

Abstract
Synthetic fluorescent analogs of the natural lipopeptide trichogin GA IV were used to investigate the peptide position and orientation in model membranes. A translocation assay based on Förster energy transfer indicates that trichogin is associated to both the outer and inner leaflet of the membrane, even at low concentration, when it is not active. Fluorescence quenching measurements, performed by using water soluble quenchers and quenchers positioned in the membrane at different depths, indicate that at low membrane-bound peptide/lipid ratios trichogin lies close to the region of polar headgroups. By increasing peptide concentration until membrane leakage takes place, a cooperative transition occurs and a significant fraction of the peptide becomes deeply buried into the bilayer. Remarkably, this change in peptide position is strictly coupled with peptide aggregation. Therefore, the mechanism of trichogin action can be envisaged as based on a two-state transition controlled by peptide concentration. One state is the monomeric, surface bound and inactive peptide, and the other state is a buried, aggregated form, which is responsible for membrane leakage and bioactivity.
INTRODUCTION
Antibiotic peptides constitute the main component of the innate defense system of all organisms, including humans (Zasloff, 2002; Bulet et al., 2004). They serve as natural barriers limiting microbial infection, and act mainly by perturbing the permeability of the cell membranes of pathogens, forming pores leading to the dissipation of electrochemical transmembrane gradients and cell death (Boman, 2003). This process does not involve the interaction with a specific receptor, but is simply driven by the physicochemical properties of the antibiotic peptides, above all their amphiphilic character and affinity for lipid bilayers (Epand and Vogel, 1999; Shai, 2002). Because this mechanism of action differs substantially from that of traditional antibiotics, these peptides have attracted a considerable interest as a possible solution to the insurgence of drug-resistant bacteria (Koczulla and Bals, 2003; Andrès and Dimarcq, 2004). However, up to now, the molecular mechanism of pore formation is still debated (Yang et al., 2001).
Recently, we characterized in detail the interaction of trichogin GA IV with model membranes (Stella et al., 2004). This lipopeptide belongs to the family of peptaibols (Peggion et al., 2003), which also includes alamethicin, one of the most studied antibiotic peptides. Peptaibols are produced by fungi of the genus Trichoderma, and are characterized by an N-terminal acyl group, a C-terminal 1,2-aminoalcohol and a high content of the nonproteinogenic α-amino acid Aib. In particular, trichogin GA IV has the following primary structure:
 |
where n-Oct is n-octanoyl, Aib α-aminoisobutyric acid, and Lol leucinol (Auvin-Guette et al., 1992).
Trichogin structure has been investigated in detail by x-ray crystallography, and by several spectroscopic methods (Peggion et al., 2003). These studies have shown that the conformation of this peptide is a partially distorted helix. The mechanism of membrane perturbation by trichogin and its analogs has also been the object of intense research (Peggion et al., 2003). However, the molecular details of this process are still debated, and different models have been proposed, including bilayer destabilization (Epand et al., 1999), channel formation (Scrimin et al., 2002), or diffusion through the membrane as an ion carrier (Milov et al., 2003). We have recently shown that trichogin forms oligomers both in water and in the membrane, and we have determined the equilibrium constants of both aggregation and partition processes. A strong correlation between the concentration of membrane-bound aggregates and membrane leakage was found, indicating that bilayer permeability is caused by these aggregates (Stella et al., 2004). However, to define the mechanism of action, structural data regarding the active state in the membrane are essential. In this work, several fluorescence quenching approaches are exploited to investigate the position of trichogin inside the lipid bilayer and its relationship with the membrane perturbing activity. To this end, we synthesized two fluorescent trichogin analogs (A3 and F10), where the fluorescent labels azulene and fluorene were introduced at position 3 and 10, respectively:
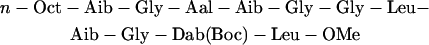 |
(A3) |
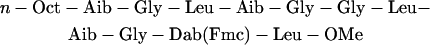 |
(F10). |
Here Aal indicates β-(1-azulenyl)-l-alanine, whereas Fmc is fluorenyl-9-methylcarbonyl, linked to the side chain of 2,4-diamino-l-butyric acid (Dab) and Boc is t-butyloxycarbonyl. These fluorophores were chosen because they act as a donor-acceptor pair in Förster energy transfer (Pispisa et al., 2002a), and were incorporated in such positions as to substitute bulky side chains to minimize perturbations of peptide structure and activity. We have already shown that this is indeed the case for F10 (Stella et al., 2004). The synthetic trichogin analog Tric-OMe, where Lol is replaced by a leucine methyl ester (Leu-OMe), was also investigated. Several studies (Peggion et al., 2003) have shown that the properties of this analog are indistinguishable from those of the natural peptide, including both the three-dimensional structure and biological activity.
MATERIALS AND METHODS
Materials
Phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL), and doxyl-labeled stearic acids, carboxyfluorescein, and Sephadex G-50 were purchased from Sigma (St. Louis, MO). Spectroscopic grade chloroform and methanol (C. Erba, Milan, Italy) were used. Polyvinyl alcohol, average molecular weight of 22,000, 88% hydrolyzed, and Triton X-100 were purchased from Acros (Geel, Belgium).
Peptide synthesis
Synthesis of peptides F10 and A3 was carried out in solution using the segment condensation approach. Peptide coupling reactions were performed by either the 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC)/(1-hydroxy-1,2,3-benzo-triazole) HOBt (König and Geiger, 1970) or by the EDC/(7-aza-1-hydroxy-1,2,3-benzotriazole) HOAt method (Carpino, 1993). The Fmc group was introduced into the Dab side chain using EDC/HOAt. Aal was kindly provided by L. Moroder (Loidl et al., 2000). Details of the synthesis and characterization of the two peptides were reported by Didonè (2001).
Liposome preparation
Large unilamellar vesicles were prepared by dissolving lipids in a chloroform/methanol solution (1:1 v/v). The solvents were evaporated under reduced argon atmosphere, until a thin film formed. Complete evaporation was ensured by applying a rotary vacuum pump for at least 2 h. The film was hydrated with a 20 mM Tris buffer (pH 7.0), containing 140 mM NaCl and 1 mM EDTA, whereas for release experiments a 30 mM carboxyfluorescein solution was used. After vigorous stirring and 10 freeze and thaw cycles, the liposome suspension was extruded for 31 times through two stacked polycarbonate membranes with 100 nm pores (Avestin, Ottawa, Ontario, Canada). Small unilamellar vesicles formed by egg phosphatidylcholine (ePC) and cholesterol (3:2 molar ratio) were prepared by sonication (Auvin-Guette et al., 1992). For release experiments, the unencapsulated fluorescent tracer was separated from the liposomes by gel filtration on a Sephadex G-50 medium column. Final phospholipid concentration was determined by the Stewart method (Stewart, 1980). All liposomes were formed by ePC and cholesterol (1:1 molar ratio), except in some leakage experiments, where dimyristoylphosphatidylcholine (DMPC) or palmitoyloleoylphosphatidylcholine (POPC) were used. Total lipid concentration (phospholipids+cholesterol) is reported.
Liposomes containing the fluorescent lipid 1-palmitoyl-2-[6-((7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)caproyl]-l-α-phosphatidylcholine (C6-NBD-PC) were prepared as follows: symmetrically labeled vesicles were obtained by adding 1% (mol/mol) C6-NBD-PC to the starting chloroform solution. Liposomes containing the fluorescent label only in the internal layer were obtained by chemically quenching with dithionite the external NBD of symmetrically labeled vesicles (McIntyre and Sleight, 1991). The quenching reaction was performed by adding an aliquot of a 1 M dithionite solution in a Tris buffer (pH 10.0) to a 4 mM liposome solution (final dithionite concentration, 80 mM). The kinetics of NBD quenching was followed by measuring fluorescence intensity. After a plateau value was reached (in ∼1 min), excess dithionite was removed by gel filtration. The dithionite concentration used was chosen to have a rapid reaction time, to minimize dithionite diffusion across the lipid bilayer. Complete quenching of external NBD was confirmed by the absence of a further fluorescence reduction after a second addition of dithionite to the final liposome solution or after an addition of bovine serum albumin (BSA), a protein able to quench NBD fluorescence by binding to lipids exposed on the outer vesicle layer (see below). The fluorescence emission of the final internally labeled liposomes was 40% of the emission intensity of the starting symmetrically labeled vesicles, indicating only a minor quenching of the C6-NBD-PC in the internal layer.
Liposomes labeled only in the external layer were prepared by adding an aliquot of a concentrated ethanolic solution of C6-NBD-PC to preformed, unlabeled vesicles, to obtain a 0.5% label molar fraction (Matsuzaki et al., 1996). The final ethanol concentration was always below 1%.
Liposome leakage
Peptide-induced membrane permeability was determined by measuring the fractional release of carboxyfluorescein entrapped inside liposomes, as already reported (Stella et al., 2004). In these and following experiments, peptides were added to water solutions from small aliquots of a concentrated methanol solution, so that the amount of methanol in the final sample was always <1%. Control experiments performed by adding methanol to carboxyfluorescein loaded vesicles clearly show that this small amount of the organic solvent does not cause any membrane perturbation, as indicated by the lack of any vesicle leakage.
Peptide translocation
Peptide translocation experiments were performed by adding F10 (final concentration, 0.5 μM) to C6-NBD-PC labeled liposomes ([lipid] = 0.2 mM). Fluorescence spectra were recorded with an excitation wavelength of 290 nm.
Lipid flip-flop
Peptide-induced lipid flip-flop was determined by measuring the percentage of fluorescence quenching induced by adding BSA to a solution of C6-NBD-PC labeled liposomes (Marx et al., 2000; Valcarcel et al., 2001). A 0.2 mM BSA solution was added to a vesicle solution corresponding to a 0.2 mM lipid concentration, which was enough to bind completely all fluorescent lipids present in the external layer of the vesicles, as shown by titration experiments. NBD fluorescence was excited at 467 nm, and BSA binding was monitored by measuring NBD emission at 522 nm.
Fluorescence quenching
Iodide quenching experiments were performed by titrating a 2 mM vesicle solution, containing 1 μM of a fluorescent peptide analog, with aliquots of a solution containing 4 M KI and 1 mM Na2SO3, prepared on the same day. Samples were excited at 280 nm, and emission was measured at 304 nm for fluorene and 382 nm for azulene. Quenching of the analogs in the absence of liposomes was also measured for comparison.
Depth-dependent quenching
The degree of labeling of doxyl-containing lipids ((1-palmitoyl-2-stearoyl(n-doxyl)-sn-glycero-3-phosphocholine) and stearic acids (5- or 16-doxyl-stearic acid) was determined by double integration of electron paramagnetic resonance (EPR) spectra (Chattopadhyay and London, 1987). Doxyl-labeled liposomes were produced by adding the labeled lipids to the initial chloroform solution (7% molar fraction). Spin label content was controlled directly on the final liposomes by double integration of the EPR spectra of an aliquot of the liposomes dissolved in isopropanol. All liposome preparations contained the same amount of spin labels, within a 10% error. The fluorescent peptide analogs were added to the different doxyl-labeled liposomes (at a lipid concentration of 0.2 or 2 mM) and to a reference unlabeled liposome solution, and steady-state fluorescence intensities were determined after a 20 min equilibration period.
Fluorescence quenching due to lipids labeled at two different depths (say position 5 and 16), as a function of peptide concentration, was analyzed according to the following equations. Assuming that two different peptide positions in the membrane are present (S for shallow and D for deep), the fluorescence intensity measured in the presence of lipids labeled at position 5 or 16 will be given by
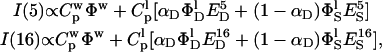 |
where  and
and 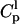 is the peptide concentration in water and in the lipid phase, respectively,
is the peptide concentration in water and in the lipid phase, respectively, 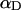 is the fraction of membrane-bound peptide located deep in the bilayer,
is the fraction of membrane-bound peptide located deep in the bilayer, 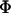 indicates the fluorophore's quantum yield in the different states and
indicates the fluorophore's quantum yield in the different states and 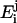 is the quenching efficiency of the fluorophore located in state i by quencher j. To eliminate the contribution of the unbound peptide, the difference between the two intensities was normalized by
is the quenching efficiency of the fluorophore located in state i by quencher j. To eliminate the contribution of the unbound peptide, the difference between the two intensities was normalized by  , the value of which can be obtained by the data previously reported (Stella et al., 2004).
, the value of which can be obtained by the data previously reported (Stella et al., 2004).
 |
Because all quantum yields and quenching efficiencies do not depend on peptide concentration, this quantity is proportional to the fraction of peptide located deep in the bilayer:
 |
where A and B are constants.
It is worth mentioning that in the above derivation we have assumed that peptide binding to the membrane is not affected by the introduction of different labeled lipids. Experiments of peptide-induced carboxyfluorescein release in vesicles containing the different quenchers (data not shown) confirm this assumption, showing that peptide activity is not altered by the introduction of 7% labeled lipids.
Resonance energy transfer
Förster energy-transfer distances were determined according to the following equation (Stella et al., 2002)
 |
where n is the refractive index of the medium,  the quantum yield of the donor,
the quantum yield of the donor,  is a constant (8.785 10−25 M cm3), J an overlap integral of the normalized fluorescence spectrum of the donor (
is a constant (8.785 10−25 M cm3), J an overlap integral of the normalized fluorescence spectrum of the donor (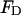 ) and the absorption spectrum of the acceptor, and
) and the absorption spectrum of the acceptor, and  is the molar extinction coefficient:
is the molar extinction coefficient:
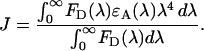 |
Quantum yield of the fluorene donor was 0.33 ± 0.01, as determined in a decanol solution by using naphthalene in cyclohexane as standard (Eaton, 1988). The molar absorptivity of peptide A3 or C6-NBD-PE (acceptors) was measured in decanol and a value of 1.4 was used for n, representing the refractive index of the membrane environment (Strahilevitz et al., 1994; Schümann et al., 1997). From our data we obtained R0 = 24 Å for fluorene-NBD and R0 = 22 Å for the fluorene-azulene pair.
Theoretical energy-transfer efficiency for a random distribution of donor and acceptor labeled peptide monomers in a membrane bilayer was evaluated assuming the peptide equidistributed between the two lipid layers, in agreement with translocation experiments (see Results). Energy-transfer efficiency was calculated using a bidimensional model (Fung and Stryer, 1978), assuming a per phospholipid surface area of 95.6 Å2 (Lis et al., 1982), and neglecting interlayer transfer (Schüman et al., 1997). Peptide diffusion in the membrane was ignored because it is not significant in the fluorescence timescale (Kusba et al., 2002). Excluded volume effects, three-dimensional models (donor and acceptor not lying on the same plane), or incomplete peptide binding to the membrane were also ignored. However, these effects would only lead to a decrease in theoretical transfer efficiency.
Fluorescence spectroscopy
Steady-state fluorescence spectra were carried out on a SPEX Fluoromax 2 fluorimeter (Edison, NJ). Time-resolved experiments were performed on a CD900 single-photon counting apparatus by Edinburgh Instruments (Edinburgh, UK). Nanosecond-pulsed excitation was obtained with a flash lamp filled with ultrapure hydrogen (0.3 bar, 30 kHz repetition rate; full width at half maximum 1.2 ns). Fluorescence intensity decays were acquired until a peak value of 104 counts was reached, and analyzed with the software provided by Edinburgh Instruments. Temperature was controlled to 25°C by a thermostated cuvette holder, except for some leakage experiments that were performed at 15°C.
To minimize peptide adsorption on cell walls, UV-grade polymethylmethacrylate cuvettes were treated overnight with a 5% (w/w) water solution of polyvinyl alcohol (Stella et al., 2004).
RESULTS
Activity of the trichogin analogs
Leakage experiments show that the fluorescent analogs exhibit a membrane perturbing activity quite similar to that of Tric-OMe (Fig. 1), so that F10 and A3 can be considered relevant models of the natural peptide.
FIGURE 1.

Peptide-induced leakage of carboxyfluorescein entrapped inside ePC/cholesterol small unilamellar vesicles (total lipid concentration 60 mM). Tric-OMe (•); F10 (▴); A3 (▪). Fractional release was determined 20 min after peptide addition.
Peptide translocation
To determine the position of trichogin into the membrane, we determined whether the peptide, added from outside the vesicles, would remain associated to the outer layer of the liposomes or be distributed in the whole bilayer. To this goal, we devised a method that takes advantage of resonance energy transfer (RET), based on the procedures previously reported by Matsuzaki et al. (1995) and Wimley and White (2000). However, our method provides a more stringent test of translocation, because three different energy-transfer efficiencies are compared, as described below.
An energy-transfer acceptor for the fluorescent analog F10 is introduced in the liposomes as a fluorescent lipid (C6-NBD-PC). We prepared three different types of vesicles: a), symmetrically labeled liposomes (DL), where the fluorescent lipid is inserted in both layers by mixing it to the other components before vesicle formation; b), outer layer labeled liposomes (OL), where the fluorescent probe is incorporated only in the outer layer by adding it to a vesicle suspension after liposome formation; c), inner layer labeled liposomes (IL), obtained by chemically quenching the label in the outer layer of DL liposomes (see Materials and Methods).
The RET phenomenon has an inverse sixth power dependence on distance, with a 50% quenching efficiency at an interprobe distance called the Förster radius (Pispisa et al., 2002b, 2003). In the case of the fluorene-NBD pair this distance is 24 Å, whereas the thickness of the bilayer is 42 Å (Lis et al., 1982). Because the fluorophore of C6-NBD-PC is located in the region of the polar headgroups (Wolf et al., 1992; Abrams and London, 1993; Mazeres et al., 1996), a peptide lying in the outer layer or a peptide distributed in the whole bilayer, when associated to the three types of liposomes described above, will be quenched quite differently.
The results of the fluorescence quenching experiments in the presence or absence of peptide translocation are schematically presented in Fig. 2. Quenching of peptide fluorescence is determined by measuring the emission intensity F in the presence of acceptor-labeled liposomes (DL, OL, or IL), and comparing it to the fluorescence measured with unlabeled vesicles (F0), where RET is absent. The increase in acceptor emission caused by RET is determined by measuring the lipid fluorescence in the presence (F) and absence (F0) of the fluorescent peptide.
FIGURE 2.

Schematic description of the translocation experiments, in which three different liposome preparations are used: double labeled (DL), with a fluorescent lipid in both layers; outer layer labeled (OL); and inner layer labeled (IL), in which the fluorophore is in one layer only. The peptide is shown as a cylinder, whereas the fluorophores are represented as a triangle (donor) or a star (acceptor). The figure describes the decrease in peptide fluorescence (F), when interacting with these three types of liposomes, with respect to a sample containing unlabeled liposomes (F0), where energy transfer is absent. Similarly, the increase in lipid fluorescence caused by peptide binding is represented. The lower panel reports the experimental results obtained in the case of trichogin analog F10, under the following experimental conditions: [F10] = 0.5 μM; [lipid] = 0.2 mM, C6-NBD-PC = 1% (DL) or 0.5% (IL and OL) mol/mol. Excitation wavelength 290 nm. The reported values are the average of duplicate experiments. DL (•); OL (▪); IL (♦).
When the peptide translocates across the membrane, it is approximately equidistributed in both layers. Therefore, half of the peptide molecules bound to IL or OL vesicles are in a layer void of acceptors (labeled lipids). For this reason, the quenching efficiency arising from these two types of vesicles will be approximately half of that caused by DL liposomes, where all peptides are surrounded by RET acceptors. On the other hand, the relative increase of lipid fluorescence due to peptide binding will be the same in all cases, because all labeled lipids will be surrounded by peptide donors.
By contrast, in the absence of translocation all peptide molecules are lying in the outer layer. In this situation IL vesicles will cause negligible quenching of peptide fluorescence, whereas the effect of OL and DL liposomes will be the same, because both these vesicles have the same amount of acceptors in the outer layer. The increase in lipid fluorescence will be different in the three cases, because OL, DL, and IL have 100%, 50%, or 0% of the acceptors surrounded by peptide donors, respectively.
This rather lengthy description is required to emphasize how our approach allows a clear-cut discrimination between the case in which the peptide is equally distributed in both layers, and that in which it is in the external layer only. The experimental results obtained for the F10 trichogin analog, reported in the lower panel of Fig. 2, clearly indicate a complete translocation of the peptide across the membrane. It is worth noting that the translocation experiments were performed at a low peptide concentration (0.5 μM) so that membrane leakage was absent. Therefore, peptide translocation cannot be related to the formation of toroidal pores, as in the case of magainin (Matsuzaki et al., 1995).
Peptide-induced lipid flip-flop
To validate the translocation experiments, we tested the effect of peptide insertion on the distribution of phospholipids. Usually, the rate of spontaneous translocation of lipids across the bilayer (lipid “flip-flop”) is so low that several days are needed until an initially asymmetric membrane (such as our IL or OL liposomes) attains a homogeneous lipid distribution in both layers (Matsuzaki et al., 1996). However, many membrane perturbing agents cause a huge enhancement of this rate, so that the flip-flop process can be completed even in a few minutes. In this case, the approach described above for the determination of peptide translocation cannot be applied, because all vesicles will become symmetrically labeled in a very short time.
To verify that lipid asymmetry was maintained at the end of our translocation experiments, we used a method based on lipid extraction by BSA. It has been shown (Marx et al., 2000; Valcarcel et al., 2001) that BSA is able to bind C6-NBD-PE by extracting it from the outer layer of vesicles. This binding leads to a significant quenching of NBD fluorescence. Fig. 3 shows the result of the addition of BSA to the vesicles, after peptide addition and after the emission intensities reported in Fig. 2 had been determined. No NBD quenching is observed for IL liposomes, demonstrating that no C6-NBD-PC is accessible from the outside, whereas a significant fluorescence decrease is observed for both OL and DL liposomes. The quenching efficiency of DL vesicles is half that of OL liposomes, in agreement with the different fraction of the labeled lipids that is in the outer layer of these vesicles. Therefore, no significant amount of lipid flip-flop is induced by trichogin at the very low concentration used, and the initial lipid asymmetry is preserved during the translocation experiments. It is worth noting, however, that preliminary experiments indicate that a peptide concentration one order of magnitude higher (5 μM) is able to induce significant perturbation of lipid asymmetry (data not shown).
FIGURE 3.

Kinetic of C6-NBD-PC extraction by BSA. Measurements were performed on samples equilibrated with peptide F10 (0.5 μM), after the translocation experiments reported in Fig. 1. BSA (0.2 mM) was added at zero time. DL liposomes (solid line); OL liposomes (dashed line); IL liposomes (dotted line). Fluorescence intensities were normalized to their value before BSA addition. Excitation wavelength 460 nm; emission wavelength 522 nm.
Peptide depth and orientation
We next determined the position and orientation of trichogin with respect to the lipid bilayer, by making use of a water-soluble and a lipid-attached quencher for fluorescence experiments.
By measuring the efficiency of water soluble quenchers, it is possible to assess the accessibility of membrane-bound fluorophores from the water phase (Castanho and Prieto, 1995). Fig. 4 shows the Stern-Volmer plots corresponding to the iodide quenching of both peptides F10 and A3, performed at a 1.0 μM peptide concentration, which ensures that the peptide is almost completely bound to the membrane (Stella et al., 2004). However, at this concentration no trichogin-induced liposome leakage takes place. For both analogs the quenching caused by iodide ions dissolved in the water phase is significantly reduced by the presence of liposomes, indicating that the peptides are inserted into the membrane, but still partially accessible to the quencher. The azulene fluorophore contained in A3 seems to be somewhat more accessible than F10, possibly because of the higher polarity of the azulenyl moiety.
FIGURE 4.

Stern-Volmer plots of iodide induced quenching. (A) Peptide F10 (1 μM) in water (▴) and in the presence of liposomes (•) ([lipid] = 2 mM). (B) Peptide A3 (1 μM) in water (▴) and in the presence of liposomes (•).
A more detailed information on the position of a fluorophore within a membrane can be obtained by the method of depth-dependent quenching (Ladokhin, 1997; London and Ladokhin, 2002). In this case, quenchers covalently bound to the phospholipid acyl chains are exploited. By varying the quencher position along the lipid acyl chain, the depth of a fluorophore within a membrane can be determined. Fig. 5 shows the results of these experiments performed with peptide F10. The quenching measurements were carried out at three different peptide concentrations, i.e., 0.5, 3.5, and 10.8 μM, which correspond to the whole activity range, causing a leakage of vesicle-entrapped carboxyfluorescein of 0, 50, and 100%, respectively, 20 min after peptide addition (Stella et al., 2004). These concentrations were chosen to investigate whether the position of the peptide in the bilayer is coupled with the membrane perturbing activity.
FIGURE 5.

Relative quenching of peptide F10 by phospholipids containing 1-palmitoyl-2-stearoyl(n-doxyl)-sn-glycero-3-phosphocholine, labeled with a doxyl moiety at different positions along its stearoyl chain. Fluorescence intensities were normalized by the intensity measured in the presence of liposomes containing lipids labeled at position 5. Peptide concentration, 0.5 μM (•); 3.5 μM (▴); 10.8 μM (♦). Lipid concentration, 0.2 mM. A lipid molecule labeled in position 5 is shown below the abscissa axis, which indicates the quencher position along the stearoyl chain. Other possible quencher positions are highlighted by solid circles in the molecular structure. λex = 290 nm, λem = 315 nm.
At 0.5 μM, the highest decrease in fluorescence intensity is caused by the quencher at position 7 (Fig. 5), which is relatively close to the polar headgroups region. As the peptide concentration increases, reaching values high enough to determine liposome leakage, the relative quenching efficiency of the deepest quencher increases significantly. These data clearly show that, upon increasing F10 concentration, the position of the peptide inside the membrane changes. Interestingly, an increase in the relative efficiency of the quencher located deep in the membrane is also observed for analog A3, in which the fluorescent label is close to the N-terminus rather than the C-terminus as in F10 (Fig. 6).
FIGURE 6.

Relative quenching of peptide A3 by phospholipids labeled with doxyl moieties at position 16 or 7. The ratio of fluorescence intensities measured for the peptide interacting with the two liposome preparations is reported. Lipid concentration, 0.2 mM. λex = 343 nm, λem = 382 nm.
To obtain an independent confirmation of this finding, we performed a similar set of experiments with different quenching lipids (see Materials and Methods), at a different lipid concentration (2 mM). As peptide concentration increases, the fluorophore is becoming, on the average, more accessible to the quencher positioned close to the center of the bilayer. This phenomenon is shown in Fig. 7, where the ratio of F10 fluorescence intensities, measured in the presence of liposomes containing stearic acids labeled at position 5 or 16 of the acyl chain, is reported.
FIGURE 7.

Relative quenching of peptide F10 by liposome containing 5- or 16-doxyl-stearic acid. The ratio of fluorescence intensities measured for the peptide interacting with the two liposome preparations is reported. Lipid concentration, 2 mM. λex = 290 nm, λem = 315 nm.
Peptide aggregation
In a previous article, time-resolved fluorescence data allowed us to prove that trichogin undergoes a two-state equilibrium inside the membrane, controlled by peptide concentration (Stella et al., 2004). The reported evidence indicated that this heterogeneity is due to aggregation phenomena. At that time, however, we were unable to discriminate the aggregation equilibrium from other effects still depending on peptide concentration, such as, for instance, an orientational equilibrium (Huang, 2000), which, in fact, does take place, as discussed in the preceding section. We thought, therefore, that it would be important to definitely establish the presence of trichogin aggregates inside the membrane. To this goal, we took advantage of the ability of analogs F10 and A3 to act as a donor-acceptor RET pair, with a Förster radius of 22 Å (see Materials and Methods). Given the strong distance dependence of RET, the transfer efficiency is expected to vary significantly on going from a random distribution of fluorophores to the clustering caused by aggregation (Strahilevitz et al., 1994; Schümann et al., 1997). Fig. 8 shows the experimental energy transfer efficiency as a function of A3 concentration, as determined in the presence of liposomes at a lipid concentration of 2 mM, corresponding to an approximately complete binding of the peptide to the membrane (Stella et al., 2004). The quenching efficiency was assessed by the decrease in average fluorescence lifetime of the donor, to avoid inner-filter effects caused by acceptor absorption (Pispisa et al., 2003). For comparison, the quenching efficiency theoretically expected in the case of a random distribution of peptides in the bilayer is also shown. As a result, the large deviation of the experimental points from the theoretical curve strongly suggests that aggregates are indeed present in the membrane.
FIGURE 8.

Intermolecular FRET efficiency between F10 and A3 trichogin analogs in the membrane (lipid concentration, 2 mM; [F10] = 2 μM) as a function of concentration of the acceptor-labeled peptide (A3). For comparison, the quenching efficiency expected for a random distribution of peptides in the bilayer (see Materials and Methods) is shown as a dashed line. λex = 265 nm, λem = 315 nm.
Effect of membrane viscosity
We next investigated the role of membrane fluidity in the mechanism of bilayer perturbation. Leakage experiments were carried out at 15°C using model membranes of quite different viscosities, i.e., DMPC or POPC, which at this temperature are below and above the gel-to-liquid crystalline thermotropic phase transition, respectively. In addition, experiments were performed with vesicles composed of ePC and cholesterol in a 1:1 molar ratio, because cholesterol is known to induce collective order in the acyl-chain conformations of lipid molecules (Thewalt and Bloom, 1992; Miao et al., 2002). As shown in Fig. 9, trichogin activity is not favored by an increase in lipid bilayer fluidity. A similar leakage is obtained in POPC and ePC/cholesterol membranes, and a somewhat higher permeability is induced in DMPC vesicles, which are in the ordered gel state.
FIGURE 9.

Tric-OMe induced leakage of carboxyfluorescein entrapped inside large unilamellar vesicles formed by DMPC (•), POPC (▪), and ePC/cholesterol (1:1 molar ratio; ▴). Fractional release was determined 20 min after peptide addition. Total lipid concentration, 0.2 mM.
DISCUSSION
The most relevant result presented in this study is the observation of a change in trichogin position in the lipid bilayer, as the membrane-bound peptide/lipid ratio increases. This effect is clearly revealed by the depth-dependent quenching experiments, reported in Figs. 5–7. At low peptide concentration, the trichogin analogs lie close to the polar headgroups region, but as the peptide concentration increases, reaching a value that is able to cause membrane leakage, the quenching efficiency of a doxyl group positioned approximately in the middle of the bilayer increases significantly, thus demonstrating that the peptide is deeply buried into the membrane. Similar results were found for the two analogs A3 and F10, with fluorophores located at the two ends of the peptide chain, indicating that the insertion has not a preferential direction.
A similar concentration-induced orientational transition has already been observed for a few other antibiotic peptides (Lee et al., 2004), as well as for the antifungal polyene nystatin (Coutinho and Prieto, 2003). This phenomenon can be explained by considering that, when the peptide binds to the surface of the bilayer, a perturbation of the surface tension of the membrane is induced (Chen et al., 2002), and that at high peptide concentration excluded volume effects come also into play (Zuckermann and Heimburg, 2001). Therefore, as the amount of peptide increases, a threshold is reached after which a transmembrane orientation becomes thermodynamically favorable. As James Joyce wrote in his Ulysses, although obviously not referring to antimicrobial peptides, “[there would be] more room if they buried them standing.”
It is worth noting that the structural features of the transmembrane arrangement depend on the charge state of the peptide. For neutral or weakly charged peptides, such as alamethicin, insertion into the lipid bilayer is feasible, whereas for highly charged peptides, such as magainin, the insertion in the apolar region of the membrane is unfavorable. In the latter case an increase in peptide concentration leads to the formation of bilayer defects (“toroidal pores”), which is another way to bring about a relaxation process for the accumulated surface tension (Yang et al., 2001; Shai, 2002).
In the case of the neutral lipopeptide trichogin, our results indicate that a high membrane-bound peptide/lipid ratio leads to the insertion of the peptide into the lipid bilayer, though some previous studies seem to contradict this finding. Epand et al. (1999) performed experiments in which the quencher amino acid 4-amino-4 carboxy-2,2,6,6-tetramethylpiperidino-1-oxyl (TOAC) was introduced in the peptide sequence, and liposomes were labeled with phosphatidylcholine analogs bearing the fluorophore 4,4-difluoro-4-bora-3a,4a-diaza-S-indacene (BODIPY) at different positions along the acyl chain. The measured fluorescence quenching was not dependent on the position of the TOAC residue along the peptide chain, nor on the peptide/lipid ratio. These results were interpreted as an indication that trichogin is lying parallel to the membrane surface at all concentrations investigated. However, it has been conclusively shown that the BODIPY group attached to phospholipids “has a clear tendency to locate in the polar headgroup region of the bilayer,” irrespective of its position along the acyl chain (Kaiser and London, 1998). We are therefore inclined to think that with the BODIPY fluorophore a transition in peptide orientation could have not been observed, even if it was present. This idea is confirmed by the absence of any clear dependence of fluorescence quenching on the BODIPY position along the lipid acyl chain (Fig. 2 of Epand et al., 1999), indicating that the fluorescent lipid analogs employed do not allow one to determine the peptide position inside the membrane. By contrast, our study does not suffer from this limitation, because the use of doxyl-labeled lipids and fatty acids is a very well-established methodology for determining the membrane position of fluorescent probes (Ladokhin, 1997; London and Ladokhin, 2002). In addition, recent structural results have shown that doxyl groups linked to phospholipid chains have a depth distribution in the membrane bilayer that is centered near the position of the probe in the chain (Vogel et al., 2003).
Finally, it must be mentioned that Monaco et al. (1999) reached conclusions similar to those reported by Epand et al. (1999), by an EPR investigation. However, they used a peptide concentration of 0.1 mM, which is definitely higher than that normally needed for antimicrobial and membrane perturbing activity. Given the complex interplay between aggregation and membrane-water partition governing trichogin activity (Stella et al., 2004), this high concentration makes a comparison between the results obtained by Monaco et al. (1999) and those reported here untenable.
In our previous study (Stella et al., 2004), we have shown that aggregation phenomena are strongly coupled with the permeability of the membrane, suggesting that peptide pores causing leakage are formed by the trichogin oligomers. Therefore, one may wonder whether all processes controlled by peptide concentration, including the transition in the position of the peptide in the membrane, are correlated.
A proper analysis of the depth dependent quenching data allowed us to obtain quantitative information regarding the fraction of peptide deeply inserted into the membrane. As shown in the Materials and Methods section, the quantity  is linearly dependent on the fraction of peptide deeply inserted into the membrane. Here,
is linearly dependent on the fraction of peptide deeply inserted into the membrane. Here,  and
and  are the fluorescence intensities of F10 measured in the presence of lipids labeled with a quencher at position 5 or 16, respectively, as derived from the data reported in Fig. 7.
are the fluorescence intensities of F10 measured in the presence of lipids labeled with a quencher at position 5 or 16, respectively, as derived from the data reported in Fig. 7. 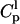 is the concentration of peptide bound to the membrane, obtained from the data reported in Stella et al. (2004). Fig. 10 compares the dependence of
is the concentration of peptide bound to the membrane, obtained from the data reported in Stella et al. (2004). Fig. 10 compares the dependence of  and of the fraction of membrane-bound aggregated peptide on peptide concentration. A remarkable correlation between the two sets of data is found, which is suggestive of the presence of two states only for trichogin in the membrane: i), a monomeric, surface bound, and inactive form and ii), a buried, aggregated state, responsible for membrane leakage. The interconversion between these two states is controlled by peptide concentration.
and of the fraction of membrane-bound aggregated peptide on peptide concentration. A remarkable correlation between the two sets of data is found, which is suggestive of the presence of two states only for trichogin in the membrane: i), a monomeric, surface bound, and inactive form and ii), a buried, aggregated state, responsible for membrane leakage. The interconversion between these two states is controlled by peptide concentration.
FIGURE 10.

Peptide concentration dependence of the fraction of membrane-bound, aggregated peptides (+), as reported in Stella et al. (2004), and the fraction of peptides deeply inserted into the membrane (○), derived from the data of the experiment reported in Fig. 7 (see Materials and Methods). The abscissa axis reports the membrane-bound peptide/lipid molar ratio, derived from the data reported in Stella et al. (2004). Lipid concentration, 2 mM.
Such coupling between membrane insertion and peptide aggregation implies the presence of a strong driving force for peptide self-association in the inserted state. Indeed, several theoretical and experimental studies show that the lipid membrane mediates attractive interactions between transbilayer inclusions (Aranda-Espinoza et al., 1996; Harroun et al., 1999; Lagüe et al., 2000). Hydrophobic mismatch, i.e., a difference between the thickness of the hydrophobic core of the bilayer and the size of the transmembrane inclusion, as is the case of trichogin, is thought to have a major role in self-association processes (de Planque and Killian, 2003). Furthermore, recent results suggest that specific interactions are probably present in the case of trichogin. The sequence motif GxxxG (where G stands for glycine and x for any amino acid) has been shown to induce specific association of transmembrane helices, helix association being even more favored if bulky residues occupy the positions just before or after the two glycines (Curran and Engelman, 2003). In this motif, both glycine residues are lying on the same face of the helix, allowing a close intermolecular packing. In addition, the possibility of hydrogen bond formation between the glycine CαH and a CO group of another helix has been suggested (Curran and Engelman, 2003). The GxxxG motif is indeed present in the trichogin GA IV primary structure, providing a possible structural basis for aggregate formation.
Finally, it is worth mentioning that Milov et al. (2003) recently reported EPR experiments showing that trichogin analogs form oligomers in apolar, membrane-mimicking solvents. They then suggested that vesicular trichogin aggregates could act as carriers within the membrane, by including polar species in their interior, thus allowing diffusion across the lipid bilayer. This model is consistent with our finding that an aggregated, inserted state is the active form, causing membrane leakage. However, the activity of ionophores operating as carriers decreases significantly with increasing membrane viscosity (Boheim et al., 1980; Pregel et al., 1995), whereas this is not the case for trichogin, as shown by the data reported in Fig. 9. Interestingly, even the activity of peptides that induce membrane leakage by forming toroidal pores in the bilayer, such as magainin, is favored by a lower membrane viscosity (Matsuzaki et al., 1991). Therefore, our observation of a slightly higher trichogin activity in the rigid DMPC membranes makes also the latter mechanism of action unlikely. By contrast, membranes modified by channel-forming peptides, such as alamethicin or gramicidin A, do not show any peculiar change in their activity at the transition temperature of the membrane (Boheim et al., 1980).
CONCLUSIONS
The results reported here show that the mechanism of membrane activity of trichogin GA IV is based on a two-state transition controlled by peptide concentration, from an inactive form, which is monomeric and lies close to the membrane surface, to a buried, aggregated state, responsible for membrane leakage. In addition, trichogin activity is not favored by an increase in membrane fluidity, and peptide translocation across the lipid bilayer is not coupled to liposome leakage. Taken together, these findings support the idea that trichogin induces membrane permeability by forming channels, rather than acting as an ion-carrier or perturbing the bilayer. Owing to the fact that trichogin is too short to span a membrane from side to side, a complex supramolecular structure is likely to form when the peptide is buried into the bilayer. Further experiments are underway to better elucidate the structural features of this complex.
Acknowledgments
We thank Prof. J. Z. Pedersen (Department of Biology, University of Rome Tor Vergata) for the EPR determinations of doxyl label content, Prof. L. Moroder (Max-Planck Institute für Biochemie, Martinsried, Germany) for providing β-(1-azulenyl)-l-alanine, Prof. G. Schwarz for helpful discussions, and C. Drago for technical assistance.
The financial support from the Italian Ministry of Education (PRIN 2002 and PRIN 2003) is acknowledged.
References
- Abrams, F. S., and E. London. 1993. Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipid polar headgroup. Biochemistry. 32:10826–10831. [DOI] [PubMed] [Google Scholar]
- Andrès, E., and J. L. Dimarcq. 2004. Cationic antimicrobial peptides: update of clinical development. J. Intern. Med. 255:519–520. [DOI] [PubMed] [Google Scholar]
- Aranda-Espinoza, H., A. Berman, N. Dan, P. Pincus, and S. Safran. 1996. Interaction between inclusions embedded in membranes. Biophys. J. 71:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auvin-Guette, C., S. Rebufatt, Y. Prigent, and B. Bodo. 1992. Trichogin A IV, an 11-residue lipopeptaibol from Trichoderma longibrachiatum. J. Am. Chem. Soc. 114:2170–2174. [Google Scholar]
- Boheim, G., W. Hanke, and H. Eibl. 1980. Lipid phase transition in planar bilayer membrane and its effect on carrier- and pore-mediated ion transport. Proc. Natl. Acad. Sci. USA. 77:3403–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman, H. G. 2003. Antibacterial peptides: basic facts and emerging concepts. J. Intern. Med. 254:197–215. [DOI] [PubMed] [Google Scholar]
- Bulet, P., R. Stöcklin, and L. Menin. 2004. Anti-microbial peptides: from invertebrates to vertebrates. Immunol. Rev. 198:169–184. [DOI] [PubMed] [Google Scholar]
- Carpino, L. 1993. 1-Hydroxy-7-azo-benzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 115:4397–4398. [Google Scholar]
- Castanho, M., and M. Prieto. 1995. Filipin fluorescence quenching by spin-labeled probes: studies in acqueous solution and in a membrane model system. Biophys. J. 69:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay, A., and E. London. 1987. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 26:39–45. [DOI] [PubMed] [Google Scholar]
- Chen, F. Y., M. T. Lee, and H. W. Huang. 2002. Sigmoidal concentration dependence of antimicrobial peptide activities: a case study on alamethicin. Biophys. J. 82:908–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho, A., and M. Prieto. 2003. Cooperative partition model of nystatin interaction with phospholipid vesicles. Biophys. J. 84:3061–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, A. R., and D. M. Engelman. 2003. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 13:412–417. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- Didonè, M. 2001. Trichogin analogs for photophysical studies in membranes. Chemistry degree thesis. University of Padua, Padua, Italy.
- Eaton, D. F. 1988. Reference materials for fluorescence measurement. Pure Appl. Chem. 60:1107–1114. [Google Scholar]
- Epand, R. F., R. M. Epand, V. Monaco, S. Stoia, F. Formaggio, M. Crisma, and C. Toniolo. 1999. The antimicrobial peptide trichogin and its interaction with phospholipid membranes. Eur. J. Biochem. 266:1021–1028. [DOI] [PubMed] [Google Scholar]
- Epand, R. M., and H. J. Vogel. 1999. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta. 1462:11–28. [DOI] [PubMed] [Google Scholar]
- Fung, B. K., and L. Stryer. 1978. Surface density determination in membranes by fluorescence energy transfer. Biochemistry. 17:5241–5248. [DOI] [PubMed] [Google Scholar]
- Harroun, T. A., W. T. Heller, T. M. Weiss, L. Yang, and H. W. Huang. 1999. Experimental evidence for hydrophobic matching and membrane-mediated interactions in lipid bilayers containing gramicidin. Biophys. J. 76:937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H. W. 2000. Action of antimicrobial peptides: two-state model. Biochemistry. 39:8347–8352. [DOI] [PubMed] [Google Scholar]
- Kaiser, R. D., and E. London. 1998. Determination of the depth of BODIPY probes in model membranes by parallax analysis of fluorescence quenching. Biochim. Biophys. Acta. 1375:13–22. [DOI] [PubMed] [Google Scholar]
- Koczulla, A. R., and R. Bals. 2003. Antimicrobial peptides: current status and therapeutic potential. Drugs. 63:389–406. [DOI] [PubMed] [Google Scholar]
- König, W., and R. Geiger. 1970. Eine neue methode zur synthese von peptiden: aktivierung der carboxylgruppe mit dicyclohexylcarbodiimid unter zusatz von 1-hydroxy-benzotriazolen. [in German] Chem Ber. 103:788–798. [DOI] [PubMed] [Google Scholar]
- Kusba, J., L. Li, I. Gryczynsky, G. Piszczek, M. Johnson, and J. R. Lakowicz. 2002. Lateral diffusion coefficients in membranes measured by resonance energy transfer and a new algorithm for diffusion in two dimensions. Biophys. J. 82:1358–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin, A. S. 1997. Distribution analysis of depth dependent fluorescence quenching in membranes: a practical guide. Methods Enzymol. 278:462–473. [DOI] [PubMed] [Google Scholar]
- Lagüe, P., M. J. Zuckermann, and B. Roux. 2000. Lipid-mediated interactions between intrinsic membrane proteins: a theoretical study based on integral equations. Biophys. J. 79:2867–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M., F. Chen, and H. W. Huang. 2004. Energetics of pore formation induced by membrane active peptides. Biochemistry. 43:3590–3599. [DOI] [PubMed] [Google Scholar]
- Lis, L. J., M. McAlister, N. L. Fuller, R. P. Rand, and V. A. Parsegian. 1982. Measurement of the lateral compressibility of several phospholipid bilayers. Biophys. J. 37:667–672. [PMC free article] [PubMed] [Google Scholar]
- Loidl, G., H. J. Musiol, N. Budisa, R. Huber, S. Poirot, D. Fourmy, and L. J. Moroder. 2000. Synthesis of β-(1-azulenyl)-L-alanine as a potential blue-colored fluorescent tryptophan analog and its use in peptide synthesis. J. Pept. Sci. 6:139–144. [DOI] [PubMed] [Google Scholar]
- London, E., and A. S. Ladokhin. 2002. Measuring the depth of amino acid residues in membrane-inserted peptides by fluorescence quenching. In Peptide-Lipid Interactions. S. Simon and T. J. McIntosh, editors. Academic Press, San Diego, CA. 89–115.
- Marx, U., G. Lassman, H. G. Holzütter, D. Wüstner, P. Müller, A. Höhlig, J. Kubelt, and A. Herrmann. 2000. Rapid flip-flop of phospholipids in endoplasmic reticulum membranes studied by a stopped-flow approach. Biophys. J. 78:2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki, K., H. Mitsunori, S. Funakoshi, N. Fujii, and K. Miyajima. 1991. Physicochemical determinants for the interactions of magainins 1 and 2 with acidic lipid bilayers. Biochim. Biophys. Acta. 1991:162–170. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., O. Murase, N. Fujii, and K. Miyajima. 1995. Translocation of a channel forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry. 34:6521–6526. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., O. Murase, N. Fujii, and K. Miyajima. 1996. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 35:11361–11368. [DOI] [PubMed] [Google Scholar]
- Mazeres, S., V. Schram, J. F. Tocanne, and A. Lopez. 1996. 7-Nitrobenz-2-oxa-1,3-diazole-4-yl-labeled phospholipids in lipid membranes: differences in fluorescence behavior. Biophys. J. 71:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre, J. C., and R. G. Sleight. 1991. Fluorescence assay for phospholipid membrane asymmetry. Biochemistry. 30:11819–11827. [DOI] [PubMed] [Google Scholar]
- Miao, L., M. Nielsen, J. Thewalt, J. H. Ipsen, M. Bloom, M. J. Zuckermann, and O. G. Mouritsen. 2002. From lanosterol to cholesterol: structural evolution and differential effects on lipid bilayers. Biophys. J. 82:1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milov, A. D., Y. D. Tsvetkov, F. Formaggio, M. Crisma, C. Toniolo, and J. Raap. 2003. Self-assembling and membrane modifying properties of a lipopeptaibol studied by CW-ESR and PELDOR spectroscopies. J. Pept. Sci. 9:690–700. [DOI] [PubMed] [Google Scholar]
- Monaco, V., F. Formaggio, M. Crisma, C. Toniolo, P. Hanson, and G. L. Millhauser. 1999. Orientation and immersion depth of a helical lipopeptaibol in membranes using TOAC as an ESR probe. Biopolymers. 50:239–253. [DOI] [PubMed] [Google Scholar]
- Peggion, C., F. Formaggio, M. Crisma, R. F. Epand, R. M. Epand, and C. Toniolo. 2003. Trichogin: a paradigm for lipopeptaibols. J. Pept. Sci. 9:679–689. [DOI] [PubMed] [Google Scholar]
- Pispisa, B., C. Mazzuca, A. Palleschi, L. Stella, M. Venanzi, F. Formaggio, C. Toniolo, and Q. B. Broxterman. 2002b. Structural features and conformational equilibria of 310-helical peptides in solution by spectroscopic and molecular mechanics studies. Biopolymers. 67:247–250. [DOI] [PubMed] [Google Scholar]
- Pispisa, B., C. Mazzuca, A. Palleschi, L. Stella, M. Venanzi, M. Wakselman, J.-P. Mazaleyrat, M. Rainaldi, F. Formaggio, and C. Toniolo. 2003. A combined spectroscopic and theoretical study of a series of conformationally restricted hexapeptides carrying a rigid binaphthyl-nitroxide donor-acceptor pair. Chem. Eur. J. 9:2–11. [DOI] [PubMed] [Google Scholar]
- Pispisa, B., A. Palleschi, C. Mazzuca, L. Stella, A. Valeri, M. Venanzi, F. Formaggio, C. Toniolo, and Q. B. Broxterman. 2002a. The versatility of combining FRET measurements and molecular mechanics results for determining the structural features of ordered peptides in solution. J. Fluoresc. 12:213–217. [Google Scholar]
- Pregel, M. J., L. Jullien, J. Canceill, L. Lacombe, and J. M. Lehn. 1995. Channel-type molecular structures. Part 4. Transmembrane transport of alkali-metal ions by bouquet molecules. J. Chem. Soc., Perkin Trans. 2. 3:417–426. [Google Scholar]
- Schümann, M., M. Dathe, T. Wieprecht, M. Beyermann, and M. Bienert. 1997. The tendency of magainin to associate upon binding to phospholipid bilayers. Biochemistry. 36:4345–4351. [DOI] [PubMed] [Google Scholar]
- Scrimin, P., P. Tecilla, U. Tonellato, A. Veronese, M. Crisma, F. Formaggio, and C. Toniolo. 2002. Zinc(II) as an allosteric regulator of liposomal membrane permeability induced by synthetic template-assembled tripodal polypeptides. Chem. Eur. J. 8:2753–2763. [DOI] [PubMed] [Google Scholar]
- Shai, Y. 2002. Mode of action of membrane active antimicrobial peptides. Biopolymers. 66:236–248. [DOI] [PubMed] [Google Scholar]
- Stella, L., C. Mazzuca, M. Venanzi, A. Palleschi, M. Didonè, F. Formaggio, C. Toniolo, and B. Pispisa. 2004. Aggregation and water-membrane partition as major determinants of the activity of the antibiotic peptide trichogin GA IV. Biophys. J. 86:936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella, L., M. Venanzi, M. Carafa, E. Maccaroni, M. E. Straccamore, G. Zanotti, A. Palleschi, and B. Pispisa. 2002. Structural features of model glycopeptides in solution and in membrane phase: a spectroscopic and molecular mechanics investigation. Biopolymers. 64:44–56. [DOI] [PubMed] [Google Scholar]
- Stewart, J. C. M. 1980. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 104:10–14. [DOI] [PubMed] [Google Scholar]
- Strahilevitz, J., A. Mor, P. Nicolas, and Y. Shai. 1994. Spectrum of antimicrobial activity and assembly of dermaseptin-b and its precursor form in phospholipid membranes. Biochemistry. 33:10951–10960. [DOI] [PubMed] [Google Scholar]
- Thewalt, J. L., and M. Bloom. 1992. Phosphatidylcholine:cholesterol phase diagrams. Biophys. J. 63:1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcarcel, C. A., M. Dalla Serra, C. Potrich, I. Bernhart, M. Tejuca, D. Martinez, F. Pazos, M. E. Lanio, and G. Menestrina. 2001. Effects of lipid composition on membrane permeabilization by sticholysins I and II, two cytolysins of the sea anemone Stichodactyla helianthus. Biophys. J. 80:2761–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, A., H. A. Scheidt, and D. Huster. 2003. The distribution of lipid attached spin probes in bilayers: application to membrane protein topology. Biophys. J. 85:1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley, W. C., and S. H. White. 2000. Determining the membrane topology of peptides by fluorescence quenching. Biochemistry. 39:161–170. [DOI] [PubMed] [Google Scholar]
- Wolf, D. E., A. P. Winiski, A. E. Ting, K. M. Bocian, and R. E. Pagano. 1992. Determination of the transbilayer distribution of fluorescent lipid analogues by nonradiative fluorescence resonance energy transfer. Biochemistry. 31:2865–2873. [DOI] [PubMed] [Google Scholar]
- Yang, L., T. A. Harroun, T. M. Weiss, L. Ding, and H. W. Huang. 2001. Barrel stave or toroidal model? A case study on melittin pores. Biophys. J. 81:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature. 415:389–395. [DOI] [PubMed] [Google Scholar]
- Zuckermann, M., and T. Heimburg. 2001. Insertion and pore formation driven by adsorption of proteins onto lipid bilayer membrane-water interfaces. Biophys. J. 81:2458–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]