

Abstract
Photosynthetic bacterial light-harvesting antenna complex LH2 was immobilized on the surface of TiO2 nanoparticles in the colloidal solution. The LH2/TiO2 assembly was investigated by the time-resolved spectroscopic methods. The excited-state lifetimes for carotenoid-containing and carotenoidless LH2 have been measured, showing a decrease in the excited-state lifetime of B850 when LH2 was immobilized on TiO2. The possibility that the decrease of the LH2 excited-state lifetime being caused by an interfacial electron transfer reaction between B850 and the TiO2 nanoparticle was precluded experimentally. We proposed that the observed change in the photophysical properties of LH2 when assembled onto TiO2 nanoparticles is arising from the interfacial-interaction-induced structural deformation of the LH2 complex deviating from an ellipse of less eccentric to a more eccentric ellipse, and the observed phenomenon can be accounted by an elliptical exciton model. Experiment by using photoinactive SiO2 nanoparticle in place of TiO2 and core complex LH1 instead of LH2 provide further evidence to the proposed mechanism.
INTRODUCTION
Immobilization of biological macromolecules on the surface of nanoparticle (Roddick-Lanzilotta and Mcquillan, 2000), carbon nanotube (Wong et al., 2004), nanoparticle film electrode (Topoglidis et al., 2000) attempting to develop biomolecular device, biocompatible implanted materials, biosensors, etc., have attracted intense current research interest (Das et al., 2004; Lee et al., 1995). For most of the protein molecules, their scaffolds exhibit more or less degree of flexibility, especially in a state of single-molecule (van Oijen et al., 1999; Bopp et al., 1999) or solution phase (Hong et al., 2004a) as have been observed for the light-harvesting complex LH2 of photosynthetic bacteria. It is expected that immobilization of the protein might cause the structural deformation of the protein, hence, their biophysical and chemical properties. Therefore, investigation of the structural deformation induced by immobilization of the proteins would have a significant importance in their potential application, as well as in the understanding of the biological function of the protein structure. It is noted in a very recent publication that nanoparticle-curvature-dependent protein conformational changes have been observed when human carbonic anhydrase I is adsorbed onto silica nanoparticle of varying size, i.e., 6 nm, 9 nm, and 15 nm in diameter. The authors found that the protein undergoes a larger conformational change when assembled onto a nanoparticle having a larger diameter (Lundqvist et al., 2004).
Photosynthetic membrane proteins play a role of biodevice for solar energy harvesting and energy conversion. Several groups have succeeded in the immobilization of the photosynthetic bacteria light-harvesting LH2 complex on mica and glass surface to investigate the single molecular fluorescence of LH2 (Bopp et al., 1997, 1999; Tietz et al., 1999; Rutkauskas et al., 2004). The crystal structure of the LH2 complex of the purple bacterium Rhodopseudomonas acidophila shows that the LH2 molecule exhibits a high symmetry with a ninefold rotational axis defined by nine αβ-dipeptides (McDermott et al., 1995; Papiz et al., 2003). The basic building block is the protein αβ-heteromer that binds three BChl a molecules and two carotenoid (Car) molecules (Papiz et al., 2003). The total 27 BChl a molecules form two concentric rings supported by the αβ-dipeptides. One ring with nine BChl a molecules of a larger interpigment separation is known as B800, another of 18 BChl a molecules with shorter interpigment separations known as B850. The striking feature of the LH2 complex is its ringlike topological structure with well-arranged two BChl a concentric rings. Another resolved LH2 crystal structure is that of Rhodospirillum molischianum, the structure of which is very closely related to that of Rps. acidophila. A remarkable difference is that this LH2 is an αβ-octamer with 16 B850 BChls and 8 B800s (Koepke et al., 1996). Since the report of the crystal structures, a large amount of work has been devoted to elucidating the function of the pigments in the energy harvesting and transferring in association with the protein structure (for reviews, see Pullerits and Sundström, 1996; Sundström et al., 1999; Hu and Schulten, 1997, van Grondelle and Novoderezhkin, 2001; Law et al., 2004). It has been shown that the ring structure of the protein complex has its unique properties in solar energy storage and transfer. One important feature is the delocalization of the excitation of the pigment molecules on the ring known as the exciton state upon the photoexcitation. Such a ringlike pigment aggregate is believed to facilitate the undirectional energy transfer (Nagarajan et al., 1996; Hu and Schulten, 1997). It is generally accepted that the excited states of B800 molecules have a monomeric character, whereas in B850 the states are delocalized over a considerable number of BChls (Trinkunas et al., 2001; Chachisvilis et al., 1997; Hofmann et al., 2003), ranging from a few or several molecules (Monshouwer et al., 1997; Novoderezhkin et al., 1999; 2003; Koolhaas et al., 1997) to the whole ring (Leupold et al., 1996, Nagarajan et al., 1996). It is generally agreed upon that these excitonic states are not completely delocalized. After the initial interlevel excitonic relaxation (Mostovoy and Knoester, 2000), the excitonic states would be trapped at a delocalization length of 4–6 molecules (for reviews, see Sundström et al., 1999; van Grondelle and Novoderezhkin, 2001; Hu et al., 2002). Another feature is the high geometrical symmetry imposed on the molecular orbital of the aggregate. Differing from the linear aggregate (J-aggregate) (Monshouwer et al., 1997), in the frame of the excitonic theory without consideration of the site energy disorder and the protein structure deformation, the lowest excited state is optically forbidden, which can preserve the excitation once relaxed to this state. Therefore, the B850 ring acts as an energy storage, as well as an efficient energy donor with an overall quantum efficiency as high as ∼95% (Hu and Schulten, 1997). However, when the flexibility of the protein and the slight different environment known as inhomogenity (site energy disorder) are considered, the exciton state cannot be extended over the whole ring and the lowest excitonic state becomes partially optical-transition allowed. Furthermore, it has been proposed that the LH2 ring can deviate from the ideally circular structure existing in the crystal phase based on the room- and low-temperature single-molecular spectroscopic studies. The single molecule spectroscopic experimental results suggest that the structure of LH2 complex would deform to an elliptical form with an eccentricity of 0.52 (van Oijen et al., 1999). The maximum deviation of LH2 from Rhodobacter sphaeroides with an eccentricity of 0.59 was observed in the solution at room temperature as revealed by the small angle x-ray diffraction experiment (Hong et al., 2004a). Both experimental and theoretical investigations show that the deformation of the protein structure would cause change in the excitonic state of B850 ring (van Oijen et al., 1999; Mostovoy and Knoester, 2000; Matsushita et al., 2001).
Meanwhile the cavity structure of LH2 stimulated the other research activity such as constructing LH2-semiconductor nanoparticle assembly to build an energy or electron donor-acceptor system (see Fig. 1). Recently we have tried assembling LH2 protein complex of Rb. sphaeroides 2.4.1, which contains pigments of both BChl a and carotenoid onto TiO2 nanoparticle in solution (Weng et al., 2003), attempting to investigate the photoinduced interfacial electron transfer between BChl a and the TiO2 nanoparticle through the protein frame. Other groups have demonstrated that the integration of electrically active photosynthetic protein complexes, i.e., reaction center from Rb. sphaeroides and photosystem I from spinach chloroplast in solid-state devices, realizing photodetectors and photovoltaic cells with internal quantum efficiencies of ∼12% (Das et al., 2004). In this work we report a further investigation of different antenna membrane protein complexes differing in cavity size (LH2 and LH1), of LH2 complexes differing in pigments, i.e., with carotenoid and without carotenoid (Gall et al., 2003; Davidson and Cogdell, 1981), when assembled onto nanoparticles of photoactive (TiO2) and photoinactive (SiO2) by means of ultrafast time-resolved absorbance-difference spectroscopy. The purpose of this study is to elucidate the nature of the change of photophysical properties of LH2 complex when immobilized onto the nanoparticles. Our results show that such a change was caused by the nanoparticle-induced protein structural deformation rather than the interfacial electron transfer between the pigments and the nanoparticles.
FIGURE 1.

Schematic diagram for the assembling between LH2 complex and the TiO2 nanoparticle.
MATERIALS AND METHODS
Sample preparation
Chemicals
The detergent sucrose monocholate was purchased from Dojindo Laboratories (Kumamoto, Japan). Luryldimethylaminoammonium oxide (LDAO) was purchased from Sigma (St. Louis, MO) and SiO2 (14 nm) colloidal solution was commercially available from Alfa (Ward Hill, MA).
Preparation of LH2 complexes
Carotenoid-containing LH2 complexes were extracted from strain Rb. sphaeroides 2.4.1, whereas carotenoidless LH2 complex was from the strain of R26.1 mutant of Rb. sphaeroides. The culture of the cells followed the reported protocols (Böse, 1963). The irradiation condition of R26.1 mutant was different from that of the others containing carotenoid; it employed glass or plastic filters to cut off the irradiation with wavelength shorter than 500 nm to prevent the mutation of R26.1 mutant back into its wild type. The carotenoid-containing LH2 complexes were obtained by solubilization of the corresponding chromatophores with 1% LDAO and by subsequent purification using DE52 ion-exchange column chromatography and stepwise elution with sodium chloride (Ohashi et al., 1996). Carotenoidless LH2 complex from R26.1 was purified by the method of sucrose gradient centrifugation, a detail procedure for purification can be found elsewhere (Limantara et al., 1998).
Preparation of LH1 complex from Rhodospirillum rubrum S1
LH1 chromatophore suspension (OD880 = 40 cm−1) was preincubated in 0.1% LDAO at 4°C for 40 min by adding 30% LDAO solution, then the suspension was further incubated in 0.3% LDAO solution at 4°C for another 70 min by adding 30% LDAO solution. Then the suspension was diluted with 20 mM Tris-HCl 10 mM sodium ascorbate buffer (pH = 7.8) to twofold, and the solution was centrifuged at 8000× g, 4°C for 15 min. The supernatant was ultracentrifuged at 105,000× g, 4°C for 1.5 h. The pellet was collected and suspended in the Tris-HCl sodium ascorbate buffer by adjusting OD880 = 40 cm−1, repeating the above extraction procedure. The supernatant of LH1 crude extract solution was dialyzed against 20 mM Tris-HCl 10 mM sodium ascorbate buffer for 4 days, while the dialysis medium was refreshed five times during this period. The dialyzed solution was centrifuged at 25,000× g at 4°C for 30 min. The dialysis pellet was collected and solubilized in 2.3% βOG (N-octyl-β-d-glucopyranoside) at 4°C for 30 min (OD880 = 28.5 cm−1) then the solution was diluted to twofold with 20 mM Tris-HCl 10 mM sodium ascorbate buffer and centrifuged at 8000× g and 40°C for 30 min. The supernatant was loaded to a 16 × 35 mm DE52 column then eluted stepwise with 0.8% βOG in 50 mM NaCl and 0.8% βOG in 250 mM NaCl, respectively.
Preparation of TiO2 nanoparticle solution
TiO2 colloidal solution was prepared by hydrolysis of Ti(iso-propoxide)4 under acidic condition (Wang et al., 2000). The size of TiO2 nanoparticle was determined by x-ray diffraction (XRD) measurement (Zhang et al., 2004) and small angle x-ray scattering (SAXS) method (Hong et al., 2004b), both of the measurements give a consistent result with an averaged diameter ∼6 nm. The pH of the colloidal solution was adjusted to 7 by addition of KOH solution before use. LH2 was mixed with TiO2 nanoparticle solution and kept stirring in the dark at 4°C under Ar gas protection for at least 48 h.
Measurement
The femtosecond time-resolved difference absorbance spectrometer has been described elsewhere (Zhang et al., 2003). Briefly it employs a 1-kHz repetition regenerative amplifier Ti:sapphire (Hurricane, Spectra Physics, Mountain View, CA) as the primary beam source. The output power of the laser is 750 mJ, with a pulse duration of 150 fs at 800 nm. The probe white light continuum was generated by focusing 800-nm beam with a power of 6 μJ/pulse onto a 2.4-mm thick sapphire plate. The 400-nm pump beam was obtained by frequency doubling of the fundamental wavelength. A dual beam configuration (probe and reference) was adopted for the detection. The probe and reference beams were collected by a pair of objective lenses coupled to a two-branched optical fiber. The spectrum was recorded by a charge-coupled device spectrometer (Acton, MA). The delay between pump and probe beam was realized by a computer-controlled translation stage. The polarization of 400-nm pump beam was set to the magic angle (54.7°) in respect to the probe beam. Picosecond time-resolved fluorescence spectra were acquired on a streak camera (C2909, Hamamatsu, Hamamatsu, Japan) combined with a polychromator; the excitation pulses were provided by a regenerative amplifier (Spitfire, Spectra Physics), whose output wavelength was converted to 400 nm with the second-harmonic generation (200 nJ/pulse, 120 fs, 1 kHz). During the measurement the samples were protected by Ar gas.
RESULTS AND DISCUSSION
TiO2 is a semiconductor with a band-gap energy of 3.2 eV. It undergoes photoinduced interfacial electron transfer reaction either by direct band-gap excitation with ultraviolet (UV)-light irradiation or dye-sensitized electron injection with visible light irradiation when coated by proper pigment molecules. By this virtue, TiO2 nanoparticle has proven to be a promising material in photocatalysis, solar energy conversion, and molecular device. Generally when the pigment molecule is assembled to TiO2 nanoparticle, an anchoring group such as carboxylate group is necessary to build a chemical link between the dye molecules and the TiO2 surface atoms (Frei et al., 1990). As to protein, it is found that the binding of protein on the TiO2 surface is dependent on the pH value of solution and the salt concentration. Specifically, binding was optimum for low salt concentrations and for a proper pH of the solution at which the protein electrostatic charges were counterpoised. The isoelectric point of TiO2 colloid is around pH = 5 (Dobson et al., 1997). For many chemical species either organic carboxylic acid or inorganic complexes having carboxylic groups on their ligand, the adsorbates would come off from the TiO2 surface when pH of the solution approaches 7. However, for protein the situation is different. It has been reported that proteins can be immobilized on the TiO2 colloidal surface at physiological condition (pH = 7–8) (Topoglidis et al., 2000). The resistance to the desorption can be partially attributed to the binding of the protein surface side carboxylic group to the titanium surface atoms of the nanoparticles. The recent x-ray crystal structure of LH2 from Rps. acidophila of better resolution (2 Å) shows that nine segments of C-terminus residues point outward on the periplasmic side (Papiz et al., 2003), which provides a good chance for the LH2 complex forming the chemical binding between the terminal carboxylic groups and surface titanium atoms on the nanoparticles, though the cavity was filled with detergent molecules or unremoved lipid molecules as shown by the neutron scattering experiment (Prince et al., 2003). Therefore, the strategy of assembling of LH2 onto the TiO2 nanoparticle by means of the C-terminal carboxylic anchoring group can be successful as shown schematically in Fig. 1. Fig. 2 displays a typical UV-visible absorption spectra of carotenoid-containing LH2 from Rb. sphaeroides 2.4.1 and the corresponding LH2/TiO2 nanoparticle solution. The concentration of LH2 was adjusted by making the optical density of LH2 at 850 nm equal 3 at 1.0-cm optical path by diluting with Tris-HCl buffer solution containing 0.1% LADO, and the concentration of TiO2 was ∼2.0 g/L. The absorption spectrum for LH2/TiO2 was corrected for the absorption of the TiO2 colloidal solution. A control LH2 sample of the same optical density was prepared and stirred under the same condition. As shown in Fig. 2 after correction for TiO2 absorption, the absorption spectrum of LH2/TiO2 is similar to that of free LH2 in the pigment absorption spectral region, which indicates that the LH2 protein complex remains undegraded when mixed with TiO2 colloidal solution. However, the B850 absorption spectra are not identical; the absorption spectrum of LH2/TiO2 is blue-shifted by 1.5 nm with respect to that of LH2, and of a smaller full-width at half-measure, 366 cm−1, compared to that of LH2 (378 cm−1) as shown in the graphic inset.
FIGURE 2.

Normalized UV-visible absorption spectra of carotenoid-containing LH2 from Rb. sphaeroides 2.4.1 (solid line) and LH2/TiO2 (2.0 g/L) colloidal solution (dotted line, after correction of the TiO2 absorption). Graphic inset shows expanded view of B850 absorption band.
Fig. 3 A presents the 852-nm probed fluorescence decay kinetics of carotenoid-containing LH2 from Rb. sphaeroides 2.4.1 and the corresponding LH2/TiO2 (2.0 g/L) colloidal solution excited at 400 nm, respectively. The fluorescence decay kinetics can be fitted by a monoexponential decay with a time constant of 1.0 ns for LH2 and 0.76 ns for LH2/TiO2, respectively. The observed decay time constant is consistent with the reported value for the lifetime of the lowest excitonic state of LH2 (Kennis et al., 1997; Freiberg et al., 1998; Monshouwer et al., 1997). Obviously the lifetime of the LH2 excited state was reduced when assembled onto TiO2 nanoparticle. Fig. 3 B shows the corresponding time-resolved fluorescence spectra acquired at a time delay of 0.8 ns. It reveals that when assembled onto the 6-nm TiO2 nanoparticle, the fluorescence spectra of LH2 exhibits a red shift of 8 nm (109 cm−1) from 854 to 862 nm.
FIGURE 3.
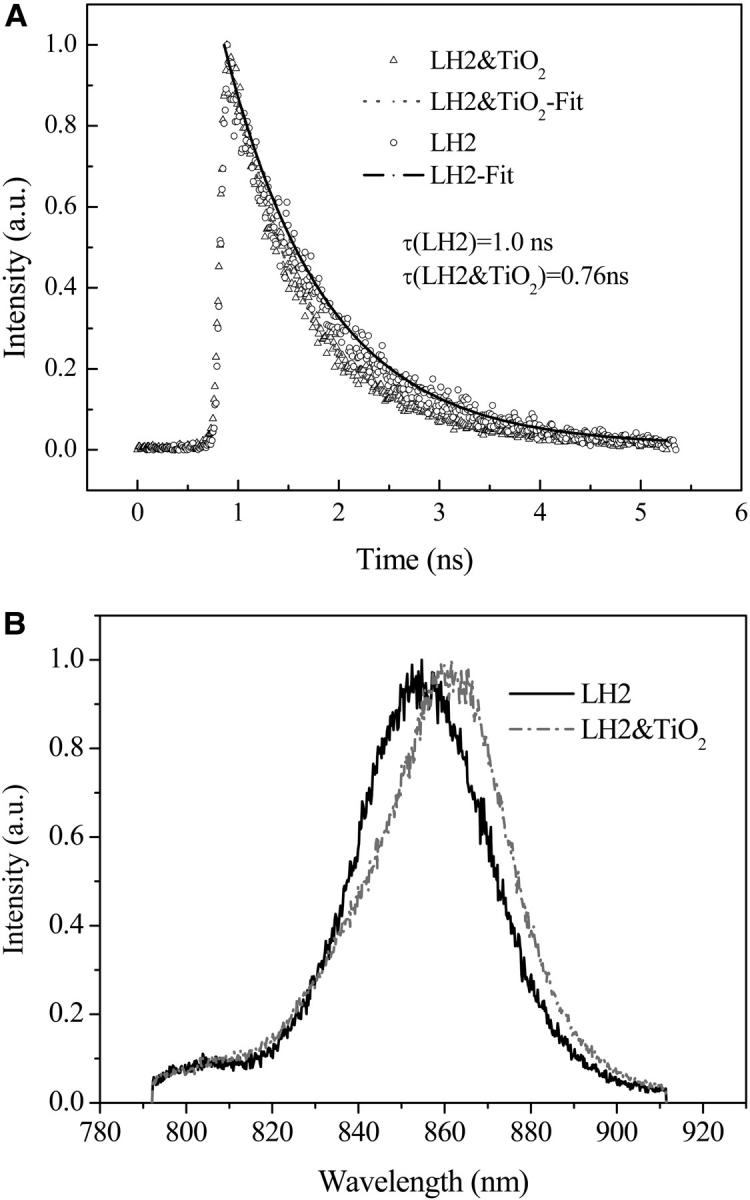
(A) Fluorescence decay kinetics of carotenoid-containing LH2 from Rb. sphaeroides 2.4.1 and the corresponding LH2/TiO2 (2.0 g/L) colloidal solution probed at 852 nm, fitted by a monoexponential decay with a time constant of 1.0 and 0.76 ns, respectively; (B) corresponding time-resolved fluorescence spectra of LH2 and LH2/TiO2.
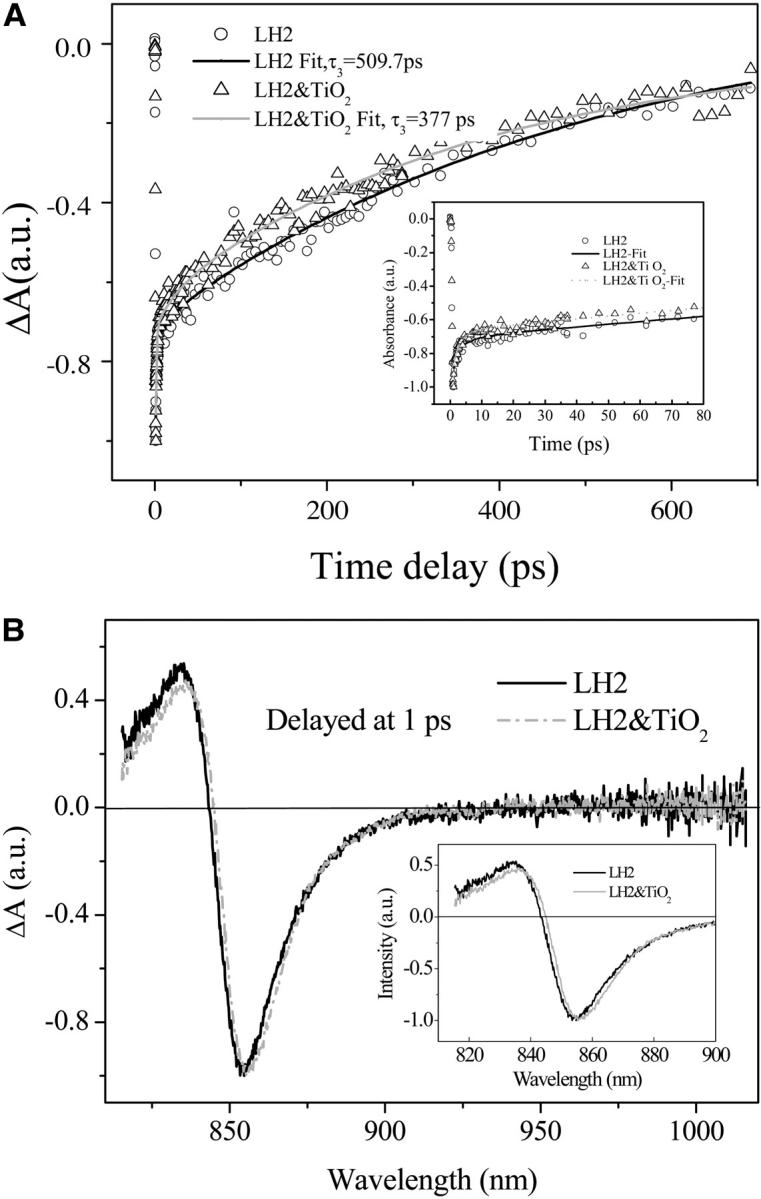
Fig. 4 A presents the B850 bleaching recovery kinetic curves of carotenoid-containing LH2 and the corresponding LH2/TiO2 (0.1 g/L) colloidal solution probed at 852 nm acquired by femtosecond time-resolved absorbance-difference spectrometer. The bleaching recovery kinetic curves can be fitted by a three-component exponential decay with time constants of τ1 = 0.79 ps (48.2%), τ2 = 17.1 ps (3.4%), and τ3 = 509.7 ps (48.2%) for LH2 and τ1 = 1.1 ps (46.0%), τ2 = 23.9 (5.5%), and τ3 = 377.0 (48.4%) for LH2/TiO2 colloidal solution. The fact indicates that the transient species or the excited state of LH2 in LH2/TiO2 assembly returns to the ground state with a faster rate than that of free LH2. At 400-nm excitation, only the pigment molecules such as BChl a and Car can be excited, whereas the direct band-gap excitation is negligible because the absorption of TiO2 nanoparticle starts at ∼380 nm. The reduction in the bleaching recovery time constant of B850 for LH2/TiO2 colloidal solution in respect to that of the free LH2 suggests that LH2 complex has been immobilized onto TiO2 nanoparticle, which in turn mediates the B850 excited-state lifetime. An obvious discrepancy between the excited-state lifetime constants of the pump-probe bleaching recovery kinetics and the fluorescence decay kinetics should be noted. The former is a multiexponential decay process whereas the latter can be described by a single exponential decay. It has been recognized that the lifetime of LH2 is excitation-pump power dependent when the excitation power exceeds the threshold for onset of nonlinear singlet-singlet (S-S) and singlet-triplet (S-T) annihilations that lead to a decrease in the measured excited-state lifetime. Therefore, special precaution should be taken in measuring the LH2 lifetime. For excitation at 800 nm, it is reported that an appropriate excitation power is ∼1 × 1013 photons cm−2pulse−1 (Pullerits et al., 1994), and even a lower excitation intensity of 5 × 1011–5 × 1012 photons cm−2pulse−1 has also been reported (Bergström et al., 1986). In our pump-probe experiment, the excitation wavelength was 400 nm, the excitation power exceeded 0.1 μJ/pulse with a focal size ∼200 μm, which corresponds to an excitation intensity large than 5 × 1012 photons cm−2pulse−1. Based on our result, the annihilation-free excitation power at 400 nm for pump-probe measurement should be lower than 5 × 1012 photons cm−2pulse−1. Surveying the literature, the reported annihilation-free fluorescence lifetime (Sebban et al., 1984, Monshouwer et al., 1997) and pump-probe lifetime (Bergström et al., 1988; Trinkunas et al., 2001) of B850 in LH2 are quite consistent, which has a value of ∼1.0 ns. When higher excitation pump power is needed in the upconversion experiment for the requirement of the signal/noise ratio, the observed fluorescence lifetime can be fitted by a three-component exponential decay kinetics with the slowest component ∼600 ps (Jimenez et al., 1996). In this case, several reasons can cause the measured lifetime being relatively shorter. The main reason is the higher pump power used for obtaining a reasonable signal/noise ratio when using CCD spectrometer, which would lead to the S-S and S-T annihilation. Another reason could be the photodamage of the sample owing to the long data-acquisition time. However, only the lifetimes of LH2 and LH2/TiO2 measured at the same excitation conditions are compared, which give a consistent tendency that the lifetime of B850 becomes shorter after being adsorbed onto TiO2 nanoparticle. The time-resolved absorbance difference spectra of LH2 and LH2/TiO2 colloidal solution are displayed in Fig. 4 B showing a slight spectral difference as illustrated in the graphic inset. The splitting between the positive (induced) absorption and the negative (bleaching) peaks are determined as 285 cm−1 for LH2 and 274 cm−1 for LH2/TiO2; the former is close to the reported value of 283 cm−1 at room temperature (Chachisvilis et al., 1997). This splitting value is believed to be related to the exciton mean free path by the relation of 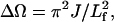 where ΔΩ is the splitting, J is the nearest-neighbor exciton intermolecular interaction, and Lf is the excitation mean free path (Meier et al., 1997). At the first glance, one may conclude that the smaller value of ΔΩ would lead to a larger delocalization size for LH2-TiO2. However, this explanation does not take into account the whole ensemble, where the smaller value of ΔΩ can also be derived from the narrower distribution of the LH2 conformers induced by interaction with TiO2 nanoparticle because the width of the B850 ground-state absorption for LH2/TiO2 becomes narrower. Thus, the correlation between ΔΩ and the extent of the excitonic delocalization may not be straightforward when the ensemble is involved.
where ΔΩ is the splitting, J is the nearest-neighbor exciton intermolecular interaction, and Lf is the excitation mean free path (Meier et al., 1997). At the first glance, one may conclude that the smaller value of ΔΩ would lead to a larger delocalization size for LH2-TiO2. However, this explanation does not take into account the whole ensemble, where the smaller value of ΔΩ can also be derived from the narrower distribution of the LH2 conformers induced by interaction with TiO2 nanoparticle because the width of the B850 ground-state absorption for LH2/TiO2 becomes narrower. Thus, the correlation between ΔΩ and the extent of the excitonic delocalization may not be straightforward when the ensemble is involved.
FIGURE 4.
(A) Bleaching recovery kinetic curves of carotenoid-containing LH2 (Rb. sphaeroides 2.4.1) and the corresponding LH2/TiO2 (0.1 g/L) colloidal solution excited at 400 nm, 0.3 μJ/pulse, and probed at 852 nm; graphic inset shows early time kinetics; (B) time-resolved absorbance difference absorption spectra of the corresponding LH2 and LH2/TiO2 colloidal solution; graphic inset shows the expanded view of the spectra.
In view of electron transfer, a very plausible mechanism for the observed reduction in the lifetime constant of bleaching recovery and fluorescence for LH2/TiO2 colloidal solution can be the photoinduced interfacial electron transfer. Because the reduction potential of B850+/B850 is estimated to be ∼0.7 V (Kropacheva and Hoff, 2001), and that of B850+/B850* is −0.76 V, whereas the conduction band edge of TiO2 is ∼−0.44 V against the standard hydrogen electrode (Mose et al., 1993), thus electron transfer from B850* to the conduction band of TiO2 is energetically favorable with a free-energy difference ΔG ≈ −0.32 V. It has been well demonstrated that natural porphyrins can inject one electron into the conduction band of TiO2 nanoparticle when it is adsorbed onto TiO2 (Kay and Grätzel, 1993). In this case, BChl a is separated from the direct contact with the nanoparticle by the polypeptides, however, the possibility of electron injection through protein cannot be ruled out. Once the interfacial electron transfer occurs, the bleaching recovery rate (krec) includes two parts, one is the rate of B850 excited state relaxing to the ground state (1/τ0); and the other is rate of the B850 oxidized radical cation being reduced to the ground-state molecule (kred), i.e., krec = 1/τ0 + kred. The latter process would happen when either an interfacial charge recombination process occurs or the radical cation is being reduced by the other pigment such as the triplet of Car (TCar). Therefore, two anticipated channels can lead to the observed lifetime shortening of the bleaching recovery kinetics: 1), a faster interfacial charge recombination; and 2), for the slow charge recombination, a fast reduction of the B850 cation by the TCar molecule as proposed in a two-step electron transfer mechanism by employing TCar as the secondary electron donor (Weng et al., 2003). It is argued that the Car molecule spans the depth of the membrane making a van der Waals contact with BChl a molecules of B850 and B800 rings. Furthermore, the reduction potential of Car is also similar to that of BChl a (Deng et al., 2000). When excited at 400 nm with high-repetition laser, TCar can be accumulated (Pullerits et al., 1994; Jimenez et al., 1996). Once B850* has transferred an electron to the TiO2 nanoparticle, it becomes an oxidized cation that is expected to be reduced by the TCar molecule at a rate of 1013 s−1(Moser et al., 1992).
Judged from the time-resolved absorption spectra of LH2/TiO2 colloidal solution, the first channel of faster charge recombination seems unlikely because there is no indication of B850 cation formation within its typical absorption spectral region (820–1000 nm; Chauvet et al., 1981), whereas the second channel remains to be examined. The generation of the triplet is related to the triplet lifetime of the Car and the repetition of the laser. In the upconversion measurement, even the repetition rate of the excitation laser was reduced to 80 kHz, S-T annihilation still occurs (Jimenez et al., 1996). To prevent disintegration of the samples and buildup of long-lived photoproducts caused by the high pulse repetition and high average power of the laser irradiation, other authors reduced the pulse repetition rate to 8–40 kHz (Pullerits et al., 1994). Though our repetition rate is 1 kHz, the excitation power is rather higher than the threshold for the onset of the S-S and S-T annihilation; thus, to examine whether the excited-state Car molecule took part in the reduction of the BChl a radical cation, carotenoidless LH2 was used instead of the Car-containing LH2 to perform the same time-resolved absorbance difference spectroscopic study. Fig. 5 A displays the femtosecond time-resolved absorbance difference spectra of carotenoidless LH2 from R26.1 mutant (Gall et al., 2003; Davidson and Cogdell, 1981) and that of the corresponding LH2/TiO2 colloidal solution, which shows the similar features with a slight different splitting between the positive absorption and the negative peaks for both the free (ΔΩ = 422 cm−1) and the immobilized LH2 (ΔΩ = 495 cm−1). Fig. 5 B presents the corresponding bleaching recovery kinetics detected at 870 nm for the carotenoidless LH2 and LH2/TiO2 colloidal solution. The parameters for the fitting of the kinetic by three-component exponential decay are τ1 = 3 ps (29%), τ2 = 30 ps (45%), and τ3 = 522 ps (26%) for LH2; and τ1 = 2.1 ps (30%), τ2 = 15 ps (42%), and τ3 = 320 ps (28%) for the corresponding LH2/TiO2 colloidal solution, respectively. Obviously the results also show a reduction in the time constant of the bleaching recovery for LH2/TiO2 colloidal solution. Thus, the observed fact unambiguously excludes the possibility that the observed reduction in lifetime is caused by a two-step electron transfer process involving the excited state of Car. Therefore, interfacial electron transfer process could not be the right cause, and the other mechanism should be considered.
FIGURE 5.
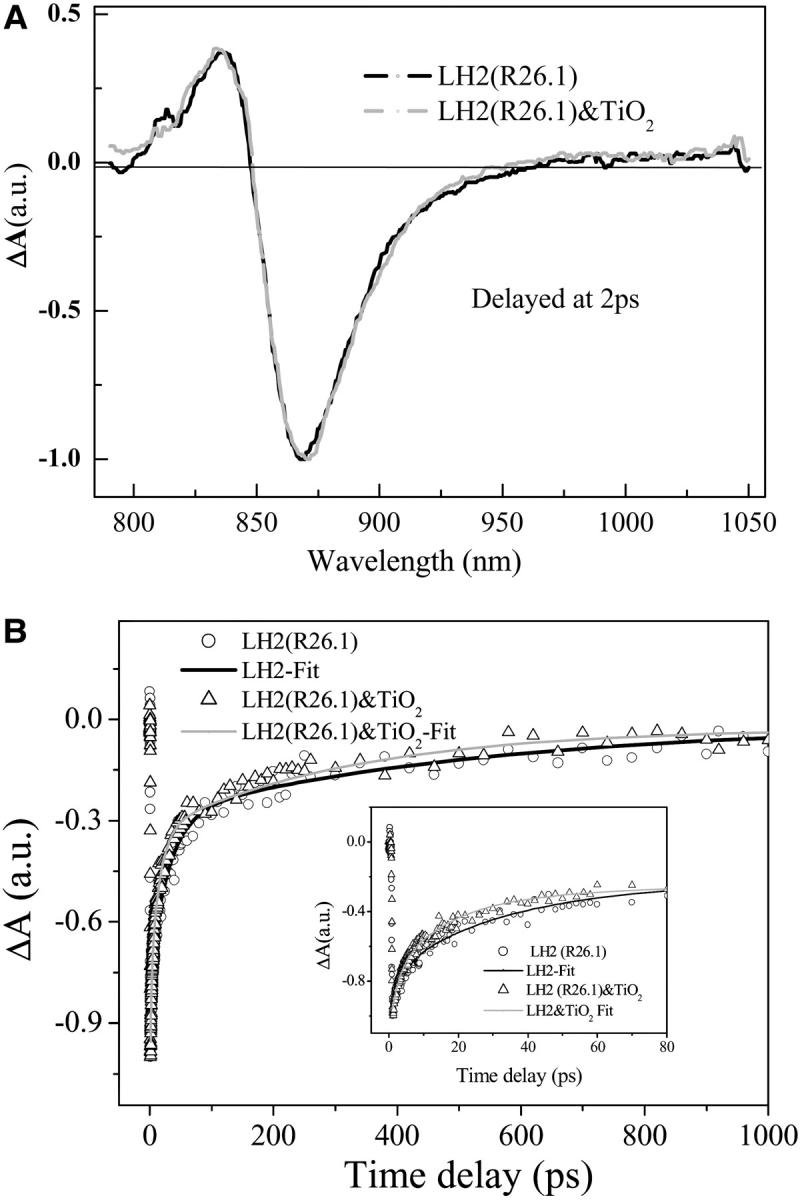
(A) Time-resolved absorbance difference spectra of carotenoidless LH2 from R26.1 mutant and that of the corresponding LH2/TiO2 (0.56 g/L) colloidal solution; (B) the corresponding bleaching recovery kinetics detected at 870 nm; graphic inset shows early time kinetics.
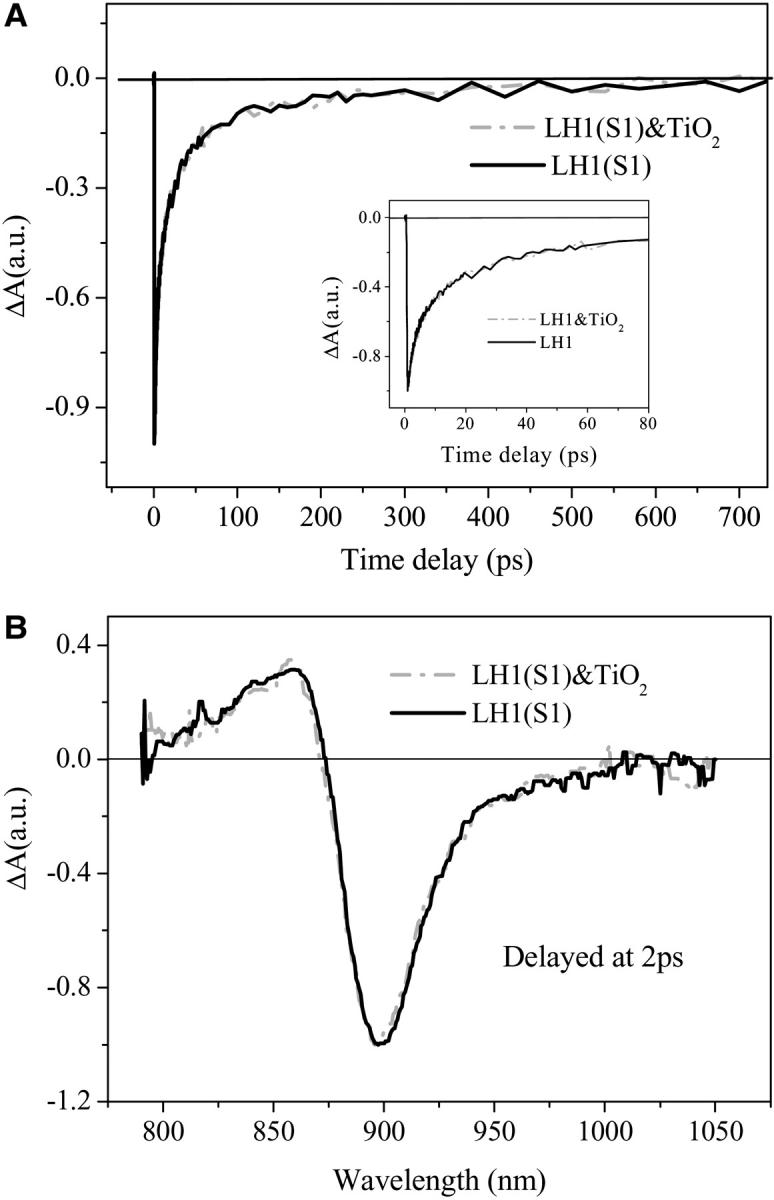
The B850 ring is constructed by nine α,β-polypeptide pairs, each polypeptide binds a BChl a molecule coupled to its two neighboring BChl a molecules. Eighteen BChl a molecules in B850 ring form nine dimers. The B850 nearest-neighbor distances of 8.9 and 9.6 Å lead to a large coupling energy of V ≈ 300 cm−1 (Sauer et al., 1996; Koolhaas et al., 1998), or more specifically single molecular spectroscopic determination gave an accurate value of 254 ± 35 cm−1 and 225 ± 35 cm−1 for the intradimer and interdimer interactions, respectively (Ketelaars et al., 2001). The Qy transitions of the individual molecules lie in the plane of the ring that is normal to the C9 axis, whereas their transition dipoles are tangential to the ring. Due to the geometric constraint, only the lowest excitation component of BChl a dimer carries essential absorption intensity, and the B850 ring can be described by a circular aggregation of nine dipoles of C9 symmetry (Freiberg et al., 1998). In an idealized excitonic model, the optical excitation is considered in n identical two-level molecules coupled to each other arranged in a ringlike structure of Cn symmetry. The intermolecular coupling leads to a splitting of the n degenerate molecular levels into a band of n discrete one-exciton levels. The delocalized one-exciton energies can be calculated as
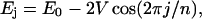 |
where E0 is the localized (monomer) excitation energy, V is the coupling energy between the neighboring molecules, and j takes n integer values as for n odd, j = 0, ± 1, ±2, …… ±(n − 1)/2; and for n even, j = 0, ± 1, ±2, …… ±n/2, n/2. By this model the oscillator strength in the plane of the ring is exclusively concentrated in the j = ±1 states, the two next lowest degenerate transitions, whereas the polarizations of the transition to j = ±1 states are mutually orthogonal. Because the wave function of j = 0 state has no node, the net transition dipole is zero. Thus, the lowest excited state is optically forbidden and does not fluoresce. However, experimental evidences such as superradiance and hole-burning spectra of LH2 at low temperature revealed that the exciton model of identical molecules is oversimplified. It has been shown experimentally that the lowest optical forbidden exciton state carries a total absorption intensity of the B850 band ranging from 2 to 10% (Monshouwer and van Grondelle, 1996; Monshouwer et al., 1997; Ketelaars et al., 2001), whereas theoretical estimation gave an expected value of 3.5–10% (Mostovoy and Knoester, 2000) or 13% (Wu and Small, 1997). van Oijen et al. noted that they have been able to identify the position of the k = 0 state in a few complexes in the single molecular spectroscopic study. Unfortunately, they do not report the position of this state relative to the main absorption peak, but their Fig. 3 B suggests that the k = 0 energy level lies ∼70 cm−1 below that of the next lowest exciton level (van Oijen et al., 1999; Mostovoy and Knoester, 2000). In fact, the 18 BChl a molecules are not identical considering their protein environment and their binding sites at α- and β-polypeptides. The heterogeneity in protein conformation leads to the heterogeneous site distribution of the molecules, as well as the heterogeneous broadening of the absorption spectra. The heterogeneous effect has been treated as energy disorder for both the diagonal and off-diagonal matrix elements of the Hamiltonian of the exciton, where the disorder for diagonal elements stands for the different site energy of the individual dimer, whereas the off-diagonal elements represent the variation of the coupling energy between the different adjacent pairs. When energy disorder effect is incorporated into the exciton theory, it leads to the mixing between the lowest exciton (j = 0) level with that of the next lowest exciton level (j = ±1), which renders the lowest exciton level being partially allowed. Wu and Small (1997) proposed symmetry-adapted basis defect patterns to analyze the effect of energy disorder on cyclic arrays of coupled chromophores (Wu and Small, 1997, 1998). They concluded that random disorder would not fully account for the spectral properties. These authors pointed out the possibility of the symmetry lowering of structural deformation to rationalize the correlated energy disorder rather than the random energy disorder invoked in the basis defect analysis; i.e., it is only the C1-symmetric component of the disorder that causes the mixing (Ketelaars et al., 2001). Deformation of the protein structure introducing a C2 modulation on the C9 symmetric ring aggregate, which not only leads to a larger splitting between the k = +1 and k = −1 states, but also reduces the C9 ring symmetry to C1 symmetry. Consequently, the upper half of the deformed ring is no longer equivalent to the lower half of the ring, rendering the lowest state more superradiant (Matsushita et al., 2001).
Later it was found that in addition to the random disorder, the observed single-molecular spectra indicate the presence of a regular modulation of the interaction between the different pigments (van Oijen et al., 1999; Bopp et al., 1999). Polarization-dependent fluorescence-excitation spectra have been performed on the single LH2 molecules (Rps. acidophila) immobilized in polyvinyl alcohol matrix at cryogenic temperature. The results unambiguously show that the B850 ring structurally deforms in their isolated form. Some of the main spectroscopic features based on the fluorescence excitation spectrum of a single LH2 complex at 1.5 K can be summarized as: 1), the two j = ±1 states split with an average separation of 110 cm−1; 2), the transition dipoles of j = +1 (stands for the higher energy level) and j = −1 (stands for the lower energy level) states are mutually orthogonal; and 3), the intensity of the j = +1 transition is weaker than the j = −1 transition, with an average ratio of 0.7. (Ketelaars et al., 2001). Thus, the structural deformation should be included in the exciton model. Matsushita et al. (2001) investigated the exciton states of an elliptically deformed ring aggregate to account for the LH2 spectroscopic properties. Three different models of elliptical deformation have been proposed. In model A, all the molecules are placed equidistantly on the ellipse. In model B and C, the intermolecular distance is modulated. The distance is shortest at the long axis and longest at the short axis of the ellipse; whereas in model C, the distance is shortest at the short axis and longest at the long axis of the ellipse (see Fig. 1, top view). The results show that only model C can reproduce the above-listed spectroscopic features. The deformation indicated by δr/r0 amounts to 7–8%, corresponding to an eccentricity of 0.49–0.52. The model also shows that the structural deformation of the protein together with the modulation of the interpigment distances lead to a part of oscillation strength of the j = −1 state being transferred to the j = 0 state at a large deformation. This leads to the optically forbidden state (j = 0) becoming partially allowed (Matsushita et al., 2001). Therefore, it is expected that structural deformation would give rise to a decrease in the excited-state lifetime of the lowest exciton state.
With the structural deformation exciton model, our observed lifetime shortening of the bleaching recovery can be rationalized. Because we have precluded the possibility of interfacial electron transfer between the pigment and the nanoparticles, the very plausible cause can be the structural deformation induced mixing of the optically forbidden lowest exciton state with the optically allowed next-lowest exciton state. When assembled onto TiO2 nanoparticles of a curved surface, both the carotenoid-containing LH2 and the carotenoidless LH2 are expected to undergo an extra structural deformation due to the interfacial interaction, which would give rise to a decrease in the observed lifetime of bleaching recovery kinetics accordingly.
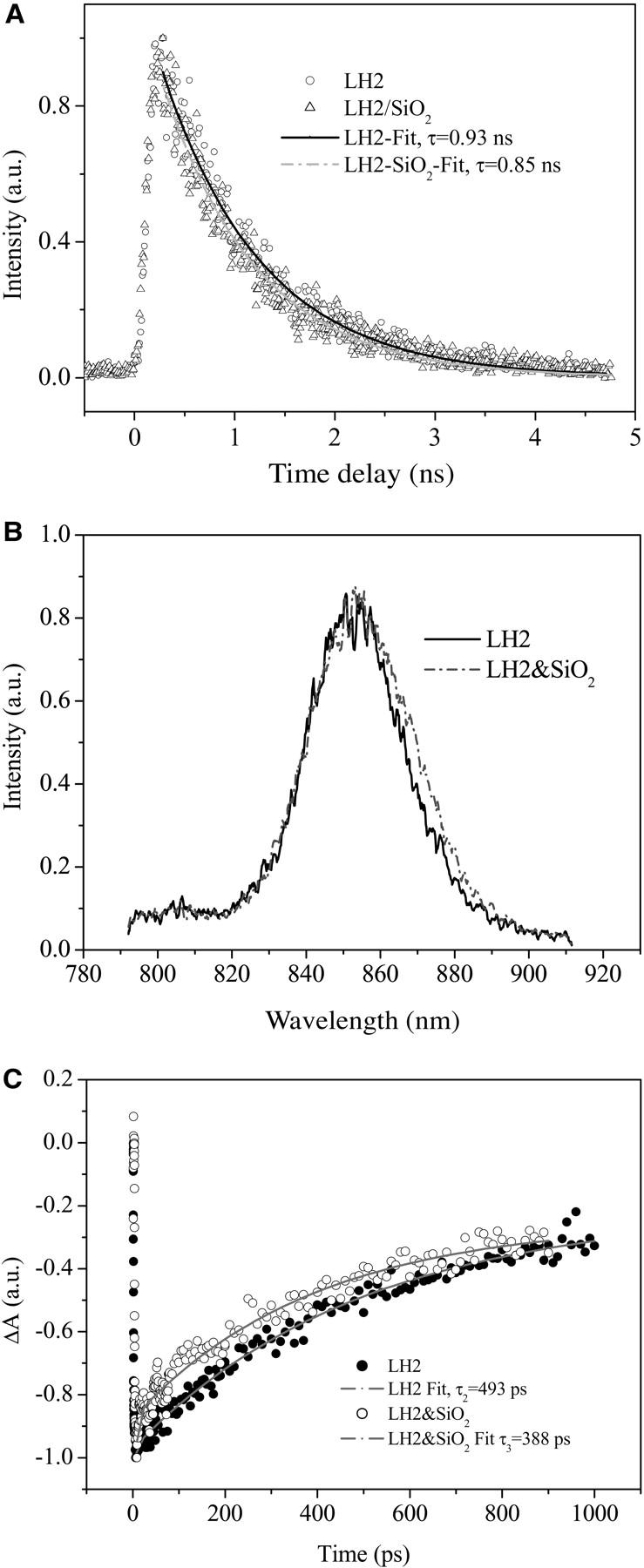
To further confirm the mechanism that the nanoparticle-induced structural deformation of LH2 protein leads to the decease in the lifetime of the lowest excitonic state, we used photochemical inactive SiO2 nanoparticle to replace the TiO2 nanoparticle, and LH1 of a larger cavity diameter to replace LH2, in which the interaction between LH1 and TiO2 nanoparticle is expected less than that between LH2 and TiO2 nanoparticle. Fig. 6 A displays the fluorescence decay kinetics of LH2 and LH2/SiO2(20.0 g/L). Fitting with monoexponential decay gives rise to a decay time constant of 0.93 and 0.85 ns, respectively, for LH2 and LH2/SiO2. An independent measurement differing in the concentration of SiO2 results in a time constant of 1.02 ns for LH2 and 0.96 ns for LH2/SiO2 (2.0 g/L). The control experiment of LH2/TiO2 (0.05 g/L) gives a time constant of 0.87 ns, indicating that the photochemical inactive nanoparticle can also lead to the decrease in the excited-state lifetime of the LH2 complex without involving any interfacial electron transfer process. Such a decrease in lifetime will be more clearly revealed in the bleaching recovery kinetics. Fig. 6 B demonstrates the time-resolved fluorescence spectra of LH2 and LH2/SiO2 (2.0 g/L) acquired at a delay time of 1.0 ns, which shows that the emission maximum of LH2/SiO2 almost remains the same as that of free LH2, whereas the spectrum is slightly broadened at the red wing. Fig. 6 C presents the B850 bleaching recovery kinetics of carotenoid-containing LH2 and the corresponding LH2/SiO2 (2.0 g/L) colloidal solution probed at 852 nm. Fitting of the experimental curves by a two-component exponential decay process leads to τ1 = 2.0 ps (50%) and τ2 = 493 ps (50%) for LH2 and τ1 = 4.5 ps (50%) and τ1 = 388 ps (50%) for LH2/SiO2(2g/L). The facts again support the mechanism of nanoparticle-induced structural deformation of LH2 leading to the decrease in the lifetime of LH2 excited state. Fig. 7 A shows bleaching recovery kinetic curves of LH1 and LH1/TiO2 excited at 400 nm and probed at 899 nm, which indicates that there is almost no difference between the two kinetic curves within the experimental errors, i.e., the phenomenon of decrease in the excited-state lifetime did not happen in the LH1/TiO2 colloidal solution. The time-resolved absorbance difference spectra of LH1 and LH1/TiO2 shown in Fig. 7 B also reveals that there is almost no difference between the two spectra within the detected region. This can be attributed to the structural difference between LH1 and LH2. For LH1 complex, the membrane protein is also constructed by the structural unit of α,β-polypeptide heterodimer with 16 subunits in total, forming a ringlike or an elliptical structure larger than that of LH2. For a ringlike structure of LH1, its diameter is of 11.5 nm, and a cavity of ∼7.0 nm in diameter (Roszak et al., 2003; Jamieson et al., 2002). Therefore, in view of molecular assembly, the 6.0-nm TiO2 nanoparticle may not be efficiently assembled to LH1 complex owing to its larger cavity, and the interaction between the nanoparticle and the protein would be weak. Therefore, the structural deformation of LH1 induced by the nanoparticle assembling would be smaller than that of LH2. This results in the exciton states of LH1 complex being less disturbed when LH1 complex is mixed with TiO2 nanoparticle colloidal solution, giving rise to a bleaching recovery lifetime constant being almost the same as that of the free LH1 complex.
FIGURE 6.
(A) Fluorescence decay kinetic curves of LH2 (Rb. sphaeroides 2.4.1) and LH2/SiO2 (20.0 g/L) colloidal solution, together with fitting curves of monoexponential decay with an individual time constant of 0.93 ns for LH2 and 0.85 ns for LH2/SiO2 colloidal solution, respectively. Pumped at 400 nm, 0.11 μJ/pulse, measured at 852 nm; (B) time-resolved fluorescence spectra of LH2 (Rb. sphaeroides 2.4.1) and corresponding LH2/SiO2 (2.0 g/L); (C) bleaching recovery kinetic curves of carotenoid-containing LH2 (Rb. sphaeroides 2.4.1) and the corresponding LH2/SiO2 (2.0 g/L) colloidal solution excited at 400 nm, 0.3 μJ/pulse, and probed at 852 nm.
FIGURE 7.
(A) Bleaching recovery kinetic curves of LH1 (Rs. rubrum S1) and LH1/TiO2 excited at 400 nm and probed at 899 nm. (B) The corresponding time-resolved absorbance difference spectra of LH1 and LH1/TiO2 colloidal solution.
Comparing the time-resolved fluorescence spectra and lifetime constants of carotenoid-containing LH2 assembled onto 6 nm TiO2 and 14 nm SiO2, it can be concluded that a larger red shift in the fluorescence spectra would correspond to a shorter excited-state lifetime. Rutkauskas et al. (2004) have investigated the fluorescence spectral fluctuation of single LH2 complexes; they showed that larger static disorder is associated with the light-induced conformers having larger free-energy barrier, and the realization of such a larger static disorder state results in a larger splitting between the k = ±1 levels, so that the lower k = −1 state becomes more populated and more emissive as compared to the k = +1 state. The lowest k = 0 state becomes more localized, more radiant, and more red-shifted (Rutkauskas et al., 2004). Judged from the emission spectra of LH2 assembled onto the above nanoparticle, the conformation states are similar to the light induced when referring to the emission spectra of the single LH2 molecule, though the extent of the spectral shift is smaller. Therefore, the time-resolved fluorescence spectra indicate that nanoparticles have induced a protein conformation change. In another single molecular spectroscopic experiment, Bopp et al. (1997) found that the lifetime of the single LH2 molecule is ∼1.0 ns, similar to that of the ensemble. After illumination, the single molecule reaches different luminescent states with lifetime ranging from 0.2 to 0.9 ps (Bopp et al., 1997). Later these authors proposed that LH2 undergoes dynamical structural deformation from a ringlike structure to an elliptical form based on the polarized fluorescence single-molecular spectroscopic study (Bopp et al., 1999). Although some extremely short lifetime such as 0.2 ns could have been attributed to the effect of excitonic annihilation and O2 photochemical effect (Bergström et al., 1986), these experiments seem to provide a certain link between the conformation change and shortening in the lifetime of the excitonic state. Our results support that the lifetime shortening of the excited state of LH2-nanoparticle assembly is caused by the nanoparticle-induced protein structural deformation. It can also be inferred from these results that the nanoparticle size is critical. For a LH2 with a cavity of 3.6 nm in diameter, 6 nm TiO2 induced a larger decrease in the excited-state lifetime than that of 14 nm SiO2, whereas for LH1 with a cavity of 7.0 nm in diameter, the 6 nm TiO2 almost has no effect on the excited-state lifetime. The facts indicate that the protein deformation depends on the extent of docking of the nanoparticle with the LH2 cavity, exhibiting a size-dependent effect. It should be noted that this size-dependent effect is different from what has been observed for human carbonic anhydrase I adsorbed on silica where the protein structural deformation depends on the total area of the protein-silica nanoparticle interacting surface (Lundqvist et al., 2004).
CONCLUSION
Assembly of light-harvesting complex LH2 of photosynthetic bacteria and TiO2 nanoparticle has been studied by time-resolved spectroscopy. Excited-state lifetimes have been measured for Car-containing and carotenoidless LH2 when assembled onto TiO2 nanoparticles with an average size of 6 nm. An obvious decrease in the excited-state lifetime of B850 was observed when LH2 was immobilized on TiO2. It is proposed that the observed change in the photophysical properties of LH2 when assembled onto TiO2 nanoparticles is arising from the interfacial-interaction-induced structural deformation of the LH2 complex. When the LH2 structure deviates from the circular symmetry, the optically forbidden lowest exciton state in the circular exciton model would mix with the two split optically allowed next-lowest exciton states to a larger extent and become more radiant, which leads to a decrease in the lifetime of the lowest exciton state. This mechanism is further verified by using photoinactive SiO2 nanoparticle in place of TiO2, and LH1 complex instead of LH2. The former also leads to the decrease of the excited-state lifetime of LH2, whereas the excited-state lifetime of the latter almost has no change when mixed with TiO2 colloidal solution owing to the weak interaction between TiO2 nanoparticle and LH1 of a larger cavity size.
Acknowledgments
We thank Junji Akahane and Hiro Ishii for help in preparation of LH1 and LH2 from R26.1 mutant and Jie Ma for artwork. We also thank the reviewers for very comprehensive and insightful comments.
This work was supported by National Hundred Talents Program, Chinese National Natural Science Fund for International Cooperation, and Japanese Society for Promotion of Science.
References
- Bergström, H., V. Sundström, R. van Grondelle, E. Åkesson, and T. Gillbro. 1986. Energy transfer within the isolated light-harvesting B800–850 pigment-protein complex of Rhodobacter sphaeroides. Biochim. Biophys. Acta. 852: 279–287. [Google Scholar]
- Bergström, H., V. Sundström, R. van Grondelle, T. Gillbro, and R. J. Cogdell. 1988. Energy transfer dynamics of isolated B800–850 and B800–820 pigment–protein complexes of Rhodobacter sphaeroides and Rhodopseudomonas acidophila. Biochim. Biophys. Acta. 936:90–98. [Google Scholar]
- Bopp, M. A., Y. Jia, L. Li, R. J. Cogdell, and R. M. Hochstrasser. 1997. Fluorescence and photobleaching dynamics of single light-harvesting complexes. Proc. Natl. Acad. Sci. USA. 94:10630–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp, M. A., A. Sytnik, T. D. Howard, R. J. Cogdell, and R. M. Hochstrasser. 1999. The dynamics of structural deformations of immobilized single light-harvesting complexes. Proc. Natl. Acad. Sci. USA. 96:11271–11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böse, S. K. 1963. Media for anaerobic growth of photosynthetic bacteria. In Bacterial Photosynthesis. H. Gest, A. San Pietro, and L. F. Vernon, editors. The Antioch Press, Yellow Springs, OH. 501–510.
- Chachisvilis, M., O. Kühn, T. Pullerits, and V. Sundström. 1997. Excitation in photosynthetic purple bacteria: wavelike motion or incoherent hopping? J. Phys. Chem. B. 101:7275–7283. [Google Scholar]
- Chauvet, J.-P., R. Viovy, R. Santos, and E. J. Land. 1981. One-electron oxidation of photosynthetic pigments in micelles, bacteriochlorophyll a, chlorophyll a, chlorophyll b, and pheophytin a. J. Phys. Chem. 85:3449–3456. [Google Scholar]
- Das, R., P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Y. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. G. Zhang, and M. Baldo. 2004. Integration of photosynthetic protein molecular complexes in solid-state electronic devices. Nano Lett. 4:1079–1083. [Google Scholar]
- Davidson, E., and R. J. Cogdell. 1981. The polypeptide composition of the B850 light-harvesting pigment-protein complex from Rhodopseudomonas sphaeroides R26.1. FEBS Lett. 132:81–84. [Google Scholar]
- Deng, Y., G. Gao, Z. He, and L. D. Kispert. 2000. Effects of polyene chain length and acceptor substituents on the stability of carotenoid radical cations. J. Phys. Chem. B. 104:5651–5656. [Google Scholar]
- Dobson, K. D., P. A. Connor, and J. McQuillan. 1997. Monitoring hydrous metal oxide surface charge and adsorption by STIRS. Langmuir. 13:2614–2616. [Google Scholar]
- Frei, H., D. J. Fitzmaurice, and M. Grätzel. 1990. Surface chelation of semiconductors and interfacial electron transfer. Langmuir. 6:198–206. [Google Scholar]
- Freiberg, A., J. A. Jackson, S. Lin, and N. W. Woodbury. 1998. Subpicosecond pump-supercontinuum probe spectroscopy of LH2 photosynthetic antenna proteins at low temperature. J. Phys. Chem. A. 102:4372–4380. [Google Scholar]
- Gall, A., R. J. Cogdell, and B. Robert. 2003. Influence of carotenoid molecules on the structure of the bacteriochlorophyll binding site in peripheral light-harvesting proteins from Rhodobacter sphaeroides. Biochemistry. 42:7252–7258. [DOI] [PubMed] [Google Scholar]
- Hofmann, C., T. J. Aartsma, H. Michel, and J. Köhler. 2003. Direct observation of tiers in the energy landscape of a chromoprotein: a single-molecule study. Proc. Natl. Acad. Sci. USA. 100:15534–15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, X. G., L. C. Du, M. P. Ye, and Y. X. Weng. 2004b. Synchrontron small angle X-ray scattering study of dye-sensitized/-unsensitized TiO2 nanoparticle colloidal solution. Chin. Phys. 13:720–724. [Google Scholar]
- Hong, X., Y. X. Weng, and M. Li. 2004a. Determination of the topological shape of integral membrane protein light-harvesting complex LH2 from photosynthetic bacteria in the detergent solution by small-angle X-ray scattering. Biophys. J. 86:1082–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X., T. Ritz, A. Damjanović, F. Autenrieth, and K. Schulten. 2002. Photosynthetic apparatus of purple bacteria. Q. Rev. Biophys. 35:1–62. [DOI] [PubMed] [Google Scholar]
- Hu, X., and K. Schulten. 1997. How nature harvests sunlight. Phys. Today. 50:28–34. [Google Scholar]
- Jamieson, S. T., Y. Wang, P. Qian, J. Y. Kirkland, M. J. Conroy, C. N. Hunter, and P. A. Bullough. 2002. Projection structure of the photosynthetic reaction center-antenna complex of Rhodospirillum rubrum at 8.5 Å resolution. EMBO J. 21:3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez, R., S. N. Dikshit, S. E. Bradforth, and G. R. Fleming. 1996. Electronic excitation transfer in the LH2 complex of Rhodobacter sphaeroides. J. Phys. Chem. 100:6825–6834. [Google Scholar]
- Kay, A., and M. Grätzel. 1993. Artificial photosynthesis. 1. Photosensitization of TiO2 solar cells with chlorophyll derivatives and related natural porphyrins. J. Phys. Chem. 97:6272–6277. [Google Scholar]
- Kennis, J. T. M., A. M. Streltsov, S. I. E. Vulto, T. J. Aartsma, T. Nozawa, and J. Amesz. 1997. Femtosecond dynamics in isolated LH2 complexes of various species of purple bacteria. J. Phys. Chem. B. 101:7827–7834. [Google Scholar]
- Ketelaars, M., A. M. van Oijen, M. Matsushita, J. Köhler, J. Schmidt, and T. J. Aartsma. 2001. Spectroscopy on the B850 band of individual light-harvesting 2 complexes of Rhodopseudomonas acidophila I. experiments and Monte Carlo simulations. Biophys. J. 80:1591–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepke, J., X. Hu, C. Muencke, K. Schulten, and H. Michel. 1996. The crystal structure of the light-harvesting complex II (B800–850) from Rhodospirillum molischianum. Structure. 4:581–597. [DOI] [PubMed] [Google Scholar]
- Koolhaas, M. H. C., R. N. Frese, G. J. S. Fowler, T. S. Bibby, S. Georgakopoulou, G. van der Zwan, C. N. Hunter, and R. van Grondelle. 1998. Identification of the upper exciton component of the B850 bacteriochlorophylls of the LH2 antenna complex, using a B800-free mutant of Rhodobacter sphaeroides. Biochemistry. 37:4693–4698. [DOI] [PubMed] [Google Scholar]
- Koolhaas, M. H. C., G. van der Zwan, R. N. Frese, and R. van Grondelle. 1997. Red shift of the zero crossing in the CD spectra of the LH2 antenna complex of Rhodopseudomonas acidophila: a structure-based study. J. Phys. Chem. B. 101:7262–7270. [Google Scholar]
- Kropacheva, T. N., and A. J. Hoff. 2001. Electrochemical oxidation of bacteriochlorophyll a in reaction centers and antenna complexes of photosynthetic bacteria. J. Phys. Chem. B. 105:5536–5545. [Google Scholar]
- Law, C. J., A. W. Roszak, J. Southall, A. T. Gardiner, N. W. Isaacs, and R. J. Cogdell. 2004. The structure and function of bacterial light-harvesting complexes (review). Mol. Membr. Biol. 21:183–191. [DOI] [PubMed] [Google Scholar]
- Lee, I., J. W. Lee, R. J. Warmack, D. P. Allison, and E. Greenbaum. 1995. Molecular electronics of a single photosystem I reaction center: studies with scanning tunneling microscopy and spectroscopy. Proc. Natl. Acad. Sci. USA. 92:1965–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leupold, D., H. Stiel, K. Teuchner, F. Nowak, W. Sander, B. Ücker, and H. Scheer. 1996. Size enhancement of transition dipoles to one- and two-exciton bands in a photosynthetic antenna. Phys. Rev. Lett. 77:4675–4678. [DOI] [PubMed] [Google Scholar]
- Limantara, L., R. Fujii, J. P. Zhang, T. Kakuno, H. Hara, A. Kawamori, T. Yagura, R. J. Cogdell, and Y. Koyama. 1998. Generation of triplet and cation-radical bacteriochlorophyll a in carotenoidless LH1 and LH2 antenna complexes from Rhodobacter sphaeroides. Biochemistry. 37:17469–17486. [DOI] [PubMed] [Google Scholar]
- Lundqvist, M., I. Sethson, and B.-H. Jonsson. 2004. Protein adsorption onto silica nanoparticles: conformational changes depend on the particles' curvature and the protein stability. Langmuir. 20:10639–10647. [DOI] [PubMed] [Google Scholar]
- Matsushita, M., M. Ketelaars, A. M. van Oijen, J. Köhler, T. J. Aartsma, and J. Schmidt. 2001. Spectroscopy on the B850 band of individual light-harvesting 2 complexes of Rhodopseudomonas acidophila II. Exciton states of an elliptically deformed ring aggregate. Biophys. J. 80:1604–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott, G., S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell, and N. W. Isaacs. 1995. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 374:517–521. [Google Scholar]
- Meier, T., V. Chernyak, and S. Mukamel. 1997. Multiple exciton coherence sizes in photosynthetic antenna complexes viewed by pump-probe spectroscopy. J. Phys. Chem. B. 101:7332–7342. [Google Scholar]
- Monshouwer, R., M. Abrahamsson, F. van Mourik, and R. van Grondelle. 1997. Superradiance and exciton delocalization in bacterial photosynthetic light-harvesting systems. J. Phys. Chem. B. 101:7241–7248. [Google Scholar]
- Monshouwer, R., and R. van Grondelle. 1996. Excitations and excitons in bacterial light-harvesting complexes. Biochim. Biophys. Acta. 1275:70–75. [DOI] [PubMed] [Google Scholar]
- Moser, C. C., J. M. Keske, K. Warncke, R. S. Farid, and P. L. Dutton. 1992. Nature of biological electron transfer. Nature. 355:796–802. [DOI] [PubMed] [Google Scholar]
- Moser, J. E., and M. Gräzel. 1993. Observation of temperature independent heterogeneous electron transfer reactions in the inverted region. Chem. Phys. 176:493–500. [Google Scholar]
- Mostovoy, M. V., and J. Knoester. 2000. Statistics of optical spectra from single-ring aggregates and its application to LH2. J. Phys. Chem. B. 104:12355–12364. [Google Scholar]
- Nagarajan, V., R. G. Alden, J. C. Williams, and W. W. Parson. 1996. Ultrafast exciton relaxation in the B850 antenna complex of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 93:13774–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoderezhkin, V., R. Monshouwer, and R. van Grondelle. 1999. Exciton (de)localization in the LH2 antenna of Rhodobacter sphaeroides as revealed by relative difference absorption measurements of the LH2 antenna and the B820 subunit. J. Phys. Chem. B. 103:10540–10548. [Google Scholar]
- Novoderezhkin, V., M. Wendling, and R. van Grondelle. 2003. Intra- and interband transfers in the B800–B850 antenna of Rhodospirillum molischianum: Redfield theory modeling of polarized pump-probe kinetics. J. Phys. Chem. B. 107:11534–11548. [Google Scholar]
- Ohashi, N., N. Ko-Chi, M. Kuki, T. Shimamura, R. J. Cogdell, and Y. Koyama. 1996. The structures of S0 spheroidene in the light-harvesting (LH2) complex and S0 and T1 spheroidene in the reaction center of Rhodobacter sphaeroides 2.4.1 as revealed by Raman spectroscopy. Biospectroscopy. 2:59–69. [Google Scholar]
- Papiz, M. Z., S. M. Prince, T. Howard, R. J. Cogdell, and N. W. Isaacs. 2003. The structure and thermal motion of the B800–850 LH2 complex from Rps. acidophila at 2.0 Å resolution and 100 K: new structural features and functionally relevant motions. J. Mol. Biol. 326:1523–1538. [DOI] [PubMed] [Google Scholar]
- Prince, S. M., T. D. Howard, D. A. Myles, C. Wilkinson, M. Z. Papiz, A. A. Freer, R. J. Cogdell, and N. W. Isaacs. 2003. Detergent structure in crystals of the integral membrane light-harvesting complex LH2 from Rhodopseudomonas acidophila strain 10050. J. Mol. Biol. 326:307–315. [DOI] [PubMed] [Google Scholar]
- Pullerits, T., M. Chachisvilis, M. R. Jones, C. N. Hunter, and V. Sundström. 1994. Exciton dynamics in the light-harvesting complexes of Rhodobacter sphaeroides. Chem. Phys. Lett. 224:355–365. [Google Scholar]
- Pullerits, T., and V. Sundström. 1996. Photosynthetic light-harvesting pigment-protein complexes: toward understanding how and why. Acc. Chem. Res. 29:381–389. [Google Scholar]
- Roddick-Lanzilotta, A. D., and A. J. Mcquillan. 2000. An in situ infrared spectroscopic study of glutamic acid and of aspartic acid adsorbed on TiO2: implications for the biocompatibility of titanium. J. Colloid Interface Sci. 227:48–54. [DOI] [PubMed] [Google Scholar]
- Roszak, A. W., T. D. Howard, J. Southall, A. T. Gardiner, C. J. Law, N. W. Isaacs, and R. J. Cogdell. 2003. Crystal structure of the RC-LH1 core complex from Rhodopseudomonas palustris. Science. 302:1969–1972. [DOI] [PubMed] [Google Scholar]
- Rutkauskas, D., V. Novoderezkhin, R. J. Cogdell, and R. van Grondelle. 2004. Fluorescence spectral fluctuations of single LH2 complexes from Rhodopseudomonas acidophila strain 10050. Biochemistry. 43:4431–4438. [DOI] [PubMed] [Google Scholar]
- Sauer, K., R. J. Cogdell, S. M. Prince, A. A. Freer, N. W. Issacs, and H. Scheer. 1996. Structure-based calculations of the optical spectra of the LH2 bacteriochlorophyll-protein complex from Rhodopseudomonas acidophila. Photochem. Photobiol. 64:564–576. [Google Scholar]
- Sebban, P., G. Jolchine, and I. Moya. 1984. Spectra of fluorescence lifetime and intensity of Rhodopseudomonas sphaeroides at room and low temperature. Comparison between the wild type, the C71 reaction center-less mutant and the B800–850 pigment-protein complex. Photochem. Photobiol. 39:247–253. [Google Scholar]
- Sundström, V., T. Pullerits, and R. van Grondelle. 1999. Photosynthetic light-harvesting: reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit. J. Phys. Chem. 103:2327–2346. [Google Scholar]
- Tietz, C., O. Chekhlov, A. Dräbenstedt, J. Schuster, and J. Wrachtrup. 1999. Spectroscopy on single light-harvesting complexes at low temperature. J. Phys. Chem. B. 103:6328–6333. [Google Scholar]
- Topoglidis, E., T. Lutz, R. L. Willis, C. J. Barnett, A. E. Cass, and J. R. Durrant. 2000. Protein adsorption on nanoporous TiO2 films: a novel approach to studying photoinduced protein/electrode transfer reaction. Faraday Discuss. 116:35–46. [DOI] [PubMed] [Google Scholar]
- Trinkunas, G., J. L. Herek, T. Polívka, V. Sundström, and T. Pullerits. 2001. Excitation delocalization probed by excitation annihilation in the light-harvesting antenna LH2. Phys. Rev. Lett. 86:4167–4170. [DOI] [PubMed] [Google Scholar]
- van Grondelle, R., and V. Novoderezhkin. 2001. Dynamics of excitation energy transfer in the LH1 and LH2 light-harvesting complexes of photosynthetic bacteria. Biochemistry. 40:15057–15068. [DOI] [PubMed] [Google Scholar]
- van Oijen, A. M., M. Ketelaars, J. Köhler, T. J. Aartsma, and J. Schmidt. 1999. Unraveling the electronic structure of individual photosynthetic pigment-protein complex. Science. 285:400–402. [DOI] [PubMed] [Google Scholar]
- Wang, Z.-S., F.-Y. Li, C.-H. Huang, L. Wang, M. Wei, L.-P. Jin, and N.-Q. Lin. 2000. Photoelectric conversion properties of nanocrystalline TiO2 electrodes sensitised with hemicyanine derivatives. J. Phys. Chem. B. 104:9676–9682. [Google Scholar]
- Wong, N., S. Kam, T. C. Jessop, P. A. Wender, and H. Dai. 2004. Nanotube molecular transporters: internalization of carbon nanotube-protein conjugates into mammalian cells. J. Am. Chem. Soc. 126:6850–6851. [DOI] [PubMed] [Google Scholar]
- Weng, Y.-X., L. Zhang, J. Yang, D.-H. Quan, L. Wang, G. Z. Yang, R. Fujii, Y. Koyama, J.-P. Zhang, J. Feng, J.-H. Yu, and B.-W. Zhang. 2003. Distance-dependent long-range electron transfer in protein: a case study of photosynthetic bacterial light-harvesting antenna complex LH2 assembled on TiO2 nanoparticle by femto-second time-resolved spectroscopy. Acta Bot. Sin. 45:488–493. [Google Scholar]
- Wu, H.-M., and G. J. Small. 1997. Symmetry adapted basis defect patterns for analysis of the effects of energy disorder on cyclic arrays of coupled chromophores. Chem. Phys. 218:225–234. [Google Scholar]
- Wu, H.-M., and G. J. Small. 1998. Symmetry-based analysis of the effects of random energy disorder on the excitonic level structure of cyclic arrays: application to photosynthetic antenna complex. J. Phys. Chem. B. 102:888–898. [Google Scholar]
- Zhang, Q.-L., L.-C. Du, Y.-X. Weng, L. Wang, H.-Y. Chen, and J.-Q. Li. 2004. Particle size-dependent distribution of carboxylate adsorption sites on TiO2 nanoparticle surface: insights into the surface modification of nanostructured TiO2 electrodes. J. Phys. Chem. B. 108:15077–15083. [Google Scholar]
- Zhang, L., J. Yang, L. Wang, G.-Z. Yang, and Y.-X. Weng. 2003. Direct observation of interfacial charge recombination to the excited-triplet state in all-trans-retinoic acid sensitized TiO2 nanoparticle by femtosecond time-resolved difference absorption spectroscopy. J. Phys. Chem. B. 107:13688–13697. [Google Scholar]