

Abstract
Microspectroscopic techniques such as Fourier transform infrared (FTIR) have played an important role in “fingerprinting” the biochemical composition of cellular components. Based on structure and function, complex biomolecules absorb energy in the mid-infrared (λ = 2–20 μm) yielding characteristic vibrational infrared (IR) spectra. However, optical detection FTIR microspectroscopy may not be suitable for IR-absorbing sample materials. Photothermal microspectroscopy (PTMS) permits the direct measurement of heat generated as a result of sample material absorbing radiation. This approach generates true absorption spectra and is implemented by interfacing a scanning probe microscope and an FTIR spectrometer. Detection is performed using a near-field ultra-miniaturized temperature sensor. Employing PTMS, IR spectra of MCF-7 cells were examined in spectral regions (900–2000 cm−1) corresponding to proteins, DNA, RNA, glycoproteins, carbohydrates, lipids, and levels of protein phosphorylation. As a cell passes through the cell cycle, its nuclear material decondenses and condenses and this has led to ambiguity as to whether the intensity of such spectral regions may be associated with the G1-, S- or G2-phases of the cell cycle. Cultured cells were tracked over a time course known to correspond to marked alterations in cell-cycle distributions, as determined using flow cytometry. Experiments were carried out in the absence or presence of lindane, a pesticide known to induce G1-arrest in MCF-7 cells. Significant (P < 0.05) elevations in spectral intensities were associated with exponentially growing cell populations, predominantly in S-phase or G2-phase, compared to more quiescent populations predominantly in G1-phase. Increases in the absorption band at 970 cm−1, associated with elevated protein phosphorylation, were observed in vibrational spectra of exponentially growing cell populations compared to those exhibiting a slowing in their growth kinetics. These results seem to suggest that intracellular bulk changes, associated with transit through the cell cycle, can be tracked using PTMS.
INTRODUCTION
Near-field photothermal microspectroscopy (PTMS) is based on the absorption of electromagnetic radiation and detection of associated heat release. At its heart is an ultra miniaturized temperature sensor in the form of a thermal probe integrated in a cantilevered structure, as used in scanning force microscopy (Hammiche et al., 2004a). This sensor consists of a 5-μm diameter, 90% platinum-10% rhodium looped filament of Wollaston wire, ∼200 μm in length (Bozec et al., 2001).
Using this technique, absorption spectra that are a characteristic fingerprint of the sample analyzed are routinely obtained with minimum or no sample preparation. Due to the damped nature of thermal waves, PTMS potentially allows higher-than-the-diffraction-limit resolution of molecular species on the surfaces of materials or living structures (Hammiche et al., 2004b).
Organized and compacted within the nucleus, the approximate length of DNA in the genome of a human somatic cell is 3 m. Within a nucleoprotein complex (chromatin) that consists of basic repeating units known as nucleosomes, this DNA is associated with histone proteins. The degree of this first-level compaction regulates the accessibility of factors that modulate the transcriptional activity of genes and ensures proper epigenetic inheritance (Wolffe, 1997; Moggs et al., 2004). As proliferating cells pass through the different growth and synthesis (e.g., DNA replication) stages of the cell cycle from interphase (G1-phase, S-phase and G2-phase) through to the mitotic (M)-phase before mitotic cell division, a significant degree of chromosomal condensation is required to reduce the volume of replicated genomes (Watrin and Legagneux, 2003). After a cell duplicates its DNA (during S-phase), loosely packed chromatin fibers condense into discrete mitotic chromosomes in M-phase; this is required to facilitate the proper segregation of genetic material between daughter cells (McHugh and Heck, 2003).
The stage of the cell cycle may contribute to the variability of IR spectra of cells within apparently similar tissues (Tobin et al., 2004). Twofold increases in the DNA/RNA infrared (IR) spectral region, obtained using synchrotron radiation based Fourier transform infrared (FTIR) microspectroscopy, have been reported in cells undergoing DNA replication in S-phase compared to cells in G1-phase (Holman et al., 2000). This would suggest that IR spectra might be an alternative tool to monitor intracellular bulk changes as a cell progresses through the cell cycle. Alternatively, chromatin condensation might render the cell genome too dense for IR absorption and therefore unobservable in IR spectra (Boydston-White et al., 1999; Diem et al., 1999). As PTMS relies on the detection of heat released after the absorption of radiation, such varying opacity within the sample material will not influence resultant spectra.
In this study, we used PTMS to investigate the spectral changes that occur in proliferating MCF-7 cells. As distinct from conventional FTIR microspectroscopy, PTMS measures heat generated after absorption of electromagnetic radiation. Thus it allows for measurements to be made with opaque materials such as a cell nucleus. An added advantage is that it requires minimum sample preparation. Experiments were carried out in the absence or presence of 10−4 M lindane (γ-hexachlorocyclohexane, γ-HCH), an agent known to induce a profound G1-phase arrest at this concentration (Kalantzi et al., 2004). Cell cycle distributions were determined in parallel experiments using flow cytometry. The aim of this approach was to ascertain whether intracellular bulk changes are spectrally observable.
MATERIALS AND METHODS
Chemicals
Chemicals were obtained from Sigma Chemical Co. (Poole, UK) unless otherwise stated. Cell culture consumables were obtained from Invitrogen Life Technologies (Paisley, UK) unless otherwise stated.
Cell culture and treatment
The human mammary carcinoma MCF-7 cell line was grown in Dulbecco's modified essential medium supplemented with 10% heat-inactivated foetal calf serum, penicillin (100 U/ml) and streptomycin (100 μg/ml) (Yared et al., 2002). Cells were grown in 5% CO2 in air at 37°C in a humidified atmosphere and disaggregated using a trypsin (0.05%)/EDTA (0.02%) solution, to form single cell suspensions before subculture or incorporation in experiments. Lindane (10−4 M) was added as a solution in dimethyl sulphoxide (DMSO) and DMSO was used as a vehicle control: DMSO concentrations did not exceed 1% (v/v).
To facilitate PTMS analysis, routinely cultured cells were disaggregated and resuspended in complete medium before seeding aliquots (5 ml, ≈105 cells) into 60-mm petri dishes containing 5-mm glass coverslips. Cells were cultured undisturbed for 24 h, 48 h, or 96 h before removal of medium and fixation with 70% ethanol (EtOH). In parallel cultures, cells were initially cultured for the first 24 h in the absence of lindane; medium was then replaced with medium containing lindane for a subsequent 24-h period giving a “48 h plus lindane” time point. To obtain a “96 h plus lindane” time point, medium from these aforementioned lindane-treated cultures was then replaced with fresh lindane-free medium for the remaining incubation period before fixation.
Flow cytometry
Cells were resuspended in aliquots of complete medium (5 ml, ≈105 cells) and seeded into 60-mm petri dishes (Davis et al., 2002). At the time points indicated (24 h, 48 h, and 96 h), cells were disaggregated and, aliquots were washed twice with phosphate-buffered saline (PBS) before fixation with ice-cold EtOH (70%, aqueous) and storage overnight at −20°C. Cell aliquots were then washed twice with PBS before incubation with RNase A (10 μg/ml) and propidium iodide (50 μg/ml) for 60 min at 37°C. DNA content of 10,000 events (cells)/treatment was analyzed using a Becton Dickinson (Franklin Lakes, NJ) FACSCaliber flow cytometer and the CELLQuest software version provided by the manufacturer. Cell cycle analysis was carried out using ModFitLT for Mac v2.0.
PTMS analysis of fixed cells
The sample material consisted of 70% EtOH-fixed cells adhered to glass coverslips. After fixation, coverslip-containing petri dishes were placed under vacuum overnight to facilitate dehydration of samples. Subsequently, dishes were individually sealed tight with adhesive tape. PTMS is implemented by integrating, using a dedicated optical interface (Specac, Orpington, UK), a scanning probe microscope (Explorer model from Veeco Instruments, Santa Barbara, CA) equipped with a Wollaston wire thermal probe (Veeco Instruments) and a FTIR spectrometer (Vector 22 model from Bruker Optics limited, Coventry, UK) (Hammiche et al., 2004b). IR spectra were acquired by placing the glass coverslips, coated with sample material, in the chamber compartment of the spectrometer. With the probe tip at the IR-focal point to maximize the signal, the tip picked up cellular material and absorption spectra were recorded. This nanosampling method was employed to enhance signal/noise ratio (SNR) by eliminating temperature reduction due to substrate heat-sinking effects. Material was acquired in an unbiased fashion from across the coverslip and variations in the quantity of sample picked up were of the order of a few tens of cells, judging from visual observation using an optical microscope. Additionally, coherent averaging (1000 scans/measurement resulting in a 20-min acquisition time) was used to further improve SNR. For each cell culture analyzed, a maximum of 10 absorption spectra from 10 different samples of cellular material were recorded per coverslip. Spectra, taken with a 16-cm−1 spectral resolution, were obtained from two measurements: one performed with cellular material attached to the probe tip, the second was a measurement of a background spectrum obtained with the probe free of any material. A first normalization was performed, by dividing the former by the later. Baseline subtraction was then applied considering zero energy absorption at 2000 cm−1: the value of the recorded signal at that wavenumber was subtracted across the entire range. Finally all spectra were normalized to the amide II peak (≈1540 cm−1). Between sample analyses, the probe was cleaned, by Joule heating to >600°C; previous investigations from our laboratory have shown that this results in pyrolysis of attached organic contaminant(s) (Pollock and Hammiche, 2001).
By sensing changes in the probe's electrical resistance, the physical parameter measured is temperature. Spectra were constructed from the measured amplitude of the temperature fluctuation at each wavelength across the entire spectrum. Factors that affect the strength of this measured signal include quantity of material analyzed and contact of the material with the probe filament. This renders quantification of absorption in terms of absolute units very difficult. Hence, the amplitude axis (y axis) of spectra presented is labeled in arbitrary units (au).
Statistical analysis
Data obtained from flow cytometry measurements were compared using either one-way ANOVA (on the effects of incubation time in untreated cultures), or two-way ANOVA (on the effects of incubation time and the addition of lindane). A probability value <5% (P < 0.05) was selected as being statistically significant (Greene and D'Oliveira, 1989).
The Mann-Whitney U test was employed to compare absorption spectra at each wavenumber for each cell culture (Greene and D'Oliveira, 1989). This is a nonparametric test that does not assume normal distribution of the numerical data. Probability diagrams were constructed for each pair of absorption spectra to be compared; thus graphically illustrating which wavenumbers exhibited the largest differences between cell cultures. Regions of spectra with a P < 0.05 are likely to represent real structural differences (Malins et al., 2003).
Multivariate (principal component) analysis was also conducted on the spectra using the Pirouette software package (Infometrix, Woodinville, WA). In multivariate data analysis, each spectrum becomes a single point in a “hyperspace,” where the number of dimensions depends on the chosen spectral resolution. The information obtained may be plotted in the form of a two-dimensional plot known as a dendrogram of similarity index. Nearness in row space, or multivariate distance, implies pattern recognition and the separation of sample clusters in the plots signifies structurally dissimilar groups.
RESULTS
Table 1 shows the changes that occur in cell-cycle distributions, measured using flow cytometry, over a 96-h period after seeding ≈105 MCF-7 cells/5-ml culture medium into 60-mm petri dishes. After disaggregation, 60.7% ± 3.5% of MCF-7 cells are in G1-phase. By 24 h, as seeded cells adhered to the base of a culture dish or coverslip and entered an exponential-growth phase, one-way ANOVA revealed that a significant (P < 0.001 compared to disaggregated cell populations) reduction in the percentage of cells in G1-phase occurred. However, at subsequent time points increases in the percentage of cells in G1-phase were observed both at 48 h (45% ± 3.5%, P < 0.003 compared to the 24-h time point) and 96 h; by this time point the percentage of cells in G1-phase (66.0% ± 4.3%) was approximately equal and not significantly different to that observed immediately after disaggregation. Corresponding time-related fluctuations in the percentages of cells in S-phase or G2-phase also exhibited a similar pattern of significant (P < 0.05) changes. Again, by 96-h incubation these percentages were comparable to levels observed immediately postdisaggregation (Table 1). Two-way ANOVA indicated that both incubation time and lindane treatment exerted a significant (P < 0.05) effect on each cell population. Furthermore, for cells in G1-phase or S-phase, the interaction of these two factors was observed to be highly significant (P < 0.001). This interaction effect is probably because lindane, an organochlorine pesticide, produces a profound G1-arrest in MCF-7 cells (Kalantzi et al., 2004). After lindane removal, treated cells escape G1-arrest and would probably re-enter an exponential growth phase. Similarly significant fluctuations in the percentages of cells in G2-phase are not observed (Table 1).
TABLE 1.
Cell cycle distributions in the absence or presence of lindane
| Incubation treatment (h) | G1-phase (%) | S-phase (%) | G2-phase (%) |
|---|---|---|---|
| Disaggregated cell population | 60.7 ± 3.5 | 30.3 ± 1.0 | 9.1 ± 2.6 |
| 24 h | 32.3 ± 2.2 | 47.7 ± 3.9 | 20.1 ± 3.3 |
| 48 h | 45.3 ± 3.5 | 39.9 ± 3.5 | 14.8 ± 2.2 |
| 24 h + 24 h lindane (48 h plus lindane) | 63.3 ± 3.6 | 24.0 ± 3.1 | 12.7 ± 4.7 |
| 96 h | 66.0 ± 4.3 | 26.4 ± 3.5 | 7.9 ± 1.7 |
| 24 h, 24 h lindane + 48 h (96 h plus lindane) | 68.5 ± 3.3 | 27.3 ± 2.9 | 4.2 ± 0.8 |
A time course of the cell-cycle distributions of MCF-7 cells post seeding. Cells were seeded and incubated, in the presence or absence of lindane, as described in Materials and Methods. DNA content of 10,000 events per incubation treatment was analyzed using a Becton Dickinson FACSCaliber flow cytometer, as described in Materials and Methods. Results are the mean ± SD of triplicate measurements from three separate experiments. Each experiment concerned triplicate measurements for each incubation treatment; these were combined to give a mean that contributed to the overall mean.
Fig. 1 shows a typical absorption spectrum of a sample of MCF-7 cells after 24-h culture. Vibrational bands, associated with spectral regions, where significant differences of interest might occur include the amide I (≈1650 cm−1), amide II (≈1540 cm−1), glycoproteins and acyl chain of lipids (δs CH3, ≈1380 cm−1), amide III (≈1260 cm−1), carbohydrates (δs C-O, ≈1155 cm−1), RNA (≈1120 cm−1), phosphodiester of nucleic acids ( ), νs CO-O-C (≈1050 cm−1), and glycogen (νs C-O, ≈1030 cm−1) (Jackson and Mantsch, 1996; Gazi et al., 2004). Also of reported interest is the absorption peak at 970 cm−1; this may be indicative of the level of intracellular protein phosphorylation (Tobin et al., 2004).
), νs CO-O-C (≈1050 cm−1), and glycogen (νs C-O, ≈1030 cm−1) (Jackson and Mantsch, 1996; Gazi et al., 2004). Also of reported interest is the absorption peak at 970 cm−1; this may be indicative of the level of intracellular protein phosphorylation (Tobin et al., 2004).
FIGURE 1.

Spectral regions of interest, annotated in a typical absorption spectrum (2000–900 cm−1) of MCF-7 cells after 24-h incubation. Cells were seeded into 60-mm petri dishes containing glass coverslips and fixed before analysis, as described in the Materials and Methods.
Fig. 2, a–c, shows the median IR spectra (2000–900 cm−1) of a maximum of 10 spectra recorded per cell culture sample. Marked variations in the DNA/RNA spectral region (≈1490–1000 cm−1) that correlate with alterations in cell-cycle distributions are apparent. A time-related reduction in spectral intensity was observed in this region suggesting that, as cells transit from the exponential-growth phase (S- and G2/M-phase) into the more quiescent G1-phase (Table 1), bulk changes in intracellular biomolecules are spectrally observable (Fig. 2 a). Further reductions in intensity in the presence of lindane after 48 h and 96 h incubation compared to median spectra obtained in the absence of this test agent at the same time points were observed (Fig. 2, b and c); again, primarily in the DNA/RNA spectral region (≈1490–1000 cm−1).
FIGURE 2.
(a) Median IR spectra (2000–900 cm−1) of a maximum of 10 recorded per cell culture sample over a time course of 24 h, 48 h, and 96 h, as indicated. Cells were seeded into 60-mm petri dishes containing glass coverslips and fixed before analysis, as described in the Materials and Methods. (b) Median IR spectra after 48-h incubation in the absence or presence of 24-h treatment with 10−4 M lindane. Cells were treated after an initial 24-h incubation in lindane-free medium; whereupon they were immediately fixed before analysis. (c) Median IR spectra after 96-h incubation in the absence or presence of 24-h treatment with 10−4 M lindane. After an initial 24-h incubation in lindane-free medium, cells were treated with the agent for 24 h after which they were incubated in fresh medium for a further 48 h. At the end of this 96-h incubation, they were immediately fixed before analysis.
Intrasample spectral variations are shown in Fig. 3, a–e. In exponentially growing cell populations (Fig. 3 a) marked variations in spectral intensities were observed at 24 h, which are most apparent in the intensity of the amide I band (≈1650 cm−1) and the centroid position of the amide II band (≈1540 cm−1). Such intrasample variations in IR signatures diminish in a time-related fashion with incubation (48 h and 96 h). Reductions in spectral intensity and variation across the 1490–1000 cm−1 band appear to be accelerated at 48 h after 24-h treatment with 10−4 M lindane (Fig. 3, b and c). Although lindane-treated 96-h cultures exhibit the greatest overall reductions in spectral intensity, an increase in spectral variation was observed compared to untreated 96-h cultures (Fig. 3, d and e). Again, this is primarily in the DNA/RNA spectral region (≈1490–1000 cm−1) and may be the result of longer-term lindane-induced cytotoxic effects.
FIGURE 3.

(a) Intrasample spectral variations obtained after 24-h incubation of MCF-7 cells seeded at a density of 105/5 ml into 60-mm petri dishes, as described in the Materials and Methods. Cellular material was fixed before recording absorption spectra with a Wollaston wire probe tip. (b) Intrasample spectral variations obtained after 48-h incubation. (c) Intrasample spectral variations obtained after 48-h incubation in the presence of a 24-h treatment with lindane. (d) Intrasample spectral variations obtained after 96-h incubation. (e) Intrasample spectral variations obtained after 96-h incubation in the presence of a 24-h treatment with lindane.
The band intensity at 970 cm−1 becomes less pronounced as cells transit from the exponential-growth phase (S- and G2/M-phase) into the more quiescent G1-phase (Fig. 3, a–e). A peak at this band has been associated with protein phosphorylation (Sanchez-Ruiz and Martinez-Carrion, 1988; Tobin et al., 2004); as such it could be expected to be elevated in actively dividing compared to resting cells.
By comparing absorption spectra using the Mann-Whitney U test to generate probability diagrams, significant differences (P < 0.05) in different regions of the spectra were observed after the analysis of different incubation treatments (Fig. 4, a–c). Fig. 4 a suggests that, although absorption spectra from exponentially growing cells after 24-h and 48-h incubation are mostly similar, significant differences are observed throughout the spectrum as cell growth slows down. Such significant differences point to true structural alterations within exponentially growing cells when compared to quiescent populations. Further significant differences were observed after incubation in the presence of lindane at 48 h, primarily in the DNA/RNA spectral region (≈1490–1000 cm−1), and 96 h (≈2000–900 cm−1) compared to corresponding untreated cultures (Fig. 3, b and c).
FIGURE 4.
(a) Plot depicting the statistical significance, as calculated by the Mann-Whitney U test, of differences at each wavenumber in the spectra for 24 h vs. 48 h and 48 h vs. 96 h incubation time, as indicated. (b) Plot depicting the statistical significance in the spectra for 48 h vs. 48 h plus lindane. (c) Plot depicting the statistical significance in the spectra for 96 h vs. 96 h plus lindane. A P < 0.05 was selected as indicating significance.
Hierarchical cluster analysis revealed marked separation of various spectrum groups (Fig. 5, a–d). This was especially evident in Fig. 5, b and c, 48 h vs. 96 h and 48 h vs. “48 h plus lindane” incubation treatments, respectively: except for a single spectrum, perfect separation was observed between these treatment groups. Although some clustering was also seen when comparing 24 h vs. 48 h and 96 h vs. “96 h plus lindane” incubation treatments, separation appeared to be less evident (Fig. 5, a and d).
FIGURE 5.
(a) Dendrogram showing hierarchical cluster analysis (HCA), based on a Euclidean distance metric to nearest neighbor, of absorption spectra derived from analysis of cellular material after 24-h incubation (A-1 to A-6, in black) compared to 48-h incubation (B-1 to B-10, shaded). Individual spectra are numbered on the ordinate axis. (b) Dendrogram showing HCA of cellular material after 48-h incubation (B-1 to B-10, in black) compared to 96-h incubation (D-1 to D-8, shaded). (c) Dendrogram showing HCA of cellular material after 48-h incubation (B-1 to B-10, in black) compared to 48-h incubation in the presence of a 24-h treatment with lindane (C-1 to C-8, shaded). (d) Dendrogram showing HCA of cellular material after 96-h incubation (D-1 to D-8, in black) compared to 96-h incubation in the presence of a 24-h treatment with lindane (E-1 to E-8, shaded).
DISCUSSION
Mid-IR spectroscopy, commonly achieved using FTIR spectrometers, is an established analytical method for the identification and characterization of molecular species in biological systems (Jackson and Mantsch, 1996). Conventionally, the IR beam is detected optically in the far-field after it has interacted with the sample (Lasch et al., 2002). Analytical results are associated with a database of characteristic electromagnetic radiation energies or absorption wavelengths (Malins et al., 1997; Cohenford and Rigas, 1998; Argov et al., 2002). This information is inferred indirectly across the entire electromagnetic spectrum by comparing detected energies (transmitted through, scattered by or reflected from the sample) with the range of energies contained in the incident beam. Usually, samples require some preparation before measurements.
The requirements for sample preparation are far less strict in photothermal, as compared to conventional, methods. The underlying principle of photothermal methods is that absorption of electromagnetic radiation results in heat generation, which can be measured by sensing the resulting surface temperature increase. This approach has been mainly limited to the photoacoustic technique (Wang, 2003). As the radiation is amplitude modulated, thermal waves are generated internally in the sample being examined. On reaching the surface, these thermal waves generate acoustic waves, which propagate inside a photoacoustic cell to reach a microphone, in the far-field, where they are detected (Sikorska et al., 2001). Our method permits a more direct measurement of temperature. Furthermore, because detection is in the near-field, it allows, potentially, for a spatial resolution that is better than the diffraction limit. Even in applications where spatial discrimination is not required; our method has other advantages over the various optical detection methods. For example, samples that are opaque to IR radiation may be difficult to analyze in transmission mode. For instance, cell nuclei may be strongly IR-absorbing (Tobin et al., 2004). Reflection/absorption methodologies require that samples be deposited on special slides (Cohenford and Rigas, 1998; Argov et al., 2002, Tobin et al., 2004). Although another spectroscopic method, attenuated total reflection (ATR) FTIR, allows for spectrum collection regardless of sample thickness, it requires that the sample be sufficiently smooth or soft, so that the surface of the measuring crystal can be brought within range of the evanescent wave (Chan and Kazarian, 2003).
Microspectroscopy has the potential to objectively identify abnormalities associated with particular disease states; deviations from characteristic biochemical fingerprints (vibrational spectra) of particular tissues would point to the presence of abnormality (Hammiche et al., 2004a). If applied to large cell numbers such objective analysis could, hypothetically, eliminate the potential for human error that sometimes occurs during diagnostic screening. The delivery of such an approach could have profound consequences in terms of screening for diseases such as cervical cancer (Cohenford and Rigas, 1998; Arbyn et al., 2004) and the elimination of false negatives, i.e., where disease is unrecognized and subsequently becomes clinically invasive. Many disease states require close monitoring over time and an archived tissue resource would be useful for intraexperimental comparative purposes. The first priority for such a diagnostic tool is preparation to prevent self-destruction or autolysis of tissue; required to preserve tissue integrity, it should not be confused with sample preparation for analysis. Fixation of samples before analysis, itself an artifact, is necessary to archive tissues before transport to diagnostic laboratories. Previously published studies have adopted elaborate methodologies, such as tissues from paraffin-embedded blocks that were sectioned and mounted on ZnSe or BaF2 windows before dewaxing (Argov et al., 2002; Gazi et al., 2004), tissues frozen on resection and cryomicrotomed onto such windows before air-drying (Tobin et al., 2004), exfoliative cytology in preservative before application to windows (Cohenford and Rigas, 1998), or even the biomonitoring of viable cells (Holman et al., 2000) or cells air-dried (Salman et al., 2003) on ZnSe windows. In contrast, we have simply used 70% EtOH to fix cells in situ. Based on staining quality and preservation of tissue architecture, EtOH- and formalin-fixed tissues have been noted to retain their architectural features in a superior fashion to frozen tissues (Gillespie et al., 2002). More importantly for this study, Gillespie et al. (2002) noted that EtOH-fixation, when compared to formalin-fixation, consistently allowed for better visual identification, using microscopy, of prostate-epithelial-cell nuclear detail; this permitted more accurate grading of hyperplastic, premalignant, and tumor cell nuclei. EtOH fixation through reversible precipitation appears not to induce denaturing effects (Sainte-Marie, 1962) and will rapidly dehydrate and preserve the integrity of cellular material. In our hands, analysis of cellular material using PTMS suggested that our samples were stable over long periods of time (∼6 months, data not shown).
The use of photothermal probes potentially allows higher-than-the-diffraction-limit nanospatial resolution of molecular species (Hammiche et al., 2004b). The first objective of our study was to determine whether clear vibrational IR spectra could be obtained after the analysis of cellular material using PTMS. Figs. 2 and 3 clearly show this to be the case and appear to be the first report of its kind in the literature. The origin of apparent spectral changes associated with cell-cycle kinetics remains ambiguous (Holman et al., 2000; Tobin et al., 2004) and to resolve this issue breast carcinoma MCF-7 cells were examined using PTMS over a time course passing from an exponential-growth phase to a quiescent state (Figs. 2 and 3). Clear elevations in the intensity of DNA/RNA spectral regions and a pronounced peak at 970 cm−1 were associated with cell populations predominantly in S- or G2/M-phase in comparison to less-proliferative G1 cells. The further reductions in spectral intensity seen after examination of cells treated with 10−4 M lindane is further proof that cell-cycle effects may be successfully monitored using PTMS.
Resistance thermometry is based on the temperature dependence of the resistance of a suitable sensing element. The platinum-rhodium wire is characterized by the following probe resistance versus temperature function:
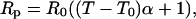 |
where α = 0.00165 ′Ω/Ω-°C is the temperature coefficient of 90% platinum-10% rhodium material, T0 is room temperature, R0 is resistance of the element at room temperature and Rp the measured resistance. The element resistance is determined by passing a current through it and measuring the voltage induced across it. Each °C increase results in an increase in resistance of 0.00165 × R0. The corresponding voltage is 0.00165 × R0 × I. Therefore sensitivity is: 0.00165°C/Ω (probe resistance) per milliamp (bias current). Sensitivity can therefore be improved by increasing the length of the filament and/or by increasing bias current. If spatial resolution is not of importance, the size of the filament can be increased (although Johnson noise has to be taken into account). Bias current can be increased up to the point where Joule heating does not affect the sample material. It should also be noted that heating increases with power dissipated, therefore with both resistance (hence size of the element) and current. As an illustration, the sensitivity of the probe used here (2.5 Ω resistance) is 4 mV per °C per milliamp bias current. The overall sensitivity of a probe made of the same material is 1.6 mV per °C per ohm (probe resistance) per milliamp (bias current).
Although these results were derived from a relatively small number of measurements, our experiment was set up using a relatively insensitive probe (individual spectra of some tens of cells with a maximum of 10 spectra per cell culture contributing to each median spectra). The highly significant differences in spectral regions (Fig. 4) and the discrimination between clusters (Fig. 5) strongly suggest that significant structural changes within exponentially growing cells compared to quiescent cells are observable using PTMS. Future plans for the development of a methodology for high resolution IR analysis (subcellular) will require the use of probes with a much smaller sensing element than that used in this initial study, i.e., of the order of a few tens of nanometers. Micromachined probes with a sensing element 150 nm in size have been used but small probe size is not sufficient. Spatial resolution also depends on thermal diffusion length, therefore on IR beam modulation frequency. For instance, an organic material such as a typical polymer is characterized by a thermal diffusion length of ∼700 nm at 100 kHz modulation frequency and ∼200 nm at 1 MHz. However, the higher the modulation frequency the smaller the amplitude of the temperature to be measured. Hence the need to use a bright source to achieve acceptable SNR at these operating frequencies. To achieve subcellular spatial resolution, PTMS is being further developed by using a micromachined sensor probe, adding an extra high frequency modulation and adapted to accommodate a much brighter IR light source from a synchrotron ring.
The characterization of conformational changes associated with transcription and replication is of great importance (Khorasanizadeh, 2004). Our findings provide the first evidence that PTMS may be used to examine cell cycle-associated intracellular structural changes. Such findings would constitute a novel approach in the use of microspectroscopy.
Acknowledgments
We thank Dr. Nigel F. Fullwood for helpful discussions.
This study was primarily supported by the Engineering and Physical Sciences Research Council, UK. Funding from the North West Cancer Research Fund, UK (R.H.) is also acknowledged.
References
- Arbyn, M., F. Buntinx, M. Van Ranst, E. Paraskevaidis, P. Martin-Hirsch, and J. Dillner. 2004. Virologic versus cytologic triage of women with equivocal Pap smears: a meta-analysis of the accuracy to detect high-grade intraepithelial neoplasia. J. Natl. Cancer Inst. 96:280–293. [DOI] [PubMed] [Google Scholar]
- Argov, S., J. Ramesh, A. Salman, I. Sinelnikov, J. Goldstein, H. Guterman, and S. Mordechai. 2002. Diagnostic potential of Fourier-transform infrared microspectroscopy and advanced computational methods in colon cancer patients. J. Biomed. Opt. 7:248–254. [DOI] [PubMed] [Google Scholar]
- Boydston-White, S., T. Gopen, S. Houser, J. Bargonetti, and M. Diem. 1999. Infrared spectroscopy of human tissue. V. Infrared spectroscopic studies of myeloid leukemia (ML-1) cells at different phases of the cell cycle. Biospectroscopy. 5:219–227. [DOI] [PubMed] [Google Scholar]
- Bozec, L., A. Hammiche, H. M. Pollock, M. Conroy, J. M. Chalmers, N. J. Everall, and L. Turin. 2001. Localized photothermal infrared spectroscopy using a proximal probe. J. Appl. Phys. 90:5159–5165. [Google Scholar]
- Chan, K. L., and S. G. Kazarian. 2003. New opportunities in micro- and macro-attenuated total reflection infrared spectroscopic imaging: spatial resolution and sampling versatility. Appl. Spectrosc. 57:381–389. [DOI] [PubMed] [Google Scholar]
- Cohenford, M. A., and B. Rigas. 1998. Cytologically normal cells from neoplastic cervical samples display extensive structural abnormalities on IR spectroscopy: implications for tumor biology. Proc. Natl. Acad. Sci. USA. 95:15327–15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, C., S. Bhana, A. J. Shorrocks, and F. L. Martin. 2002. Oestrogens induce G1 arrest in benzo[a]pyrene-treated MCF-7 breast cells whilst enhancing genotoxicity and clonogenic survival. Mutagenesis. 17:431–438. [DOI] [PubMed] [Google Scholar]
- Diem, M., S. Boydston-White, and L. Chiriboga. 1999. Infrared spectroscopy of cells and tissues: shining light onto a novel subject. Appl. Spectrosc. 53:148A–161A. [Google Scholar]
- Gazi, E., J. Dwyer, N. Lockyer, P. Gardner, J. C. Vickerman, J. Miyan, C. A. Hart, M. Brown, J. H. Shanks, and N. Clarke. 2004. the combined application of FTIR microspectroscopy and ToF-SIMS imaging in the study of prostate cancer. Faraday Discuss. 126:41–59. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. W., C. J. Best, V. E. Bichsel, K. A. Cole, S. F. Greenhut, S. M. Hewitt, M. Ahram, Y. B. Gathright, M. J. Merino, R. L. Strausberg, J. I. Epstein, S. R. Hamilton, G. Gannot, G. V. Baibakova, V. S. Calvert, M. J. Flaig, R. F. Chuaqui, J. C. Herring, J. Pfeifer, E. F. Petricoin, W. M. Linehan, P. H. Duray, G. S. Bova, and M. R. Emmert-Buck. 2002. Evaluation of non-formalin tissue fixation for molecular profiling studies. Am. J. Pathol. 160:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, J. and M. D'Oliveira. 1989. Learning to Use Statistical Tests in Psychology. Open University Press, Milton Keynes, UK.
- Hammiche, A., L. Bozec, M. J. German, J. M. Chalmers, N. J. Everall, G. Poulter, M. Reading, M. Reading, D. B. Grandy, F. L. Martin, and H. M. Pollock. 2004a. Mid-infrared microspectroscopy of difficult samples using near-field photothermal microspectroscopy. Spectroscopy. 19:20–42. [Google Scholar]
- Hammiche, A., L. Bozec, H. M. Pollock, M. German, and M. Reading. 2004b. Progress in near-field photothermal infra-red microspectroscopy. J. Microsc. 213:129–134. [DOI] [PubMed] [Google Scholar]
- Holman, H. Y, M. C. Martin, E. A. Blakely, K. Bjornstad, and W. R. McKinney. 2000. IR spectroscopic characteristics of cell cycle and cell death probed by synchrotron radiation based Fourier transform IR spectromicroscopy. Biopolymers. 57:329–335. [DOI] [PubMed] [Google Scholar]
- Jackson, M., and H. H. Mantsch. 1996. Biomedical infrared spectroscopy. In Infrared Spectroscopy of Biomolecules. H. H. Mantsch and D. Chapman, editors. Wiley-Liss, New York. 311–340.
- Kalantzi, O. I., R. Hewitt, K. J. Ford, L. Cooper, R. E. Alcock, G. O. Thomas, J. A. Morris, T. J. McMillan, K. C. Jones, and F. L. Martin. 2004. Low-dose induction of micronuclei by lindane. Carcinogenesis. 25:613–622. [DOI] [PubMed] [Google Scholar]
- Khorasanizadeh, S. 2004. The nucleosome: from genomic organization to genomic regulation. Cell. 116:259–272. [DOI] [PubMed] [Google Scholar]
- Lasch, P., A. Pacifico, and M. Diem. 2002. Spatially resolved IR microspectroscopy of single cells. Biopolymers. 67:335–338. [DOI] [PubMed] [Google Scholar]
- Malins, D. C., P. M. Johnson, E. A. Barker, N. L. Polissar, T. M. Wheeler, and K. M. Anderson. 2003. Cancer-related changes in prostate DNA as men age and early identification of metastasis in primary prostate tumors. Proc. Natl. Acad. Sci. USA. 100:5401–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malins, D. C., N. L. Polissar, and S. J. Gunselman. 1997. Infrared spectral models demonstrate that exposure to environmental chemicals leads to new forms of DNA. Proc. Natl. Acad. Sci. USA. 94:3611–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh, B., and M. M. S. Heck. 2003. Regulation of chromosome condensation and segregation. Curr. Opin. Genet. Dev. 13:185–190. [DOI] [PubMed] [Google Scholar]
- Moggs, J. G., J. I. Goodman, J. E. Trosko, and R. A. Roberts. 2004. Epigenetics and cancer: implications for drug discovery and safety assessment. Toxicol. Appl. Pharmacol. 196:422–430. [DOI] [PubMed] [Google Scholar]
- Pollock, H. M., and A. Hammiche. 2001. Micro-thermal analysis: techniques and applications. J. Phys. D Appl. Phys. 34:R23–R53. [Google Scholar]
- Sainte-Marie, G. 1962. A paraffin embedding technique for studies employing immunofluorescence. J. Histochem. Cytochem. 10:250–256. [Google Scholar]
- Salman, A., J. Ramesh, V. Erukhimovitch, M. Talyshinsky, S. Mordechai, and M. Huleihel. 2003. FTIR microspectroscopy of malignant fibroblasts transformed by mouse sarcoma virus. J. Biochem. Biophys. Methods. 55:141–153. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ruiz, J. M., and M. Martinez-Carrion. 1988. A Fourier-transform infrared spectroscopic study of the phosphoserine residues in hen egg phosvitin and ovalbumin. Biochemistry. 27:3338–3342. [DOI] [PubMed] [Google Scholar]
- Sikorska, A., B. J. Linde, and J. I. Kukielski. 2001. The effect of solvent polarity on photo-acoustic spectra of alkyl-cyanobiphenyl derivatives. J. Opt. A: Pure Appl. Op. 3:S71–S76. [Google Scholar]
- Tobin, M. J., M. A. Chesters, J. M. Chalmers, F. J. M. Rutten, S. E. Fisher, I. M. Symonds, A. Hitchcock, R. Allibone, and S. Dias-Gunasekara. 2004. Infrared microscopy of epithelial cancer cells in whole tissues and in tissue culture, using synchrotron radiation. Faraday Discuss. 126:27–39. [DOI] [PubMed] [Google Scholar]
- Wang, L. H. V. 2003. Ultrasound-mediated biophotonic imaging: A review of acousto-optical tomography and photo-acoustic tomography. Dis. Markers. 19:123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrin, E., and V. Legagneux. 2003. Introduction to chromosome dynamics in mitosis. Biol. Cell. 95:507–513. [DOI] [PubMed] [Google Scholar]
- Wolffe, A. P. 1997. Chromatin: Structure and Function. Academic Press, New York.
- Yared, E., T. J. McMillan, and F. L. Martin. 2002. Genotoxic effects of oestrogens in breast cells as detected by the micronucleus assay and the Comet assay. Mutagenesis. 17:345–352. [DOI] [PubMed] [Google Scholar]