-ray structures and other experimental studies.
FIGURE 6.

Interaction consistency in the free-energy landscape of the C-peptide system. Red points represent conformations with D_FH < 6 Å, green points those with D_Salt < 3 Å, and blue points those that have more than six helical residues defined by DSSP (Kabsch and Sander, 1983). Only the configurations whose total potential energy is <−12,500 kcal/mol are considered. See Table 2 footnote for the definitions.
FIGURE 7.

The representative structures at the global-minimum free-energy state (a, GM) and the two local-minimum states (b, LM2; c, LM3). As for the peptide structures, besides the backbone structure, side chains of only Glu−-2, Phe-8, Arg+-10, and His+-12 are shown in ball-and-stick model.
TABLE 3.
Properties of the x-ray structure of C-peptide and the representative structures found in the free-energy landscape at 300 K
| PC1* | PC2 | Rg (Å) | RMSD (Å) | D_Salt (Å) | D_FH (Å) | |
|---|---|---|---|---|---|---|
| X-ray | −0.29 | 0.84 | 6.15 | 0.0 | 3.86 | 5.03 |
| GM | −0.09 | 0.92 | 6.20 | 1.79 | 2.78 | 5.27 |
| LM2 | 1.89 | −1.14 | 5.90 | 5.54 | 23.5 | 5.83 |
| LM3 | −1.49 | −0.07 | 6.13 | 3.80 | 2.92 | 10.83 |
Different role of the side-chain interactions in stabilization of C-peptide
These two side-chain interactions found in the global-minimum free-energy state in the landscape at 300 K as well as in the experimental measurements have quite different characteristics. The interaction between Glu−-2 and Arg+-10 can be considered as long-range electrostatic interactions, whereas the interaction between Phe-8 and His+-12 is a short-range interaction mainly caused by hydrophobic and weak electrostatic interactions. In LM2 and LM3, the peptide is trapped in one of the collapsed local-minimum states where one of the major stabilizing factors is lost. Thus, the free-energy landscape of C-peptide reveals a funnel-like shape where all the important interactions consistently exist only in the global-minimum free-energy state. Unless they are consistent, a short peptide like C-peptide cannot fold into stable conformations but becomes a random-coil state.
To understand the role of each side-chain interaction on the stabilization of C-peptide, we examine Fig. 6 in more detail. We see that the green points (salt bridge) and the blue points (helix) do not overlap except for the region around GM. On the other hand, the red points (D_FH < 6.0 Å) and the blue points have significant overlaps. Because His+-12 is four residues apart from Phe-8 in sequence, their side chains come close to each other when α-helix is formed between the two residues (note that one turn of α-helix corresponds to 3.6 residues). We thus claim that the major factor to stabilize the C-terminal helix of C-peptide is not the salt-bridge formation but the side-chain interaction of Phe-8 and His+-12. The salt-bridge formation may not contribute to the stabilization of the α-helix structure of C-peptide directly, but its major role is to prevent the α-helix structure from extending to the N-terminus.
Dependence on the force-field parameters
Recent studies by ours and other groups highlighted that the small differences in the force-field parameters for proteins, in particular, the backbone torsion parameters, significantly affect the secondary-structure propensities for small peptides (Yoda et al., 2004a,b; Zhou, 2003). For instance, AMBER parm94 is generally known to favor α-helix structures too much. Our previous results indicated that the helical content generated by the simulation in explicit solvent with AMBER parm99 becomes much less than that with AMBER parm94 (Yoda et al. 2004a,b), whereas another study based on GB/SA models indicated that AMBER parm99 is also significantly biased toward helical structures (Zhou, 2003). The discrepancy is presumably due to the difference of the solvent models, because GB/SA model tends to favor helical structures more than the explicit solvent model (Nymeyer and Garcia, 2003; Zhou, 2003). We have compared the secondary-structure contents of C-peptide and the C-terminal β-hairpin of protein G in explicit water for six force-field parameters, namely, AMBER parm94, AMBER parm96, AMBER parm99, CHARMM22, OPLS-AA/L, and GROMOS96. We found that the results with AMBER parm99 and CHARMM22 were the most consistent with the experimental implications for C-peptide in the sense that AMBER parm94 created too much α-helix, whereas AMBER parm96, OPLS-AA/L, and GROMOS96 produced too little amount of α-helix. After finishing all the calculations for C-peptide, several updates for the backbone torsion parameters in these force-field parameters have been proposed (Garcia and Sanbonmatsu, 2002, Duan et al., 2003, Sakae and Okamoto, 2003, MacKerell et al., 2004). Although the simulations using these updated parameters may demonstrate better results on the secondary-structure contents, we believe that the current study with AMBER parm99 is meaningful and also contributes to our understanding of folding and stability for C-peptide, at least, on the role of side-chain interactions, because these interactions are determined mainly by the atomic parameters in the side chains. Toward the complete understanding for protein folding with computer simulations, efficient conformational sampling methods like the generalized-ensemble approaches and accurate force-field parameters are still required.
CONCLUSIONS
In this article we have studied the folding mechanism of an analog of the C-peptide of ribonuclease A in explicit water by a replica-exchange multicanonical molecular dynamics simulation based on all-atom models. Using this efficient conformational sampling method, we observed the free-energy landscape of this peptide at different temperatures. In the free-energy landscape at room temperature, three distinct free-energy minima were found, and the global free-energy minimum structure has an α-helix structure with a distortion near the N-terminus, which is in good agreement with the observations by CD or NMR experiments. The global-minimum free-energy structure has two major side-chain interactions, namely, that between Glu−-2 and Arg+-10 and that between Phe-8 and His+-12. The former interaction works as a restraint to prevent the α-helix structure from extending to the N-terminus, whereas the latter interaction greatly enhances the stability of helical structure toward the C-terminal ends. All these interactions consistently exist only in the global-minimum free-energy structure. The consistency principle or the principle of minimal frustration is thus satisfied even with the small foldable peptide.
Acknowledgments
Our simulations were performed on the computers at the Research Center for Computational Science, Okazaki National Research Institutes.
This work was supported, in part, by grants from the Research for the Future Program of the Japan Society for the Promotion of Science (JSPS-RFTF98P01101) and from the NAREGI Nanoscience Project (Y.O.), and Grants-in-Aid for Scientific Research on Priority Areas (“Membrane Interface” (Y.S.) and “Water and Biomolecules” (Y.O.)) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- Anfinsen, C. B. 1973. Principles that govern folding of protein chains. Science. 181:223–230. [DOI] [PubMed] [Google Scholar]
- Armstrong, K. M., R. Fairman, and R. L. Baldwin. 1993. The (i, i+4) Phe-His interaction studied in an alanine-based alpha-helix. J. Mol. Biol. 230:284–291. [DOI] [PubMed] [Google Scholar]
- Baldwin, R. L. 1995. The nature of protein-folding pathways - the classical versus the new view. J. Biomol. NMR. 5:103–109. [DOI] [PubMed] [Google Scholar]
- Bartels, C., and M. Karplus. 1998. Probability distributions for complex systems: adaptive umbrella sampling of the potential energy. J. Phys. Chem. B. 102:865–880. [Google Scholar]
- Berg, B. A., and T. Neuhaus. 1991. Multicanonical algorithms for 1st order phase-transitions. Phys. Lett. B. 267:249–253. [DOI] [PubMed] [Google Scholar]
- Berg, B. A., and T. Neuhaus. 1992. Multicanonical ensemble - a new approach to simulate 1st-order phase-transitions. Phys. Rev. Lett. 68:9–12. [DOI] [PubMed] [Google Scholar]
- Bruschweiler, R., D. Morikis, and P. E. Wright. 1995. Hydration of the partially folded peptide Rn-24 studied by multidimensional NMR. J. Biomol. NMR. 5:353–356. [DOI] [PubMed] [Google Scholar]
- Cheung, M. S., A. E. Garcia, and J. N. Onuchic. 2002. Protein folding mediated by solvation: water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc. Natl. Acad. Sci. USA. 99:685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikenji, G., M. Kikuchi, and Y. Iba. 1999. Multi-self-overlap ensemble for protein folding: ground state search and thermodynamics. Phys. Rev. Lett. 83:1886–1889. [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, J. Merz, K. M., D. M. Ferguson, D. Spellmeyer, T. Fox, J. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Dill, K. A., and H. S. Chan. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4:10–19. [DOI] [PubMed] [Google Scholar]
- Ding, H.-Q., N. Karasawa, and I. W. A. Goddard. 1992. Atomic level simulations on million particles: the cell multipole method for Coulomb and London nonbond interactions. J. Chem. Phys. 97:4309–4315. [Google Scholar]
- Dobson, C. M., A. Sali, and M. Karplus. 1998. Protein folding: a perspective from theory and experiment. Angew. Chem. Int. Ed. Engl. 37:868–893. [DOI] [PubMed] [Google Scholar]
- Duan, Y., C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, and T. Lee. 2003. A point-charge force field for molecular mechanics simulations of proteins. J. Comput. Chem. 24:1999–2012. [DOI] [PubMed] [Google Scholar]
- Evans, D. J., and G. P. Moriss. 1983. The isothermal isobaric molecular-dynamics ensemble. Phys. Lett. A. 98:433–436. [Google Scholar]
- Feig, M., A. D. MacKerell, and C. L. Brooks. 2003. Force field influence on the observation of pi-helical protein structures in molecular dynamics simulations. J. Phys. Chem. B. 107:2831–2836. [Google Scholar]
- Ferrenberg, A. M., and R. H. Swendsen. 1988. New Monte-Carlo technique for studying phase-transitions. Phys. Rev. Lett. 61:2635–2638. [DOI] [PubMed] [Google Scholar]
- Ferrenberg, A. M., and R. H. Swendsen. 1989. Optimized Monte-Carlo data-analysis. Phys. Rev. Lett. 63:1195–1198. [DOI] [PubMed] [Google Scholar]
- Fincham, D. 1992. Leapfrog rotational algorithms. Mol. Sim. 8:165–178. [Google Scholar]
- Garcia, A. E. 1992. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 68:2696–2699. [DOI] [PubMed] [Google Scholar]
- Garcia, A. E., and K. Y. Sanbonmatsu. 2001. Exploring the energy landscape of a beta hairpin in explicit solvent. Proteins. 42:345–354. [DOI] [PubMed] [Google Scholar]
- Garcia, A. E., and K. Y. Sanbonmatsu. 2002. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA. 99:2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go, N. 1983. Theoretical studies of protein folding. Annu. Rev. Biophys. Bioeng. 12:183–210. [DOI] [PubMed] [Google Scholar]
- Hansmann, U. H., M. Masuya, and Y. Okamoto. 1997. Characteristic temperatures of folding of a small peptide. Proc. Natl. Acad. Sci. USA. 94:10652–10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmann, U. H., and Y. Okamoto. 1997. Generalized-ensemble Monte Carlo method for systems with rough energy landscape. Phys. Rev. E. 56:2228–2233. [Google Scholar]
- Hansmann, U. H., Y. Okamoto, and F. Eisenmenger. 1996. Molecular dynamics, Langevin and hybrid Monte Carlo simulations in a multicanonical ensemble. Chem. Phys. Lett. 259:321–330. [Google Scholar]
- Hansmann, U. H., Y. Okamoto, and J. N. Onuchic. 1999. The folding funnel landscape for the peptide Met-enkephalin. Proteins. 34:472–483. [DOI] [PubMed] [Google Scholar]
- Hansmann, U. H. E., and Y. Okamoto. 1999. Effects of side-chain charges on alpha-helix stability in C-peptide of ribonuclease A studied by multicanonical algorithm. J. Phys. Chem. B. 103:1595–1604. [Google Scholar]
- Higo, J., N. Ito, M. Kuroda, S. Ono, N. Nakajima, and H. Nakamura. 2001. Energy landscape of a peptide consisting of alpha-helix, 3(10)-helix, beta-turn, beta-hairpin, and other disordered conformations. Protein Sci. 10:1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover, W. G., A. J. C. Ladd, and B. Moran. 1982. High-strain-rate plastic flow studied via non-equilibrium molecular dynamics. Phys. Rev. Lett. 48:1818–1820. [Google Scholar]
- Hukushima, K., and K. Nemoto. 1996. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 65:1604–1608. [Google Scholar]
- Jang, S. M., Y. Pak, and S. M. Shin. 2002. Multicanonical ensemble with Nose-Hoover molecular dynamics simulation. J. Chem. Phys. 116:4782–4786. [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhara, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- Kamiya, N., J. Higo, and H. Nakamura. 2002. Conformational transition states of a beta-hairpin peptide between the ordered and disordered conformations in explicit water. Protein Sci. 11:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao, A., and N. Go. 1999. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 9:164–169. [DOI] [PubMed] [Google Scholar]
- Kitao, A., S. Hayward, and N. Go. 1998. Energy landscape of a native protein: jumping-among-minima model. Proteins. 33:496–517. [DOI] [PubMed] [Google Scholar]
- Kitao, A., F. Hirata, and N. Go. 1991. The effects of solvent on the conformation and the collective motions of protein: normal mode analysis and molecular-dynamics simulations of melittin in water and in vacuum. Chem. Phys. 158:447–472. [Google Scholar]
- Kolinski, A., and J. Skolnick. 1994. Monte Carlo simulations of protein folding.1. Lattice model and interaction scheme. Proteins. 18:338–352. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. 1991. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules.1. The method. J. Comput. Chem. 13:1011–1021. [Google Scholar]
- Kuwajima, K., and M. Arai. 1999. Old and new views of protein folding. In The 24th Taniguchi International Symposium. K. Kuwajima and M. Arai, editors. Elsevier, Kisarazu, Japan.
- La Penna, G., A. Mitsutake, M. Masuya, and Y. Okamoto. 2003. Molecular dynamics of C-peptide of ribonuclease A studied by replica-exchange Monte Carlo method and diffusion theory. Chem. Phys. Lett. 380:609–619. [Google Scholar]
- Lyubartsev, A. P., A. A. Martsinovski, S. V. Shevkunov, and P. N. Vorontsovvelyaminov. 1992. New approach to Monte-Carlo calculation of the free-energy method of expanded ensembles. J. Chem. Phys. 96:1776–1783. [Google Scholar]
- MacKerell, A. D., M. Feig, and C. L. Brooks III. 2004. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126:698–699. [DOI] [PubMed] [Google Scholar]
- Marinari, E., and G. Parisi. 1992. Simulated tempering: a new Monte-Carlo scheme. Europhys. Lett. 19:451–458. [Google Scholar]
- Merritt, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505–524. [DOI] [PubMed] [Google Scholar]
- Mitsutake, A., Y. Sugita, and Y. Okamoto. 2001. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers. 60:96–123. [DOI] [PubMed] [Google Scholar]
- Mitsutake, A., Y. Sugita, and Y. Okamoto. 2003a. Replica-exchange multicanonical and multicanonical replica-exchange Monte Carlo simulations of peptides. I. Formulation and benchmark test. J. Chem. Phys. 118:6664–6675. [Google Scholar]
- Mitsutake, A., Y. Sugita, and Y. Okamoto. 2003b. Replica-exchange multicanonical and multicanonical replica-exchange Monte Carlo simulations of peptides. II. Application to a more complex system. J. Chem. Phys. 118:6676–6688. [Google Scholar]
- Morikami, K., T. Nakai, A. Kidera, M. Saito, and H. Nakamura. 1992. PRESTO (protein engineering simulator): a vectorized molecular dynamics program for biopolymers. Comput. Chem. 16:243–248. [Google Scholar]
- Nakajima, N., H. Nakamura, and A. Kidera. 1997. Multicanonical ensemble generated by molecular dynamics simulation for enhanced conformational sampling of peptides. J. Phys. Chem. B. 101:817–824. [Google Scholar]
- Nymeyer, H., and A. E. Garcia. 2003. Simulation of the folding equilibrium of alpha-helical peptides: a comparison of the generalized Born approximation with explicit solvent. Proc. Natl. Acad. Sci. USA. 100:13934–13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo, Y. Z., and C. L. Brooks. 2003. Exploring Flory's isolated-pair hypothesis: statistical mechanics of helix-coil transitions in polyalanine and the C-peptide from RNase A. Proc. Natl. Acad. Sci. USA. 100:13916–13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, Y., M. Fukugita, T. Nakazawa, and H. Kawai. 1991. Alpha-helix folding by Monte-Carlo simulated annealing in isolated C-peptide of ribonuclease-A. Protein Eng. 4:639–647. [DOI] [PubMed] [Google Scholar]
- Onuchic, J. N., Z. LutheySchulten, and P. G. Wolynes. 1997. Theory of protein folding: the energy landscape perspective. Annu. Rev. Phys. Chem. 48:545–600. [DOI] [PubMed] [Google Scholar]
- Osterhout, J. J., R. L. Baldwin, E. J. York, J. M. Stewart, H. J. Dyson, and P. E. Wright. 1989. H-1-NMR studies of the solution conformations of an analog of the C-peptide of ribonuclease-A. Biochemistry. 28:7059–7064. [DOI] [PubMed] [Google Scholar]
- Pande, V. S., A. Y. Grosberg, T. Tanaka, and D. S. Rokhsar. 1998. Pathways for protein folding: is a new view needed? Curr. Opin. Struct. Biol. 8:68–79. [DOI] [PubMed] [Google Scholar]
- Pitera, J. W., and W. Swope. 2003. Understanding folding and design: replica-exchange simulations of “Trp-cage” fly miniproteins. Proc. Natl. Acad. Sci. USA. 100:7587–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, Y. M., and V. S. Pande. 2003. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys. J. 84:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae, Y., and Y. Okamoto. 2003. Optimization of protein force-field parameters with the Protein Data Bank. Chem. Phys. Lett. 382:626–636. [Google Scholar]
- Schaefer, M., C. Bartels, and M. Karplus. 1998. Solution conformations and thermodynamics of structured peptides: molecular dynamics simulation with an implicit solvation model. J. Mol. Biol. 284:835–848. [DOI] [PubMed] [Google Scholar]
- Shea, J. E., and C. L. Brooks. 2001. From folding theories to folding proteins: a review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 52:499–535. [DOI] [PubMed] [Google Scholar]
- Shoemaker, K. R., R. Fairman, D. A. Schultz, A. D. Robertson, E. J. York, J. M. Stewart, and R. L. Baldwin. 1990. Side-chain interactions in the C-peptide helix: Phe 8…His 12+. Biopolymers. 29:1–11. [DOI] [PubMed] [Google Scholar]
- Shoemaker, K. R., P. S. Kim, E. J. York, J. M. Stewart, and R. L. Baldwin. 1987. Tests of the helix dipole model for stabilization of alpha-helices. Nature. 326:563–567. [DOI] [PubMed] [Google Scholar]
- Sugita, Y., and A. Kitao. 1998. Improved protein free energy calculation by more accurate treatment of nonbonded energy: application to chymotrypsin inhibitor 2, V57A. Proteins. 30:388–400. [PubMed] [Google Scholar]
- Sugita, Y., A. Kitao, and Y. Okamoto. 2000. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 113:6042–6051. [Google Scholar]
- Sugita, Y., and Y. Okamoto. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314:141–151. [Google Scholar]
- Sugita, Y., and Y. Okamoto. 2000. Replica-exchange multicanonical algorithm and multicanonical replica-exchange method for simulating systems with rough energy landscape. Chem. Phys. Lett. 329:261–270. [Google Scholar]
- Sugita, Y., and Y. Okamoto. 2002. Free-energy calculations in protein folding by generalized-ensemble algorithms. In Computational Methods for Macromolecules: Challenges and Applications. T. Schlick and H. H. Gan, editors. Springer-Verlag, Berlin. 304–332.
- Tilton, R. F., J. C. Dewan, and G. A. Petsko. 1992. Effects of temperature on protein-structure and dynamics: x-ray crystallographic studies of the protein ribonuclease-A at 9 different temperatures from 98 K to 320 K. Biochemistry. 31:2469–2481. [DOI] [PubMed] [Google Scholar]
- Torrie, G. M., and J. P. Valleau. 1977. Non-physical sampling distributions in Monte-Carlo free-energy estimation: umbrella sampling. J. Comp. Phys. 23:187–199. [Google Scholar]
- Wang, J., P. Cieplak, and P. A. Kollman. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules. J. Comput. Chem. 21:1049–1074. [Google Scholar]
- Yoda, T., Y. Sugita, and Y. Okamoto. 2004a. Comparisons of force fields for proteins by generalized-ensemble simulations. Chem. Phys. Lett. 386:460–467. [Google Scholar]
- Yoda, T., Y. Sugita, and Y. Okamoto. 2004b. Secondary-structure preferences of force fields for proteins evaluated by generalized-ensemble simulations. Chem. Phys. 307:269–283. [Google Scholar]
- Young, M. R., and V. S. Pande. 2003. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys. J. 84:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, R., B. J. Berne, and R. Germain. 2001. The free energy landscape for beta hairpin folding in explicit water. Proc. Natl. Acad. Sci. USA. 98:14931–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, R. 2003. Free energy landscape of protein folding in water: explicit vs. implicit solvent. Proteins. 53:148–161. [DOI] [PubMed] [Google Scholar]