

Abstract
Although light microscopy and three-dimensional image analysis have made considerable progress during the last decade, it is still challenging to analyze the genome nano-architecture of specific gene domains in three-dimensional cell nuclei by fluorescence microscopy. Here, we present for the first time chromatin compaction measurements in human lymphocyte cell nuclei for three different, specific gene domains using a novel light microscopic approach called Spatially Modulated Illumination microscopy. Gene domains for p53, p58, and c-myc were labeled by fluorescence in situ hybridization and the sizes of the fluorescence in situ hybridization “spots” were measured. The mean diameters of the gene domains were determined to 103 nm (c-myc), 119 nm (p53), and 123 nm (p58) and did not correlate to the genomic, labeled sequence length. Assuming a spherical domain shape, these values would correspond to volumes of 5.7 × 10−4 μm3 (c-myc), 8.9 × 10−4 μm3 (p53), and 9.7 × 10−4 μm3 (p58). These volumes are ∼2 orders of magnitude smaller than the diffraction limited illumination or observation volume, respectively, in a confocal laser scanning microscope using a high numerical aperture objective lens. By comparison of the labeled sequence length to the domain size, compaction ratios were estimated to 1:129 (p53), 1:235 (p58), and 1:396 (c-myc). The measurements demonstrate the advantage of the SMI technique for the analysis of gene domain nano-architecture in cell nuclei. The data indicate that chromatin compaction is subjected to a large variability which may be due to different states of genetic activity or reflect the cell cycle state.
INTRODUCTION
The supramolecular architecture of the genome is not random and the organization is of functional significance (Cremer et al., 2000, 2001, 2003; Cremer and Cremer, 2001; Dundr and Misteli, 2001; Kozubek et al., 2002). Individual chromosomes occupy distinct territories that are subdivided into distinct domains and functional subunits in a hierarchical manner (Cremer and Cremer, 2001; van Driel et al., 2003). Molecular labeling by fluorescence in situ hybridization (FISH) (Solovei et al., 2002b) as well as in vivo labeling approaches (Zink and Cremer, 1998; Tsukamoto et al., 2000; Tumbar and Belmont, 2001; Bubulya and Spector, 2004) using fluorescent proteins, for instance, have been applied to visualize the compartments of the genome architecture by fluorescence light microscopy. By means of confocal laser scanning microscopy or other three-dimensional (3D) imaging techniques, positions, and distances of fluorescence labeled chromosome territories and domains as for instance centromeres (e.g., Cremer et al., 2001), telomeres (e.g., Amrichova et al., 2003), or specific gene loci (e.g., Bartova et al., 2002), were measured. However, using conventional, diffraction limited light microscopy such as confocal laser scanning microscopy or epifluorescence light microscopy, the optical resolution is limited to ∼200 nm in the lateral direction and to several times worse in the axial direction (Pawley, 1995; Stelzer, 1998; Edelmann et al., 1999). Distances considerably smaller than the optical resolution can be measured after labeling with two or even more spectral signatures, e.g., by fluorescence resonance energy transfer (FRET) (Jares-Erijman and Jovin, 2003), or spectral precision distance microscopy (SPDM) (Esa et al., 2000, 2001).
Functional models suggest that the location and chromatin compaction of individual genes is correlated to gene activity and determines the accessibility for macromolecules (van Driel et al., 2003; Spector, 2003). Although the size of artificially introduced tandem repeats has been determined using conventional light microscopy (Tsukamoto et al., 2000), so far this has not been possible for individual small gene domains in “native”, genetically unmodified cells such as human lymphocytes. To analyze gene compaction under such “natural” circumstances, appropriate microscopic systems are required that cover the nanoscale in their range of analysis. Atomic force microscopy (AFM) (Horber and Miles, 2003) or scanning near-field optical microscopy (SNOM) (Richards, 2003) are routine techniques for the quantitative analysis of cell surfaces (e.g., Perner et al., 2002), metaphase chromosomes (e.g., Winkler et al., 2003) or isolated chromatin (e.g., Kepert et al., 2003) with a resolution of some 10 nm. However, these scanning probe techniques are not suitable for the analysis of structures inside cell nuclei in three dimensions. The situation is similar for electron microscopy (EM). EM has the advantage of superior resolution but again it is not applicable to intact nuclei. Therefore, novel techniques for fluorescence light microscopy breaking the conventional diffraction limit imposed to structural resolution using narrowed or altered point spread functions compared to conventional light microscopy, e.g., 4Pi-microscopy (Hell and Stelzer, 1992; Kano et al., 2001), stimulated-emission-depletion (STED) microscopy (Hell and Wichmann, 1994; Hell, 2003), spatially modulated illumination (SMI) microscopy (Schneider et al., 1999; Albrecht et al., 2002; Failla et al., 2002c; Martin et al., 2004), standing wave-field microscopy (Bailey et al., 1993; Freimann et al., 1997) etc., have been developed and applied to biological objects. Although the principle feasibility of biological applications and the gain of resolution of these systems were demonstrated, they are individual laboratory setups and biological routine applications are still challenging or not available.
Here, we show for the first time systematic measurements of the sizes of specific, fluorescence-labeled gene domains in 3D cell nuclei of human peripheral blood by SMI microscopy. In contrast to Martin et al. (2004), who measured small labels in cryosections of ∼0.14 μm thickness, we did the SMI measurements within intact nuclei of ∼5–10 μm height.
MATERIAL AND METHODS
Cell preparation
Human peripheral blood lymphocytes of a healthy donor were stimulated and cultivated for 48 h at 37°C in chromosome medium B, containing phytohaemaglutinine at 2.5 mg/l (Biochrom, Berlin, Germany) to obtain unsynchronized cells. For G1 enriched cultures, the cells were arrested in metaphase by adding 200 μl of Colcemid for 24 h at 37°C. The cells were washed twice in fresh medium and then incubated for another 4 h to proceed in G1 phase. Cell cycle distribution was controlled by flow cytometry after DNA staining. After treatment with 75 mM KCl, the cells were fixed in ice-cold methanol/acetic acid (3:1) twice for 1 h each. The cells were then dropped on slides. In some experiments also unsynchronized stimulated cells were used according to the same preparation.
DNA probes
The gene domains of p53, p58, and c-myc were targeted with DNA probes of different sizes (Qbiogene, Irvine, CA). All probes were labeled with digoxigenin (DIG) and detected via an FITC labeled rabbit-anti-DIG antibody (Qbiogene). In the case of double labeling an additional goat-anti-rabbit antibody was used carrying Alexa 647 as a red dye (Molecular Probes, Eugene, OR).
Fluorescence in situ hybridization
The slides were incubated in 2× SSC for 30 min at 37°C. After treatment by an ethanol series (70%:80%:90%) for 2 min each at room temperature, the slides were air-dried. Then the specimens were denatured with 70% formamide in 2× SSC for 2 min at 72°C, followed by an additional ethanol treatment on ice and air drying. 10 μL of each denatured DNA probe were added to the target specimen, covered by a plastic coverglass, and incubated for 24 h at 37°C in a humidified chamber. Washing was performed with 2× SSC for 5 min at 72°C and 1× PBD for 2 min at room temperature. 50 μL of the FITC labeled anti-DIG antibody were added and incubated for 40 min at 37°C in the dark. After washing twice in 1× PBD for 5 min each, the second antibody treatment inclusive washing was done (in some cases only). Then the cells were embedded in 30 μl Vectashield with DAPI (50 μg/μl) and covered with a coverglass.
SMI microscopy
SMI microscopy was performed by means of the laboratory setup as described in detail elsewhere (Schneider et al., 1999; Albrecht et al., 2002). The instrument was equipped with an Ar+ laser for 488 nm illumination (FITC) and a Kr+ laser for 647 nm illumination (Alexa 647). Specimen illumination by a standing wave field was performed via two Plan APO-chromatic objective lenses (100×, 0.7–1.4 NA; Leica, Bensheim, Germany). Signal detection took place via one of these lenses and a highly sensitive cooled CCD camera (PhaseHL, Lübeck, Germany). The cells were imaged in a 400 slice stack taken along the common optical axis of the two objective lenses. For this purpose the specimen slide was moved by a piezo driven temperature compensating carrier in steps of 20 nm (see abscissa of Figs. 3 B and 4 B). The acquisition time for each image slice was ∼2–3 s resulting in a total acquisition time for the whole 3D image of 7–10 min. Typically 150–200 image slices around the hybridization signals were acquired for each cell nucleus. After image acquisition the data sets of the cells were further analyzed with a newly developed program for SMI measurements with a low signal/noise ratio.
FIGURE 3.

(A) Projection image of a 3D-image stack of a lymphocyte cell nucleus (G1-phase) recorded by SMI microscopy along the optical axis. Since no counterstaining was applied, the nucleus is only visible due to the fluorescence background after FISH. The dashed line indicates the nuclear border schematically. Two gene domains for p53 on chromosome 17 (arrows) were labeled by a FISH probe (FITC, green) and additionally detected by a secondary antibody reaction (Alexa 647, red). Due to the simultaneous double color staining, the FISH “spots” were identified and discriminated from possible background signals (e.g., arrowheads). The modulation curves were measured for each color. Note: due to a slight lateral chromatic shift between green and red in the SMI detection pathway, the labeling sites did not exactly colocalize. Since this shift did not impede the gene domain identification and was not used to extract information, no attempt was made for correction. (B) Modulation curves of a labeled gene domain (a, green; b, red) indicated by the thick arrow in A. For a 3D data set, 200 images (“optical sections”) were recorded with an axial image distance of 20 nm (abscissa unit). The curves show the relative intensity of the AID at the labeled domain in each axial “optical section” along the z axis (abscissa). Note: the modulation curves are a direct measure of the spot size. In contrast, the size of the image spot in A appears to be different because the smallest object size in lateral dimensions is given by the lateral PSF of the microscope lens and the visible signal to background ratio.
FIGURE 4.

(A) Projection image of a 3D-image stack of a lymphocyte cell nucleus recorded by SMI-microscopy. Since no counterstaining was applied, the nucleus is only visible due to the fluorescence background after FISH. The dashed line indicates the nuclear border schematically. Gene domains for p58 on chromosome 1 were labeled by a FISH probe (FITC, green). Besides a spatially isolated domain (a), two additional closely neighbored domains (b and c) were observed compatible with the assumption that in this nucleus one domain had already fully replicated whereas the other had not. (B) Modulation curves for the three domains a, b, and c obtained from image stacks of 20 nm slice distance (abscissa unit). For further details, see Fig. 3 B. Note: the modulation curves shown here are representative in such a way that they show the variability that may occur within one nucleus. They are not representative for the mean diameter of the p58 gene domain.
Data analysis
For data analysis of SMI modulation curves of biological objects, a special interactive computer program was written. The unspecific background of the data stacks was identified as the overall average of intensity in a 9 × 9 pixel surrounding and subtracted. Usually SMI modulation curves of biological objects were not symmetric (see, for example, Fig. 4 B). Therefore, a background value right and left of the maximum was determined. From these values an optimized, global background value was calculated for each individual modulation curve and suggested to the user. The user can select or modify this background. Then the modulation contrast was calculated from the global maximum and all local minima. The later were identified as those that were lying closest to the local minima in an expected ideal periodic pattern.
RESULTS AND DISCUSSION
The intensity signal of a subwavelength sized fluorescent gene domain was obtained by the determination of the intensity distribution through the entire image stack of a cell nucleus along the optical axis (axial intensity distribution, AID). The AID modulates in a typical way that can be described by the convolution of the object with the SMI point spread function (SMI-PSF) obtained by the product of the illumination modulation (illumination PSF) and the PSF of the detecting microscope lens along the optical axis (detection PSF) (Failla et al., 2002a).
In Fig. 1, a schematic AID signal of an object with an axial extension below the resolution limit (“extended point object”) is shown. The modulation width (axial distance between the maxima of two neighboring fringes) is determined by
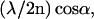 |
with λ the wavelength of the illumination laser beam, n the refraction index of the medium, and α the angle between the interfering laser beams and the optical axis. The enveloping shape of the pattern is given by the detection PSF. Ideal point objects in a background free environment would not show any modulation “background”, i.e., the modulation minima would reach zero. However, extended objects lead to a modulation background as shown in Fig. 1, where the modulation minima do not reach zero. The reason for this typical shape is that the fluorescence signal is a convolution of the fluorescence of the object and the point spread function of the SMI microscope. The modulation contrast (MC) is defined by the quotient of the maximum of the nonmodulating (background) part of the signal and the intensity maximum of the whole signal (= MInmp/MIAID). The MC is related to the size of the object in the direction of the z axis. The MC value is equal for objects of the same size but different total fluorescence intensity. Assuming a homogeneous fluorophore distribution in the fluorescent object, the relation between MC and the actual size at a given excitation wavelength can be calculated and this curve can be used as a calibration curve for absolute object nano-sizing (Fig. 2).
FIGURE 1.

Theoretical axial intensity distribution (AID) of an “extended point object” with MIAID being the maximal intensity of the AID and MInmp being the maximal intensity of the nonmodulating part. The modulation contrast MC is defined by MInmp/MIAID.
FIGURE 2.

Calibration curve between MC and absolute size in nanometers for an illumination wavelength of 488 nm calculated by virtual SMI microscopy computer simulation, assuming a homogenous fluorescence distribution.
FISH was applied to methanol/acetic acid fixed human lymphocytes enriched in G1-phase. Small DNA probes for the gene domains of p53, p58, and c-myc were available and covered a genomic length of 45 kb (p53), 85 kb (p58), and 120 kb (c-myc) (Table 1). The probes were labeled with digoxigenin (DIG) and detected by a FITC labeled anti-DIG antibody (green). In several cases those gene domain sequences were additionally labeled by a secondary antibody against FITC carrying Alexa 647 as a red dye, to discriminate true and false (= background spots) FISH “spots” and to explore antibody effects on the gene domain size (Fig. 3 A). Additionally, also unsynchronized stimulated cells were used to demonstrate the capacity of the instrument to measure highly differently sized gene domains within the same nucleus as being expected in S- or G2-phase (Fig. 4 A).
TABLE 1.
Summary of SMI gene domain measurements
| Gene locus | Number of loci analyzed | Probe length (kb) | Mean diameter ± SD (nm) | Spherical volume (10−4μm3) | Compaction factor |
|---|---|---|---|---|---|
| p53 | 41 | 45 | 119 ± 14 | 8.9 | 1: 129 |
| p58 | 28 | 85 | 123 ± 10 | 9.7 | 1: 235 |
| c-myc | 28 | 120 | 103 ± 12 | 5.7 | 1: 396 |
The sizes (diameter in the axial direction) of the green (FITC) FISH “spots” were usually considered for further statistical evaluation. In the case of two color labeling, both colocalizing fluorescence signals (FITC, green; Alexa 647, red) were independently evaluated for each gene domain. Although additional antibodies were used for two color labeling, the mean spot sizes did not differ significantly (green, 109 ± 16 nm; red, 109 ± 23 nm). Nevertheless concerning an individual gene domain, both types of results were possible, e.g., coincidence (green, 106 nm; red, 108 nm) and difference (green, 106 nm; red, 130, nm).
Between 28 and 41 individual domains were recorded per gene. Figs. 3 and 4 show typical examples of a cell nucleus with the p53 or p58 gene domain labeled. Highly (Figs. 3, Ba and b, and 4 Ba) and lowly modulating (Fig. 4, Bb and c) curves were obtained depending on the signal/noise ratio and the spot size. From the green-labeled sites, the MC was determined and rescaled into nanometer size according to the calibration curve in Fig. 2. The mean values of the diameter were calculated to 103 nm (c-myc), 119 nm (p53), and 123 nm (p58). The data show remarkably small standard deviations of 10–14 nm which is compatible to the size of a few nucleosomes only.
Assuming spherical domain shapes, the volumes were estimated from the SMI data to be 5.7 × 10−4 μm3, 8.9 × 10−4 μm3 and 9.7 × 10−4 μm3 (Table 1), which is ∼2 orders of magnitude smaller than the observation volume of ∼0.15 μm3 (given by the ellipsoidal full width at half maximum of the 3D-PSF) (Bornfleth et al., 1998) in a confocal laser scanning microscope using a high numerical aperture objective lens. Furthermore, if in a confocal laser scanning microscope one would estimate the size of the gene domains (in a first coarse approximation supposed to be a spherical object) by means of their apparent lateral extension only (at least 250 nm), the SMI volume estimate is still smaller by an order of magnitude. It may be noted that using these figures to estimate the total volume of the ∼2 × 30,000 gene domains in a diploid nucleus, values in the order of ∼4,000–9,000 μm3 would be obtained using a spherical approximation of the confocal observation volume as domain size. Using the SMI volume estimate, and assuming all gene domains of similar size, a total volume of only 34–58 μm3 would result. Although the first (confocal) values exceed by far the nuclear volume available in normal human somatic cells (∼500 μm3), the second (SMI) values would be fitting even for small nuclei such as in cells of the retina.
So far no direct correlation between the genomic size (i.e., the probe size) and the geometric size (i.e., diameter or volume) was found for the gene domains studied. From the methodological point of view, this may be interpreted to reflect no or only slight variations in structure of the labeled gene domains by the FISH probes in the sequence length range studied. Otherwise if probe binding would have a dominant influence on the gene domain size, a direct genomic/geometric size correlation would be expected. From the biological point of view, this supports the finding (Solovei et al., 2002a), that disrupting effects of fixation and denaturing FISH procedures do not induce major effects on gene domain sizes beyond a scale of ∼100 nm. Moreover, the comparison of the p53 and p58 data may contradict to a possible reduction of the z dimension for larger objects (dimensions in the order of 100 nm) by methanol/acetic acid fixation.
The mean chromatin compaction was estimated from the measured diameter and the probe size. With 1 kb linear DNA having a length of 340 nm, chromatin compaction factors between 1:129 (p53), 1:235 (p58), and 1:396 (c-myc) were found.
The results presented here show that SMI microscopy (Schneider et al., 1999; Albrecht et al., 2002) is a powerful tool to measure nano-sizes by far field light microscopy and hence to estimate volumes of individual compact fluorescence objects (Failla et al., 2002c) in the hundred and subhundred nanometer range with high precision and reproducibility. In contrast to scanning probe techniques (Horber and Miles, 2003; Richards, 2003) this can be done inside 3D objects like cell nuclei in a noninvasive way. So far the MC of several specific gene domains were measured and rescaled into size values by using one theoretically calculated calibration curve. The precision of the size measurements may, however, be further increased if variations of individual specimen conditions could be considered. This may for instance be possible, if one would add particles of exactly known size to the specimen as an internal reference standard, which can be measured to select the most appropriate theoretical calibration curve (Wagner et al., 2005).
One may argue that with a precision of 10 nm corresponding to the size of one nucleosome, the fixation step as well as FISH labeling would have a deep influence on the measurement. Methanol/acetic acid fixation may distore nuclear structures because the cell nuclei are flat and the lateral dimensions are increased. Thermal denaturation during the FISH procedure and the probe may increase the gene domain size because of swelling up the gene. To overcome this problem, COMBO-FISH (COMbinatorial Oligo FISH) (Hausmann et al., 2003) is presently under development. This technique makes use of the specific colocalization of some 10 fluorescence labeled oligomeres of only 15–30 nucleotides in a given gene domain. These oligomeres are able to form a triple DNA strand so that denaturation of the target DNA strand is negligible. Moreover the applicability to vital cells is possible without any fixation. Due to the smallness of the oligomeres and their binding mechanism it can be assumed that this technique may be a further improvement for gene domain size measurements as preliminary results indicate (data not shown).
Another methodological aspect may be an incomplete probe attachment during FISH. This may cause different sizes of the same gene domain and partly reason the variability of the individual measurements. This may be overcome by increasing the number of nuclei analyzed or by COMBO-FISH with oligo-probes of different colors that may allow a spectral control of the completeness of the label.
The general goal of this article was to demonstrate the methodological power of SMI microscopy for nano-sizing of individual “native” gene domains in 3D human cell nuclei. Nevertheless, these data show that chromatin compaction on the gene level is subjected to a large variability which might be correlating to the gene activity or accessibility for macromolecule complexes, as for instance “transcription factories” (Martin et al., 2004). So far only the methodological progress is obvious. However, discriminating gene domains according to their nano-size and correlating it to their potential transcriptional activity (Failla et al., 2002b) may be a future procedure also for routine applications in molecular tumor diagnostics and early prognostic which is still a challenging task for molecular pathology (Stankiewicz and Lupski, 2002). Additionally, the technique is not limited to the analysis of nucleic acids via FISH labeling, but is also capable to analyze the size of protein complexes after immunohistochemical staining.
Acknowledgments
The authors thank Heike Dittmar, IMB, Jena, for technical assistance and Dr. Jutta Finsterle, KIP, Heidelberg, for her continuous support and many fruitful discussions.
The financial supports of the German Federal Ministry of Education and Research (BMBF), of the Deutsche Forschungsgemeinschaft (DFG), and of the European Union (EU) are gratefully acknowledged. G.H. got a scholarship of the Graduiertenkolleg “Tumordiagnostik und -therapie unter Einsatz dreidimensionaler radiologischer und lasermedizinischer Verfahren” (DFG).
References
- Albrecht, B., A. V. Failla, A. Schweitzer, and C. Cremer. 2002. Spatially modulated illumination microscopy allows axial distance resolution in the nanometer range. Appl. Opt. 41:80–87. [DOI] [PubMed] [Google Scholar]
- Amrichova, J., E. Lukasova, S. Kozubek, and M. Kozubek. 2003. Nuclear and territorial topography of chromosome telomeres in human lymphocytes. Exp. Cell Res. 289:11–26. [DOI] [PubMed] [Google Scholar]
- Bailey, B., D. L. Farkas, D. L. Taylor, and F. Lanni. 1993. Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature. 366:44–48. [DOI] [PubMed] [Google Scholar]
- Bartova, E., S. Kozubek, P. Jirsova, M. Kozubek, H. Gajova, E. Lukasova, M. Skalnikova, A. Ganova, I. Koutna, and M. Hausmann. 2002. Nuclear topography and gene activity in human differentiated cells. J. Struct. Biol. 139:76–89. [DOI] [PubMed] [Google Scholar]
- Bornfleth, H., K. Sätzler, R. Eils, and C. Cremer. 1998. High-precision distance measurements and volume-conserving segmentation of objects near and below the resolution limit in three-dimensional confocal microscopy. J. Microsc. 189:118–136. [Google Scholar]
- Bubulya, P. A., and D. L. Spector. 2004. “On the move”ments of nuclear components in living cells. Exp. Cell Res. 296:4–11. [DOI] [PubMed] [Google Scholar]
- Cremer, M., J. von Hase, T. Volm, A. Brero, G. Kreth, J. Walter, C. Fischer, I. Solovei, C. Cremer, and T. Cremer. 2001. Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome Res. 9:541–567. [DOI] [PubMed] [Google Scholar]
- Cremer, M., K. Küpper, B. Wagler, L. Wizelman, J. von Hase, Y. Weiland, L. Kreja, J. Diebold, M. R. Speicher, and T. Cremer. 2003. Inheritance of gene density-related higher order chromatin arrangements in normal and tumor cell nuclei. J. Cell Biol. 162:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer, T., and C. Cremer. 2001. Chromosome territories, nuclear architecture and gene regulatin in mammalian cells. Nat. Rev. Genet. 2:292–301. [DOI] [PubMed] [Google Scholar]
- Cremer, T., G. Kreth, H. Koester, R. H. A. Fink, R. Heintzmann, M. Cremer, I. Solovei, D. Zink, and C. Cremer. 2000. Chromosome territories, inter chromatin domain compartment and nuclear matrix: an integrated view on the functional nuclear architecture. Crit. Rev. Eukaryot. Gene Expr. 10:179–212. [PubMed] [Google Scholar]
- Dundr, M., and T. Misteli. 2001. Functional architecture in the cell nucleus. Biochem. J. 356:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann, P., A. Esa, M. Hausmann, and C. Cremer. 1999. Confocal laser-scanning fluorescence microscopy: In situ determination of the confocal point-spread function and the chromatic shifts in intact cell nuclei. Optik. 110:194–198. [Google Scholar]
- Esa, A., A. E. Coleman, P. Edelmann, S. Silva, C. Cremer, and S. Janz. 2001. Conformational differences in the 3d nanostructure of the immunglobulin heavy-chain locus, a hotspot of chromosomal translocations in B-lymphocytes. Cancer Genet. Cytogenet. 127:168–173. [DOI] [PubMed] [Google Scholar]
- Esa, A., P. Edelmann, L. Trakhtenbrot, N. Amariglio, G. Rechavi, M. Hausmann, and C. Cremer. 2000. 3D-Spectral Precision Distance Microscopy (SPDM) of chromatin nanostructures after triple-colour DNA labelling: A study of the BCR-region on chromosome 22 and the Philadelphia chromosome. J. Microsc. 199:96–105. [DOI] [PubMed] [Google Scholar]
- Failla, A. V., A. Cavallo, and C. Cremer. 2002a. Subwavelength size determination by spatially modulated illumination virtual microscopy. Appl. Opt. 41:6651–6659. [DOI] [PubMed] [Google Scholar]
- Failla, A. V., B. Albrecht, U. Spoeri, A. Schweitzer, A. Kroll, M. Bach, and C. Cremer. 2002b. Nanotopology analysis using spatially modulated illumination (SMI) microscopy. Complexus. 1:29–40. [Google Scholar]
- Failla, A. V., U. Spoeri, B. Albrecht, A. Kroll, and C. Cremer. 2002c. Nanosizing of fluorescent objects by spatially modulated illumination microscopy. Appl. Opt. 41:7275–7283. [DOI] [PubMed] [Google Scholar]
- Freimann, R., S. Pentz, and H. Horler. 1997. Development of a standing-wave fluorescence microscope with high nodal plane flatness. J. Microsc. 187:193–200. [DOI] [PubMed] [Google Scholar]
- Hausmann, M., R. Winkler, G. Hildenbrand, J. Finsterle, A. Weisel, A. Rapp, E. Schmitt, S. Janz, and C. Cremer. 2003. COMBO-FISH: specific labeling of nondenatured chromatin targets by computer-selected DNA oligonucleotide probe combinations. Biotechniques. 35:564–577. [DOI] [PubMed] [Google Scholar]
- Hell, S. W. 2003. Toward fluorescence nanoscopy. Nat. Biotechnol. 21:1347–1355. [DOI] [PubMed] [Google Scholar]
- Hell, S. W., and E. H. K. Stelzer. 1992. Properties of a 4Pi confocal fluorescence microscope. J. Opt. Soc. Am. A. 9:2159–2166. [Google Scholar]
- Hell, S. W., and J. Wichmann. 1994. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19:780–782. [DOI] [PubMed] [Google Scholar]
- Horber, J. K., and M. J. Miles. 2003. Scanning probe evolution in biology. Science. 302:1002–1005. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman, E. J., and T. M. Jovin. 2003. FRET imaging. Nat. Biotechnol. 11:1387–1396. [DOI] [PubMed] [Google Scholar]
- Kano, H., S. Jakobs, M. Nagorni, and S. W. Hell. 2001. Dual-color 4Pi-confocal microscopy with 3D-resolution in the 100 nm range. Ultramicroscopy. 90:207–213. [DOI] [PubMed] [Google Scholar]
- Kepert, J. F., K. F. Toth, M. Caudron, N. Mucke, J. Langowski, and K. Rippe. 2003. Conformation of reconstituted mononucleosomes and effect of linker histone H1 binding studied by scanning force microscopy. Biophys. J. 85:4012–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozubek, S., E. Lukasova, P. Jirsova, I. Koutna, M. Kozubek, A. Ganova, E. Bartova, M. Falk, and R. Pasekova. 2002. 3D Structure of the human genome: order in randomness. Chromosoma. 111:321–331. [DOI] [PubMed] [Google Scholar]
- Martin, S., A. V. Failla, U. Spoeri, C. Cremer, and A. Pombo. 2004. Measuring the size of biological nanostructures with Spatially Modulated Illumination Microscopy. Mol. Biol. Cell. 15:2449–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawley, J. 1995. Handbook of Biological Confocal Microscopy. Plenum Press, New York.
- Perner, B., A. Rapp, C. Dressler, L. Wollweber, J. Beuthan, K. O. Greulich, and M. Hausmann. 2002. Variations in cell surfaces of estrogen treated breast cancer cells detected by a combined instrument for far-field and near-field microscopy. Anal. Cell. Pathol. 24:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, D. 2003. Near-field microscopy: throwing light into the nanoworld. Philos. Transact. Ser. A Math. Phys. Eng. Sci. 361:2843–2857. [DOI] [PubMed] [Google Scholar]
- Schneider, B., I. Upmann, I. Kirsten, J. Bradl, M. Hausmann, and C. Cremer. 1999. A dual-laser, spatially modulated illumination fluorescence microscope. Eur. Microsc. Anal. 57:5–7. [Google Scholar]
- Solovei, I., A. Cavallo, L. Schermelleh, F. Jaunin, C. Scasselati, D. Cmarko, C. Cremer, S. Fakan, and T. Cremer. 2002a. Spatial preservation of nuclear chromatin architecture during three-dimensional fluorescence in situ hybridization (3D-FISH). Exp. Cell Res. 276:10–23. [DOI] [PubMed] [Google Scholar]
- Solovei, I., J. Walter, M. Cremer, F. Habermann, L. Schermelleh, and T. Cremer. 2002b. FISH on three-dimensionally preserved nuclei. In FISH: A Practical Approach. J. Squire, B. Beatty, and S. Mai, editors. Oxford University Press, Oxford. 119 – 157.
- Spector, D. L. 2003. The dynamics of chromosome organization and gene regulation. Annu. Rev. Biochem. 72:573–608. [DOI] [PubMed] [Google Scholar]
- Stankiewicz, P., and J. R. Lupski. 2002. Genome architecture, rearrangements and genomic disorders. TIG. (Trends Genet). 18:74–82. [DOI] [PubMed] [Google Scholar]
- Stelzer, E. H. K. 1998. Contrast, resolution, pixelation, dynamic range and signal-to-noise ratio: fundamental limits to resolution in fluorescence microscopy. J. Microsc. 189:15–24. [Google Scholar]
- Tsukamoto, T., N. Hashiguchi, S. M. Janicki, T. Tumbar, A. S. Belmont, and D. L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871–878. [DOI] [PubMed] [Google Scholar]
- Tumbar, T., and A. S. Belmont. 2001. Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat. Cell Biol. 3:134–139. [DOI] [PubMed] [Google Scholar]
- van Driel, R., P. F. Fransz, and P. J. Verschure. 2003. The eukaryotic genome: a system regulated at different hierarchical levels. J. Cell Sci. 116:4067–4075. [DOI] [PubMed] [Google Scholar]
- Wagner, C., U. Spöri, and C. Cremer. 2005. High-precision SMI microscopy size measurements by simultaneous frequency domain reconstruction of the axial point spread function. Optik. 116:15–21. [Google Scholar]
- Winkler, R., B. Perner, A. Rapp, M. Durm, C. Cremer, K. O. Greulich, and M. Hausmann. 2003. Labeling quality and chromosome morphology after low temperature FISH analyzed by scanning far-field and scanning near-field optical microscopy. J. Microsc. 209:23–33. [DOI] [PubMed] [Google Scholar]
- Zink, D., and T. Cremer. 1998. Cell nucleus: chromosome dynamics in nuclei of living cells. Curr. Biol. 8:R321–R324. [DOI] [PubMed] [Google Scholar]