

Abstract
Oxidation and reduction (redox) are known to modulate the function of a variety of ion channels. Here, we report a redox regulation of the function of ClC-0, a chloride (Cl−) channel from the Torpedo electric organ. The study was motivated by the occasional observation of oocytes with hyperpolarization-activated Cl− current when these oocytes expressed ClC-0. We find that these atypical recording traces can be turned into typical ClC-0 current by incubating the oocyte in millimolar concentrations of reducing agents, suggesting that the channel function is regulated by oxidation and reduction. The redox control apparently results from an effect of oxidation on the slow (inactivation) gating: oxidation renders it more difficult for the channel to recover from the inactivated states. Introducing the point mutation C212S in ClC-0 suppresses the inactivation state, and this inactivation-suppressed mutant is no longer sensitive to the inhibition by oxidizing reagents. However, C212 is probably not the target for the redox reaction because the regulation of the inactivation gating by oxidation is still present in a pore mutant (K165C/K165 heterodimer) in which the C212S mutation is present. Taking advantage of the K165C/K165 heterodimer, we further explore the oxidation effect in ClC-0 by methane thiosulfonate (MTS) modifications. We found that trimethylethylammonium MTS modification of the introduced cysteine can induce current in the K165C/K165 heterodimer, an effect attributed to the recovery of the channel from the inactivation state. The current induction by MTS reagents is subjected to redox controls, and thus the extent of this current induction can serve as an indicator to report the oxidation state of the channel. These results together suggest that the inactivation gating of ClC-0 is affected by redox regulation. The finding also provides a convenient method to “cure” those atypical recording traces of ClC-0 expressed in Xenopus oocytes.
INTRODUCTION
The members of the ClC family transport Cl− ions across cell membranes and play important roles in various physiological functions, including regulation of the membrane potential, transepithelial Cl− transport, and, in the case of bacteria, the resistance to acidic environments (Jentsch et al., 1999, 2002; Maduke et al., 2000; Iyer et al., 2002; Accardi and Miller, 2004). The structural architecture of the transmembrane domains of these molecules has recently been suggested from the crystal structure of two bacterial ClC homologs (Dutzler et al., 2002, 2003). Significant conservation of the functionally important channel sequences among ClC family members suggests that the structure of bacterial ClCs may serve as a model for understanding the structure and function of vertebrate ClC channels (Chen and Chen, 2003; Lin and Chen, 2003). Indeed, the presence of two ion-transport pathways in these family members, which was predicted two decades ago based on functional electrophysiological studies carried out in the Torpedo ClC-0 channel (Miller, 1982; Hanke and Miller, 1983; Miller and White, 1984), was confirmed by the bacterial ClC structure.
Although high-resolution structures of bacterial ClC proteins provided a rich suggestion for vertebrate ClC channel functions, recent studies showed that the bacterial ClC protein is not an ion channel but a transporter (Accardi et al., 2004; Accardi and Miller, 2004). This finding indicates that studying vertebrate ClC channels is necessary for understanding the channel functions, namely the gating and permeation properties of these proteins. For functional studies of ClC channels, Xenopus oocytes have been widely used, and ClC-0 is the best-studied member of the entire ClC family. However, in expressing this prototypic member of the ClC family in Xenopus oocytes, we encounter a wide spectrum of current recording traces. Fig. 1 illustrates the variation of current obtained from two-electrode voltage-clamp recordings. Frequently, typical ClC-0 current traces such as those shown in the literature are obtained, with a characteristic “crossover” at the negative voltage range (Fig. 1, left). However, it is also quite common to encounter oocytes expressing atypical current, which normally would have been considered “bad” oocytes without ClC-0 current (Fig. 1, middle). Between these two extremes, there are also oocytes that produce the ClC-0-like current at depolarized potentials, but when the membrane potentials are negative enough—for example, −140 or −160 mV—the hyperpolarization-activated current appears as if the recorded current is contaminated by the leak current or by the endogenous oocyte channels (Fig. 1, right).
FIGURE 1.

Variations of the ClC-0 current in different oocytes. Three oocytes that were not pretreated with oxidizing or reducing reagents were recorded using protocol 2. Test voltages were from +80 mV to −160 mV in −20-mV steps. The −120 mV voltage step was shown only for the final 10–20 ms. On the left are typical ClC-0 current traces, with the first crossover occurring between −80 and −100 mV. The traces in the middle and right panels are oxidized: the current is small and a short pulse (125 ms) at −140 or −160 mV activated the current. On the other hand, for the typical recordings (left), the same negative voltage pulse that was twice as long did not elicit the hyperpolarization-induced current, indicating that most of the channels were not in the deep inactivated state during the recording. After treatment of these three oocytes with β-ME, the current measured at +80 mV in the middle and right panels increases to ∼10 and 13 μA, respectively, whereas the current shown in the left panel did not increase.
Because Xenopus oocytes are valuable for functional studies of ClC channels, we were motivated to investigate the variation of the ClC-0 current. We discovered by serendipity that such a variation was largely due to the oxidation state of the oocyte. Oxidation of oocytes by externally applied copper phenanthroline (CuPhe) not only rendered the ClC-0 current traces atypical, but also led to an inhibition of the current. On the other hand, incubating the oocytes in β-mercaptoethanol (β-ME) or dithiothretol (DTT) turned atypical ClC-0 recordings into typical traces. Oxidation and reduction are known to affect the functions of many proteins, including ion channels (Feng et al., 2000; Pessah and Feng, 2000; Xia et al., 2000; Tang et al., 2001; Zeidner et al., 2001). To understand more about this redox effect, we first examined whether any of the 12 endogenous cysteine residues in ClC-0 could be the target of oxidation and reduction. We found that mutation of an inactivation-suppressed mutant C212S (Lin et al., 1999) is resistant to the oxidation inhibition by CuPhe. However, a heterodimeric mutant, K165C/K165, which was constructed in the C212S mutation background, also exhibited a prominent regulation by oxidation. Thus, it is likely that the oxidation causes the channel to stay in the deep inactivation state. Experiments using methane thiosulfonate (MTS) reagents to modify the introduced cysteine in the K165C/K165 heterodimer are also consistent with this assertion, because the more inactivated the channel, the more current is induced by MTS reagents.
MATERIALS AND METHODS
Mutagenesis and channel expression
The wild-type (WT) and mutant ClC-0 were all constructed in the expression vector pBluescript. The construction of the K165C/K165 heterodimer in the background of the C212S mutation was described previously (Lin and Chen, 2000). Construction of the other heterodimers followed the same approach. Briefly, the two monomers (that is, K165C/C212S and C212S) were linked together by proper restriction sites, with four amino acid residues (GTTS) inserted between (Lin and Chen, 2000). No appreciable difference between the two versions of the heterodimer (e.g., K165-K165C or K165C-K165) was noticed. Thus, they will simply be called “K165C heterodimer” in the following text. For the other various cysteine mutants, the WT ClC-0 constructed in pBluescript vector was used as a mother plasmid. Mutations of the 12 endogenous cysteines (single, double, or triple mutants) were made using PCR mutagenesis approaches. All mutations were confirmed by commercially available DNA sequence services. Synthesis of RNAs from the cDNA constructs and the injection of RNAs into Xenopus oocytes were performed according to previously described methods (Chen, 1998; Lin et al., 1999; Lin and Chen, 2000).
Preparation of the sulfhydryl reactive reagents
DDT and β-ME were purchased from Pharmacia Biotech (Piscataway, NJ) and Bio-Rad (Hercules, CA), respectively. For the oxidizing reagents, CuPhe was made by mixing 150 mM CuSO4 with 500 mM 1,10-phenanthroline (Sigma Chemical, St. Louis, MO) in 1:4 ethanol/water mixture (Fahlke et al., 1998). Hydrogen peroxide (H2O2) was from EM Science/Merck (Darmstadt, Germany). After RNA injection, oocytes were kept in a high Ca2+ ND96 solution of (in mM) 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES, pH 7.5. To manipulate the oxidation or reduction conditions of the oocytes, the oxidizing and reducing reagents were directly added to the incubation buffer. Upon recording, the redox controlling reagents were removed by transferring the oocytes to a fresh ND96 medium. For oocytes in the mildly oxidized condition, no redox reagent was added to the incubation buffer. As shown in Results, and by others (Gross and MacKinnon, 1996; Zeidner et al., 2001), the plain ND96 solution has a mild oxidizing capability.
MTS reagents were purchased from Toronto Research Chemicals (North York, Ontario). Stock solutions of 0.1–0.3 M were prepared in distilled water and stored at −70°C. The working solutions containing indicated concentrations of the MTS reagents were made immediately before use.
Macroscopic current recordings
Whole-oocyte currents were measured by a two-electrode, voltage-clamp amplifier (725C, Warner Instruments, Hamden, CT). Control of the membrane potential and data acquisition were performed by a Pentium PC computer equipped with Digidata 1200 analog interface, using pClamp6 software (Axon Instruments, Union City, CA). The standard recording (extracellular) solution was ND96 solution, containing (in mM) 96 NaCl, 2 KCl, 1 MgCl2, 0.3 CaCl2, and 5 HEPES, pH 7.5. Except where indicated, two voltage protocols were used throughout the study. In one (protocol 1), the membrane potential of the oocyte was clamped at −30 mV, and the current was monitored with a +40-mV voltage step for 50 or 100 ms followed by a −150-mV voltage step for 100 ms. Since the fast gate of the channel usually has an open probability close to unity at the voltage of +40 mV, protocol 1 is convenient to monitor the slow gating of ClC-0 in a continuous recording. The voltage pulses were given once every 2, 4, or 6 s. The higher the pulse frequency was (the more time the channel experienced the negative voltage, −150 mV), the faster was the recovery of the channel from the inactivation state. The oocyte current at the end of the +40-mV voltage step was used to monitor the slow-gating process.
Examination of the fast-gate open probability (Po) from macroscopic current recordings followed a protocol (protocol 2) that applied a family of different voltage steps to the oocyte (Chen, 1998; Lin et al., 1999; Lin and Chen, 2000). To start this voltage protocol, the current of the oocyte was first activated by protocol 1. Usually, it took ∼50 pulses (given once every 2 s) for the slow gating to reach a steady state (see Fig. 2 C). Protocol 2 (see Fig. 2, B and D) started with a negative voltage step of −120 mV for 100–200 ms to further ensure the opening of the slow gate. A positive prepulse (at +60 or +80 mV) was then given to open the fast gate, followed by different test voltages from +60 or +80 mV to −160 mV in −20-mV steps. The tail current was measured at −100 mV, and the extrapolated value to the beginning of the tail current was normalized to that obtained after the most depolarized test pulse.
FIGURE 2.

Effect of β-ME on the oxidized WT ClC-0 current. Whole-oocyte current was compared before (A and B) and after (C and D) 5 mM β-ME treatment for 1 h. Temperature was ∼20°C. (A) Time course of current activation by protocol 1, with one pulse every 2 s. The current at the end of +60 mV (○) and −150 mV (•) are plotted against time. The recording traces within the inset box are taken from 0-, 58-, and 118-s time points, respectively. The oocyte current did not increase significantly, even though a −150 mV voltage step for 100 ms was applied every 2 s. (B) After the recordings in A, the same oocyte was immediately evaluated with protocol 2, with the test voltages from +60 mV to −160 mV in −20-mV steps. (C and D) Whole-cell current of the same oocyte as that examined in A and B after the oocyte was treated with β-ME. The experimental manipulations in C and D were the same as those in A and B, respectively. Current traces in the inset box are the same time points as those described in A. Notice that the current measured at +40 mV is monotonically increased to a much higher level by the −150 mV voltage step (see ○in C), and the current deactivation at negative potentials is prominent, resulting in the typical crossover of the recording traces at negative voltages (see recording traces at −120, −140, and −160 mV in D).
Except for the experiments shown in Fig. 6 B (see Fig. 6), all recordings were conducted at room temperature (∼20–24°C). In Fig. 6 B, the temperature control was made by preincubating the solution in a water bath set at the same temperature as the adjusted room temperature. The temperature of the solution in the recording chamber was measured by a small temperature-sensitive element connected to a heater controller (TC324A; Warner Instruments).
FIGURE 6.

Current induction of the K165C heterodimer by MTSET. (A) Voltage dependence of the current induction. The oocyte current was monitored by the voltage pulse shown on the right. The indicated voltages were the potentials applied during the pulse. Each circle represents the current amplitude measured at the end of the +40 mV voltage step. Arrows indicate the time point at which 30 μM MTSET was applied. (B) Temperature dependence of the current induction. Oocyte current was monitored by the pulsing protocol as in the bottom panel of A, and each circle is the current measured at the end of the +40 mV voltage step. All oocytes in A and B were incubated in ND96 without β-ME for at least 2 h before the experiments. (C) Comparison of the fast-gate Po-V curves of the MTSET and β-ME-treated K165C heterodimers. Data points were the average of three independent measurements. MTSET, 100 μM; β-ME, 2 mM. The solid and dotted curves were drawn according to a Boltzmann equation: Po = Pmin + (1 − Pmin)/[1 + exp(−zF(V − V1/2)/RT)], with Pmin = 0.15. The values of V1/2 and z were: −83 mV and 0.77, respectively, for β-ME (circles), and −86 mV and 0.89 for MTSET (squares).
For the experiments involving solution exchanges, the solutions containing the required reagents (such as MTS reagents, β-ME, or CuPhe) were made immediately before use. The volume of the recording chamber was ∼1 ml, and the flow rate of the solution was usually more than 15–20 ml/min, making the solution exchange time constant <5 s. The effects of β-ME and CuPhe were much slower than the solution exchange speed. In the current-induction experiments by 2-aminotheyl MTS (MTSEA) or trimethylethylammonium MTS (MTSET), the concentrations of the MTS reagents were chosen to give an apparent induction time constant at least fivefold larger than the solution exchange time.
Data analysis
An archive of electrophysiological experiments has shown that ClC-0 contains two identical pores, as suggested from the three equally spaced current levels in single-channel recording traces (Miller, 1982; Hanke and Miller, 1983; Miller and White, 1984; Bauer et al., 1991; Chen and Miller, 1996; Ludewig et al., 1996; Middleton et al., 1996; Saviane et al., 1999; Lin and Chen, 2000; Chen and Chen, 2003). These two pores are regulated by two gating mechanisms, fast gating and slow gating. At the macroscopic current level, the fast gating is represented by the current relaxation at the millisecond time range, as shown by the current deactivation at the negative voltage range in Fig. 1 (left). This current deactivation results from a decrease of the fast-gate Po upon membrane hyperpolarization. On the other hand, the slow-gate opening is favored when the membrane potential is hyperpolarized (Pusch et al., 1997; Chen, 1998). Thus, in an experiment using protocol 1, the current at +40 mV will gradually increase because the channel recovers from the inactivation state when the oocyte experiences the negative potential (−150 mV) in each voltage sweep. With protocol 2, the reduction of fast-gate Po at large negative voltages usually surpasses the increase of the driving force for permeant ions, resulting in “typical” recording traces (Fig. 1, left) with a characteristic “crossover” at the negative voltage range (Pusch et al., 1995; Ludewig et al., 1996; Chen, 1998; Maduke et al., 1998; Chen and Chen, 2001).
To compare the recorded current we adopted an empirical method that quantifies the pattern of the recording traces. From the current traces activated by protocol 2, we measured the steady-state current at the most depolarized voltage (+60 or +80 mV) and counted the number of traces that crossed the one obtained at the 20-mV, more depolarized voltage. For example, the crossover numbers for recordings in Fig. 1 (left, middle, and right) are 4, 0, and 0, respectively. In our entire set of data performed in ∼100 mM extracellular Cl−, the largest crossover number is 4. Since each voltage step differs by 20 mV, and the most hyperpolarized voltage step in protocol 2 is −160 mV, the first crossover occurs between the trace at −80 and that at −100 mV, or at more hyperpolarized voltages when the recording traces become atypical.
Statistical information was collected using Origin software (OriginLab, Northampton, MA). The results in Table 1, and the average value in Figs. 3 and 4 A, are given as mean ± SD to better show the distribution of the values. All other results are presented as mean ± SE.
TABLE 1.
Variation of the WT ClC-0 current activated by protocol 2
| Before β-ME
|
After β-ME
|
||||
|---|---|---|---|---|---|
| Frog | No. of Oocytes | No. of crossover (mean ± SD) | I (μA) (mean ± SD) | No. of crossover (mean ± SD) | I (μA) (mean ± SD) |
| 1 | 25 | 2.3 ± 1.0 | 11.7 ± 7.6 | 3.1 ± 0.8* | 13.0 ± 8.9 |
| 2 | 7 | 3.4 ± 0.5 | 20.7 ± 5.3 | 3.4 ± 0.5 | 23.3 ± 7.2 |
| 3 | 10 | 2.9 ± 1.1 | 8.5 ± 4.4 | 3.4 ± 0.5 | 10.4 ± 4.2 |
| 4 | 12 | 3.2 ± 0.4 | 17.7 ± 6.7 | 3.3 ± 0.5 | 20.1 ± 6.9 |
| 5 | 9 | 3.2 ± 0.4 | 11.6 ± 4.1 | 3.2 ± 0.4 | 14.6 ± 4.2 |
| 6† | 16 | 2.0 ± 1.3 | 14.0 ± 4.7 | 2.8 ± 0.8* | 19.2 ± 6.1* |
| 7† | 16 | 1.9 ± 1.0 | 7.6 ± 3.8 | 3.1 ± 0.8* | 14.9 ± 5.5* |
| 8 | 17 | 3.4 ± 0.6 | 14.8 ± 3.6 | 2.8 ± 0.7 | 21.1 ± 4.7* |
| 9 | 22 | 3.6 ± 0.6 | 9.4 ± 3.0 | N.D. | N.D. |
| 10 | 24 | 3.1 ± 0.5 | 11.7 ± 3.8 | N.D. | N.D. |
| 11† | 14 | 0.1 ± 0.3 | 2.5 ± 1.1 | 2.9 ± 0.9* | 10.6 ± 2.8* |
| 12† | 15 | 0.3 ± 0.5 | 1.7 ± 0.7 | 3.5 ± 1.1* | 3.9 ± 1.5* |
| 13† | 4 | 0.3 ± 0.5 | 7.5 ± 4.3 | 2.3 ± 1.0* | 9.4 ± 4.3* |
| 14† | 20 | 3.1 ± 0.6 | 8.4 ± 3.4 | 3.4 ± 0.7 | 11.9 ± 3.7* |
Test voltages were from +60 or +80 mV to −160 mV in −20-mV steps. The same oocyte was recorded before and after a 1-h treatment with 5 mM β-ME. The number of trace crossovers and the current from the most depolarized voltage are shown. Frogs 1–7 were operated for the first time, whereas frogs 8–14 had received at least one surgery before the oocytes were harvested.
A statistical difference (pair t-test, p < 0.01) from the control value obtained before β-ME treatment, suggesting that the oocytes might be at least partially oxidized.
Frogs that had been kept longer than 3 months before oocytes were harvested.
FIGURE 3.

Oxidation effects on the WT ClC-0. (A) Effects of 10 μM CuPhe on ClC-0. All current traces in the left, middle, and right panels were from the same oocyte. Recordings on the left were made after the oocyte was treated with 5 mM β-ME for 1 h. Recordings in the middle were made after the oocyte was treated with 10 μM CuPhe. At right are recordings made after the oocyte was again treated with 5 mM β-ME after the CuPhe treatment. The number of crossovers and the current at the most depolarized voltage (both presented as mean ± SD) for 20 oocytes are shown. The asterisks indicate a significant difference from the corresponding value on the left panel (pair t-test with the significance level at p = 0.01). (B) Recordings of WT ClC-0 in another 19 oocytes. The recording procedure was the same as that shown in A, except that the recordings in the middle panel were made after 1 h of treatment with 10 μM CuPhe + 5 mM β-ME. (C) Effects of CuSO4 on WT ClC-0. The recording procedure was the same as that shown in A and B, except that the recordings in the middle panel were made after 1 h of treatment with 10 μM CuSO4 (n = 7). (D) Effects of H2O2 on WT ClC-0. The experimental procedure was the same as those shown in A–C, except that the recordings in the middle panel were made after 1 h of treatment with 0.01% H2O2 (n = 5).
FIGURE 4.

Effects of CuPhe on various cysteine mutants of ClC-0. (A) Effects of CuPhe on C212S mutant. The oocyte was not pretreated with β-ME before the recording shown on the left. The recording on the right was made after the same oocyte was treated with 10 μM CuPhe for 1 h. From nine oocytes, the number of crossovers and the current at +60 mV were also shown (mean ± SD). There is no statistical difference before and after CuPhe at a significant level of p = 0.01 (pair t-test). (B) Effects of CuPhe on various mutants of ClC-0. The endogenous cysteine residues were mutated as indicated. The oocyte current was compared before and after 10 μM CuPhe treatment for 1 h using protocol 2. The current amplitude at the most depolarized voltage (+60 or +80 mV) after CuPhe treatment was normalized to the current before CuPhe. The data of WT channels were from a different set of oocytes from that shown in Fig. 3 A.
RESULTS
Redox control of WT ClC-0 current
The expression of WT ClC-0 in Xenopus oocytes is robust in that Cl− current is usually observed 24–48 h after mRNA injection. The pattern of the whole-oocyte current, however, is highly variable from different batches of oocytes, and sometimes even within the same batch of oocytes. In some recordings, the current is outwardly rectified at steady state, with deactivation when the membrane potential is stepped to a negative voltage, as expected from the voltage dependence of fast-gate open probability (Fig. 1, left). It is, however, quite common to encounter mRNA-injected oocytes with currents that appear to be contaminated by the endogenous hyperpolarization-activated current (Fig. 1, middle and right). Such an atypical ClC-0 current can be converted into the typical ClC-0 current by incubating oocytes in reducing reagents, as illustrated by the experiments shown in Fig. 2. For this experiment, the oocyte was incubated in ND96 without reducing reagents, and was recorded 2 days after mRNA injection. This oocyte had a very small outward current, and a short (100-ms) negative pulse to −150 mV was long enough to activate a hyperpolarization-induced current (Fig. 2 A, inset). Delivering a voltage family (from +60 to −160 mV) using protocol 2 did not elicit current traces crossing each other (Fig. 2 B). After the experiments shown in Fig. 2, A and B, the same oocyte was incubated in 5 mM β-ME for 1 h, and the same voltage protocol activated a very different pattern of current traces. With a 100-ms voltage pulse to −150 mV continuously applied at 0.5 Hz, the outward current (and also the instantaneous inward current) was gradually activated (Fig. 2 C). Using protocol 2, the activated current showed the hallmark of crossover at negative voltages (Fig. 2 D). These experiments indicate that oxidation and reduction of the oocyte may affect the function of the WT ClC-0.
The different patterns in the WT ClC-0 current expressed in Xenopus oocytes appeared to come from natural variation, most likely related to the quality of oocytes. The most atypical current (at +80 mV) was usually small, and there was no crossover of traces at negative voltage ranges. The typical recordings, on the other hand, usually revealed 3–4 crossovers in our recording conditions. There were also recordings with 1–2 crossovers, and they were considered as intermediate recordings between the typical and atypical traces. Table 1 lists a summary of WT ClC-0 current before and after a 1-h treatment with 5 mM β-ME in oocytes taken from 14 frogs. If the number of crossovers or the mean current increases after β-ME treatment, we consider that the oocytes might at least be partially oxidized. Overall, the oocytes from freshly adopted frogs produced oxidized current only occasionally. However, after the frogs were kept for several months, it appeared that more oocytes expressed atypical current (Table 1).
As the atypical current can be converted into typical recordings by β-ME, we suspect that oxidation may confer an opposite effect; that is, converting the typical ClC-0 current into atypical traces. Fig. 3 A (left) shows the recording of an oocyte that has been pretreated with reducing agents. There are three crossovers in this recording at −120, −140, and −160 mV (traces at these three voltages cross with those at −100, −120, and −140 mV, respectively). After this recording, the same oocyte is subjected to oxidation in the presence of 10 μM CuPhe. In 1 h, ∼70% of the current is inhibited, and the recordings do not show any crossover (Fig. 3 A, middle). Application of 5 mM β-ME on the same oocyte then completely recovers the current (Fig. 3 A, right). The average number of crossovers and the current amplitude at +80 mV in 20 oocytes (from three frogs) are also shown for each condition. It can be seen that 10 μM CuPhe treatment invariably inhibits the current. Furthermore, none of the 20 CuPhe-treated oocytes shows any crossover of recording traces (Fig. 3 A, middle).
Because the ClC-0 inactivation gating is known to be facilitated by extracellular zinc ions (Chen, 1998), it might be possible that the inhibition by CuPhe does not come from an effect of oxidation but from a direct effect of copper ions in facilitating the slow gating of the channel. To assure that the effect was not simply from copper ions, we conducted control experiments in which the oxidation power of 10 μM CuPhe was removed while copper ions were present in the incubation buffer. Incubating oocytes in the mixture of CuPhe and β-ME, though resulting in a slight decrease of current at +80 mV, has no effect on the number of crossovers (Fig. 3 B). Treating oocytes with 10 μM copper sulfate (CuSO4) for 1 h does not result in alterations of the crossover number and the open channel current (Fig. 3 C). Using a metal-ion-free reagent, H2O2, on the other hand, gives a similar oxidation effect as that produced by CuPhe. Incubating oocytes in 0.01% H2O2 for 1 h suppresses WT ClC-0 current by nearly 70%, and reduces the number of crossovers as well (Fig. 3 D).
Inactivation-suppressed mutant C212S is insensitive to oxidation
The inhibition of the WT current by oxidizing reagents occurs slowly, and the oxidized channel appears to be more difficult to activate by hyperpolarization (Fig. 2). This suggests that oxidation may render it more difficult for the channel to recover from the inactivation state. If this is the case, we expect that oxidation will have a small effect, or even no effect, on the channel whose slow (inactivation) gate is locked open. The inactivation-suppressed mutant C212S (Lin et al., 1999) usually shows current traces with three to four crossovers at negative voltages (Fig. 4 A, left). To examine the oxidation effect on the C212S mutant, we applied 10 μM CuPhe to the oocyte. Fig. 4 A (right) shows that the C212S mutant is resistant to the inhibition of 10 μM CuPhe. With a 15-fold higher concentration (150 μM), <10% of the current was inhibited in 1 h (data not shown).
To further examine whether any of the other 11 endogenous cysteine residues in the WT ClC-0 was also important for the redox control described here, we tested the sensitivity of various cysteine mutants to the CuPhe inhibition. Fig. 4 B summarizes the effect of oxidation (10 μM CuPhe for 1 h) on the mutants in which single or multiple endogenous cysteine residues were mutated. Except for the mutants including the C212 mutation (C212S single mutant or the C212A/C213A/C229S triple mutant), all the cysteine mutants showed sensitivity to CuPhe inhibition approximately similar to that of the WT channel. On the other hand, mutants lacking a cysteine at position 212 appeared to be resistant to the inhibition by oxidizing reagents.
C212 is probably not the target for the redox effect on the inactivation gating
To understand the functional role of C212 in the aforementioned redox control on the channel's function, we studied the effects of CuPhe and β-ME on several C212S-related mutants (Fig. 5). If the resistance of C212S to CuPhe was due to a prevention of disulfide-bond formation between the two C212 residues, a C212-C212S heterodimer should also be resistant to CuPhe inhibition. This, however, was not the case (Fig. 5 A). Another mutant, the K165C heterodimer in the presence of C212S background mutation, provides further insight into the role of C212S mutation. This mutant was shown to have prominent inactivation gating, although the C212 residue was replaced with serine in both subunits (Lin and Chen, 2000). In Fig. 5 B (upper left), an oocyte expressing the K165C heterodimer (with C212S background) was first incubated in 5 mM β-ME for more than 2 h. The oocyte was then transferred to the recording chamber, and the current was monitored by pulsing protocol 1. The current measured at +40 mV was slowly decreased when β-ME was removed from the bath, indicating an oxidation capability of the plain ND96 solution. The oxidation power was further increased by adding 1.5 μM CuPhe to the ND96 solution (Fig. 5 B, upper panel). Furthermore, both the spontaneously oxidized current and the CuPhe-inhibited current can be recovered by β-ME treatment with similar time courses (Fig. 5 B, bottom panel), suggesting that the spontaneous oxidation in ND96 solution and the CuPhe treatment may exert the same effect in inhibiting the K165C heterodimer. Besides the K165C heterodimer, we also examined the K165E heterodimer in the C212S background mutation, and this mutant was also sensitive to CuPhe inhibitions (Fig. 5 C). These results suggest that the residue C212 is probably not the target of the redox reaction responsible for the functional consequence described above.
FIGURE 5.

Redox control of the C212S-related mutants. (A) Effects of CuPhe on the C212-C212S heterodimer. Recordings at left were made before, and recordings at right after, treatment with CuPhe (10 μM) for 1 h. Dotted lines represent zero-current level. The number of trace crossovers is 4 on the left and 0 on the right. Five other oocytes showed similar effects. (B) Redox control of the slow-gate open probability of the K165C heterodimer (in C212S background). Shown at upper left is the current reduction of the heterodimer in ND96 with or without 1.5 μM CuPhe. The oocyte was first incubated in β-ME for at least 1 h. The recording was initially made in the same β-ME-containing solution. The arrow indicates the time when the solution was replaced with pure ND96 solution, or ND96 solution with 1.5 μM CuPhe. The current was measured at +40 mV using protocol 1, and the amplitudes were normalized to the current amplitudes just before the solution exchange. The graph in the upper right corner gives a comparison of the current reduction rate in the absence or presence of 1.5 μM CuPhe. Time constants were derived from a single-exponential fit of the recordings like those shown on the left. The bottom panel shows how inhibition of the K165C heterodimer, both by spontaneous oxidation and by the applied CuPhe, could be reversed by the reducing reagent β-ME. The dotted line represents zero-current level. The oocyte was initially recorded in ND96, and the redox reagents were applied as shown by the horizontal lines. (C) Redox control of K165-K165E heterodimer (in C212S background). Recordings at left were made before, and recordings at right after, 1 h of CuPhe (10 μM) treatment. Three other oocytes showed similar effects. In addition, five oocytes expressing K165-K165H heterodimer (also in C212S background) also showed similar effects of CuPhe.
Current induction of the K165C heterodimer by MTS reagents reports the redox state
The redox control of the ClC-0 gating can be further demonstrated through the modification of K165C mutants by thiol-specific, MTS reagents. In the K165C homodimer (in C212S background), MTSET is able to react with the introduced cysteine, but the ClC-0-like current cannot be induced (Lin and Chen, 2000). However, the functional consequence of MTSET modification in the K165C heterodimer (also in C212S background) is different. Fig. 6 shows that MTSET can induce current in the K165C heterodimer. This effect is due to the modification of the only introduced cysteine residue at position 165 because MTSEA is unable to induce more current after the channel is treated with MTSET (data not shown). The rate of current induced by MTSET in the K165C heterodimer is voltage-sensitive—the apparent modification rate is faster when the monitoring pulse contains a negative voltage step (Fig. 6 A). Furthermore, the apparent MTSET modification rate is also temperature sensitive. With an ∼10°C increase in the bath temperature, the current induction rate is increased by ∼6-fold in a nominal MTSET concentration of 30 μM (Fig. 6 B). The Q10 value of the MTSET induction rate in Fig. 6 B may be severely underestimated because of a very high temperature dependence of the hydrolysis rate of MTS reagents. These observations point to the possibility that although the K165C-containing pore in the heterodimer does not become functional after MTSET modification—that is, the fast gate is not open—the MTSET modification may increase the slow-gate open probability of the heterodimer, thus making more current flow through the other pore hosting the K165 residue.
If the MTSET modification of K165C only affects the slow gating of the heterodimeric channel and does not open the fast gate of the MTSET modified pore, the fast-gate Po-V curve of the MTSET-modified heterodimer must be the same as that of the channel without MTSET modification. Fig. 6 C compares the fast-gate Po-V curves of the heterodimer between the oocytes treated with MTSET and those reduced with β-ME. Both curves have a V1/2 similar to that of the C212S background channel, suggesting that the current of the oocyte after MTSET modification comes from the WT, K165-containing pore. The other pore containing the MTSET-modified K165C is not functional, a conclusion consistent with the modification of MTSET on the K165C homodimer. These results together suggest that the induction of the current in the K165C heterodimer by MTSET is due to the increase of the slow-gate open probability.
The modification of the K165C heterodimer with MTSET and the other MTS reagent, MTSEA, also depends on the redox state of the oocyte. Fig. 7 compares the current induction by MTS reagents in the K165C heterodimer with or without β-ME pretreatment. After the oocyte is incubated in β-ME for more than 30–60 min, the slow gate of the K165C heterodimer is more easily opened by the negative voltage step of protocol 1. Under such conditions, MTSET cannot further induce current (Fig. 7 A, right). By comparison, when the oocyte was not pretreated with reducing reagents, MTSET could induce current from the K165C heterodimer (Fig. 7 A, left; for averaged data, see Fig. 7 C). Modification of the introduced cysteine in the K165C heterodimer by MTSEA makes the K165C-containing pore functional (Lin and Chen, 2000), but the fold increase of the current induced by MTSEA depends on the redox state of the oocyte as well (Fig. 7 B; for averaged data see Fig. 7 D). When the oocyte is preincubated with β-ME, the current after MTSEA treatment is usually ∼2-fold that before the MTSEA modification (Fig. 7 D), reflecting the functional revival of the other pore by MTSEA with little change of the slow-gate open probability (Fig. 7 B, right). In comparison, when oocytes are not pretreated with β-ME, the current after MTSEA modification can be as high as 10-fold (mean ± SE = 5.1 ± 0.6) that before the application of MTSEA (Fig. 7 B, left). These experiments suggest that in the absence of β-ME, oxidation of the oocyte may have rendered the slow gate of the heterodimer more difficult to open. Therefore, MTSET or MTSEA are able to induce more current because the channel has a higher slow-gate open probability after MTS modification.
FIGURE 7.

Effects of oxidation on the current induction of the K165C heterodimer by MTSET and MTSEA. (A and B) Induction of current in the K165C heterodimer by 30 μM MTSET and 3 μM MTSEA in oocytes without (left) or with (right) β-ME pretreatment. Each circle represents the current amplitude measured at the end of the +40-mV voltage step in protocol 1. (Insets) Whole oocyte current recordings before (indicated by b) and after (indicated by a) the application of MTS reagents. Each trace is an average of three traces made during the period indicated by short line segments. (C and D) Ratios of the current amplitudes of the heterodimer before and after the treatment of MTS reagents in oxidized (−β-ME) and reduced (+β-ME) conditions. Note that in the reduced condition (also see right panel in both A and B), the current of the oocyte after MTSEA treatment is ∼2-fold that before MTSEA induction (Ia/Ib ≅ 2). MTSET did not induce current (Ia/Ib ≅ 1) in reduced oocytes.
DISCUSSION
We have shown that oxidation and reduction can control the inactivation gating of ClC-0. In the oxidized condition, the channel appears to be at a “deep” inactivated state, and the channel's slow gate is more difficult to open by negative voltage pulses. Only when the membrane potential is stepped to a very negative potential, for example, −120 to −160 mV, can a small portion of the channel recover from this deep inactivated state. Thus, an oxidized oocyte would not show a nice deactivation relaxation because of two opposite effects: a reduction of the current due to the fast-gate closure and an increase of current due to the slow-gate opening by the very negative potentials. This understanding is significant in studying ClC-0 because expression of ClC-0 in Xenopus oocytes frequently generates hyperpolarization-induced current. One might consider that such atypical recordings perhaps result from the contamination of the endogenous Cl− channels of the oocyte. Our study, however, indicates that when ClC-0 is expressed in Xenopus oocytes, the hyperpolarization-induced current does not necessarily come from the endogenous Cl− channels. In our experience, incubation of the oocytes in reducing agents, such as 2–5 mM β-ME or DTT, for 30 min or longer always turns the atypical traces into typical ClC-0 recordings that show crossovers of traces at negative voltages (Fig. 2).
The oxidation effect on the slow gating of the WT ClC-0 is slow and is rather difficult to study. By simply removing reducing agents in the solution, the current of WT ClC-0 can decrease, but the extent of the current reduction usually varies quite significantly from oocyte to oocyte, depending on the variation between frogs (Table 1). Application of oxidizing reagents (CuPhe or H2O2) increases this oxidizing power. This oxidation effect can be observed in the K165C heterodimer as well as in the WT ClC-0. Through comparison of the MTS modifications between the oxidized and reduced conditions, we found that the extent of current induction in the K165C heterodimer by the MTS reagents can be used to report the redox state. This conclusion is based on the observation that MTS modification of the cysteine at position 165 alters the inactivation gating of the channel. Previous studies have argued that the side chain of residue 165 is important for the inactivation mechanism of ClC-0 because the K165C heterodimer shows prominent inactivation even though this heterodimer is constructed in the background of C212S (Lin and Chen, 2000), a mutation that suppresses the inactivation state of ClC-0 (Lin et al., 1999). The study described here further demonstrates that when the introduced cysteine of the K165C heterodimer is modified by MTSEA or MTSET, the open probability of the inactivation gate is increased (Figs. 6 and 7), as supported by the following observations. First, the MTSET modification rate of the heterodimer is both voltage- and temperature-sensitive, and the voltage and temperature dependencies corroborate with those found in the slow gating of ClC-0 (Fig. 6, A and B). Second, the fast-gate Po-V curves of the heterodimers with or without MTSET modification are the same (Fig. 6 C), a phenomenon different from the situation where the modifying reagent is MTSEA (Lin and Chen, 2000). Finally, in the manipulations that can maximally activate the slow gate—for example, recording β-ME-pretreated oocytes at low temperatures with a pulsing protocol containing a negative voltage step (protocol 1)—MTSET is unable to induce current in the heterodimer, whereas MTSEA induces exactly twofold current (Fig. 7). These results together suggest that modification of the introduced cysteine by MTSET or MTSEA increases the slow-gate open probability of the heterodimer. From the opposite angle, the different extent of current induction by MTSET and MTSEA in the oxidized and reduced conditions provides clear evidence that the inactivation gating of ClC-0 depends on the redox state of the oocyte.
The mechanisms underlying the oxidation effects on ion channels are variable. Previous studies have shown that the M448C mutant of the Shaker K+ channel is not functional when the channel is expressed in Xenopus oocytes incubated in ND96, but the mutant can be functionally recorded in reduced environments (Gross and MacKinnon, 1996). The oxidation effect is attributed to a disulfide-bond formation between the two 448C residues (Liu et al., 1996), an effect consistent with the picture in which the four residues at position 448 are close to each other in the outer vestibule of the channel pore (Doyle et al., 1998). An oxidation effect was also demonstrated in a G-protein-coupled inwardly rectifying K+ (GIRK) channel in which the GIRK current was increased when reducing agents (such as DTT) were applied (Zeidner et al., 2001). In this case, a cysteine residue at the N-terminal cytoplasmic domain is responsible for the redox regulation (Zeidner et al., 2001). For ClC-0, what is the target for the oxidation chemistry responsible for the effect on the inactivation gating?
If the side chain of residue 165 is important in controlling the gating of ClC-0, it might be possible that the oxidation-reduction occurs on the K165C residue, explaining the oxidation effect in the K165C mutant. Stable cysteine-sulfenic acid (Cys-SOH) derivatives have been observed in redox regulation of enzyme catalysis (Claiborne et al., 1999; van Montfort et al., 2003; Wang et al., 2004). It might be plausible that oxidation of K165C results in a side chain with a sulfinic (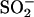 ) or sulfonic (
) or sulfonic (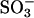 ) group, both carrying a negative charge. However, because the oxidized and reduced K165C mutants have comparable MTSEA induction rates (within twofold difference, data not shown), we think this possibility is less likely because a reaction of the above oxidized sulfonyl groups with MTSEA should be much slower than the MTS modification of a free thio group. Furthermore, the K165E heterodimer suggests that a cysteine at K165 position is not required for the oxidation effect (Fig. 5 C). We therefore suggest that oxidation should occur at sites other than the K165C residue. Cysteine mutation experiments showed that only mutants lacking the cysteine at position 212 were resistant to CuPhe inhibition (Fig. 4). However, the oxidation effect should not be due to a disulfide bond formation between these two cysteines because a C212S-C212 heterodimer is still sensitive to CuPhe inhibition (Fig. 5 A). The residues of the bacterial ClC molecule that correspond to C212 of ClC-0 are buried in the middle of each of the two protein subunits, and they are tens of Å away from each other. Therefore, structural information from bacterial ClCs argues that the disulfide bond formation between the two C212 residues is rather unlikely. We further suggest that the oxidation target may not be the residue C212, because both the K165C and K165E heterodimers are constructed in the C212S background mutation, and the redox effects in these mutants are still present. Therefore, the resistance of the C212S mutant to CuPhe inhibition most likely results from the absence of inactivation state instead of the elimination of the oxidation target. This conclusion is analogous to the explanation for the resistance of the C212S mutant to extracellular zinc inhibition (Lin et al., 1999).
) group, both carrying a negative charge. However, because the oxidized and reduced K165C mutants have comparable MTSEA induction rates (within twofold difference, data not shown), we think this possibility is less likely because a reaction of the above oxidized sulfonyl groups with MTSEA should be much slower than the MTS modification of a free thio group. Furthermore, the K165E heterodimer suggests that a cysteine at K165 position is not required for the oxidation effect (Fig. 5 C). We therefore suggest that oxidation should occur at sites other than the K165C residue. Cysteine mutation experiments showed that only mutants lacking the cysteine at position 212 were resistant to CuPhe inhibition (Fig. 4). However, the oxidation effect should not be due to a disulfide bond formation between these two cysteines because a C212S-C212 heterodimer is still sensitive to CuPhe inhibition (Fig. 5 A). The residues of the bacterial ClC molecule that correspond to C212 of ClC-0 are buried in the middle of each of the two protein subunits, and they are tens of Å away from each other. Therefore, structural information from bacterial ClCs argues that the disulfide bond formation between the two C212 residues is rather unlikely. We further suggest that the oxidation target may not be the residue C212, because both the K165C and K165E heterodimers are constructed in the C212S background mutation, and the redox effects in these mutants are still present. Therefore, the resistance of the C212S mutant to CuPhe inhibition most likely results from the absence of inactivation state instead of the elimination of the oxidation target. This conclusion is analogous to the explanation for the resistance of the C212S mutant to extracellular zinc inhibition (Lin et al., 1999).
Although we have documented a redox control on the inactivation gating of ClC-0, our understanding of this effect is incomplete in several respects. First, we have so far not identified the target for the oxidation-reduction reaction. Thus, we do not know if the effect is due to an oxidation of the channel protein or perhaps due to the oxidation of endogenous protein molecules, or even the lipids of the oocyte membrane. Second, we do not understand how the inactivation gating of ClC-0 is controlled by the redox state of the oocyte except for the finding that the inactivation-suppressed mutant is resistant to oxidation inhibition. Finally, although we have noted that the oxidized ClC-0 current is more frequently encountered in the oocytes from older-aged frogs, we do not know precisely how the oocyte quality is related to the variation of the current. However, even with these unanswered questions, our study illustrates the redox control on the ClC-0 inactivation, and shows that the atypical ClC-0 current in Xenopus oocytes can be “cured” by reducing reagents. This discovery should be helpful in functional studies of ClC-0, at least in the experiments using Xenopus oocytes as the expression system.
Acknowledgments
We thank Emilie Chang for technical assistance in some of the experiments. We also thank Robert Fairclough and Ebenezer Yamoah for critical reading and comments on the manuscript.
This work was supported by a National Institutes of Health research grant (GM65447).
Chia-Wei Lin's present address is Center for Research on Occupational and Environmental Toxicology, Oregon Health and Science University, Portland, OR 97201.
References
- Accardi, A., L. Kolmakova-Partensky, C. Williams, and C. Miller. 2004. Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J. Gen. Physiol. 123:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- Bauer, C. K., K. Steinmeyer, J. R. Schwarz, and T. J. Jentsch. 1991. Completely functional double-barreled chloride channel expressed from a single Torpedo cDNA. Proc. Natl. Acad. Sci. USA. 88:11052–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. F., and T. Y. Chen. 2001. Different fast-gate regulation by external Cl(−) and H(+) of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. F., and T. Y. Chen. 2003. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 122:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. Y. 1998. Extracellular zinc ion inhibits ClC-0 chloride channels by facilitating slow gating. J. Gen. Physiol. 112:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne, A., J. I. Yeh, T. C. Mallett, J. Luba, E. J. Crane 3rd, V. Charrier, and D. Parsonage. 1999. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 38:15407–15416. [DOI] [PubMed] [Google Scholar]
- Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., T. H. Rhodes, R. R. Desai, and A. L. George, Jr. 1998. Pore stoichiometry of a voltage-gated chloride channel. Nature. 394:687–690. [DOI] [PubMed] [Google Scholar]
- Feng, W., G. Liu, P. D. Allen, and I. N. Pessah. 2000. Transmembrane redox sensor of ryanodine receptor complex. J. Biol. Chem. 275:35902–35907. [DOI] [PubMed] [Google Scholar]
- Gross, A., and R. MacKinnon. 1996. Agitoxin footprinting the shaker potassium channel pore. Neuron. 16:399–406. [DOI] [PubMed] [Google Scholar]
- Hanke, W., and C. Miller. 1983. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82:25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R., T. M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- Jentsch, T. J., T. Friedrich, A. Schriever, and H. Yamada. 1999. The CLC chloride channel family. Pflugers Arch. 437:783–795. [DOI] [PubMed] [Google Scholar]
- Jentsch, T. J., V. Stein, F. Weinreich, and A. A. Zdebik. 2002. Molecular structure and physiological function of chloride channels. Physiol. Rev. 82:503–568. [DOI] [PubMed] [Google Scholar]
- Lin, C. W., and T. Y. Chen. 2000. Cysteine modification of a putative pore residue in ClC-0: implication for the pore stoichiometry of ClC chloride channels. J. Gen. Physiol. 116:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. W., and T. Y. Chen. 2003. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J. Gen. Physiol. 122:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. W., C. W. Lin, and T. Y. Chen. 1999. Elimination of the slow gating of ClC-0 chloride channel by a point mutation. J. Gen. Physiol. 114:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., M. E. Jurman, and G. Yellen. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 16:859–867. [DOI] [PubMed] [Google Scholar]
- Ludewig, U., M. Pusch, and T. J. Jentsch. 1996. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 383:340–343. [DOI] [PubMed] [Google Scholar]
- Maduke, M., C. Miller, and J. A. Mindell. 2000. A decade of CLC chloride channels: structure, mechanism, and many unsettled questions. Annu. Rev. Biophys. Biomol. Struct. 29:411–438. [DOI] [PubMed] [Google Scholar]
- Maduke, M., C. Williams, and C. Miller. 1998. Formation of CLC-0 chloride channels from separated transmembrane and cytoplasmic domains. Biochemistry. 37:1315–1321. [DOI] [PubMed] [Google Scholar]
- Middleton, R. E., D. J. Pheasant, and C. Miller. 1996. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 383:337–340. [DOI] [PubMed] [Google Scholar]
- Miller, C. 1982. Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci. 299:401–411. [DOI] [PubMed] [Google Scholar]
- Miller, C., and M. M. White. 1984. Dimeric structure of single chloride channels from Torpedo electroplax. Proc. Natl. Acad. Sci. USA. 81:2772–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah, I. N., and W. Feng. 2000. Functional role of hyperreactive sulfhydryl moieties within the ryanodine receptor complex. Antioxid. Redox Signal. 2:17–25. [DOI] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, and T. J. Jentsch. 1997. Temperature dependence of fast and slow gating relaxations of ClC-0 chloride channels. J. Gen. Physiol. 109:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Saviane, C., F. Conti, and M. Pusch. 1999. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 113:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. D., H. Daggett, M. Hanner, M. L. Garcia, O. B. McManus, N. Brot, H. Weissbach, S. H. Heinemann, and T. Hoshi. 2001. Oxidative regulation of large conductance calcium-activated potassium channels. J. Gen. Physiol. 117:253–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfort, R. L., M. Congreve, D. Tisi, R. Carr, and H. Jhoti. 2003. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 423:773–777. [DOI] [PubMed] [Google Scholar]
- Wang, Q., D. Dube, R. W. Friesen, T. G. LeRiche, K. P. Bateman, L. Trimble, J. Sanghara, R. Pollex, C. Ramachandran, M. J. Gresser, and Z. Huang. 2004. Catalytic inactivation of protein tyrosine phosphatase CD45 and protein tyrosine phosphatase 1B by polyaromatic quinones. Biochemistry. 43:4294–4303. [DOI] [PubMed] [Google Scholar]
- Xia, R., T. Stangler, and J. J. Abramson. 2000. Skeletal muscle ryanodine receptor is a redox sensor with a well defined redox potential that is sensitive to channel modulators. J. Biol. Chem. 275:36556–36561. [DOI] [PubMed] [Google Scholar]
- Zeidner, G., R. Sadja, and E. Reuveny. 2001. Redox-dependent gating of G protein-coupled inwardly rectifying K+ channels. J. Biol. Chem. 276:35564–35570. [DOI] [PubMed] [Google Scholar]