

Abstract
Fluorescence correlation spectroscopy is a potentially powerful tool for measuring protein-protein interactions directly in single living cells. We previously reported on the detection of homodimer formation in cells using molecular brightness analysis. Here, we extend the technique to detect binding between different proteins. Proteins are labeled with the fluorescent markers YFP and CFP. We first determine the coexpression ratio of both proteins by measuring the intensity ratio with a dual-color setup. The effect of fluorescence resonance energy transfer on the intensity ratio is explicitly taken into account. The brightness of cells coexpressing both proteins is measured in a single-color setup. Selecting the laser wavelength of the two-photon light source allows us to either coexcite both proteins or to selectively excite YFP-labeled proteins. This approach enables us to distinguish between homodimer and heterodimer formation. We first present the theory and then demonstrate experimental feasibility using the ligand binding domains of retinoic acid receptor (RARLBD) and of retinoid X receptor (RXRLBD). Both proteins form heterodimers, and RXRLBD also forms homodimers in the presence of its agonist. We explore binding between these proteins in the presence and absence of RXR agonist. Our results demonstrate that brightness analysis offers a quantitative method for determining protein interactions in cells.
INTRODUCTION
Two-photon fluorescence fluctuation experiments are very attractive for in vivo applications. The small optical observation volume allows our probing any location within a cell with submicron resolution (Denk et al., 1990). Fluorescently labeled proteins diffusing through the observation volume give rise to spontaneous signal fluctuations that provide dynamic and static information about the system without the need for external perturbation. The cellular environment is much more complex than test-tube conditions. Autofluorescence, spatial heterogeneity, and other factors complicate fluorescence fluctuation experiments in living cells. Nevertheless, it has been shown that fluctuation correlation spectroscopy measurements in living cells are quite feasible (Berland, 1995; Brock et al., 1998; Politz et al., 1998). We successfully demonstrated molecular brightness measurements of EGFP in cells using photon counting histogram (PCH) analysis (Chen et al., 2002). Molecular brightness characterizes the average detected photon count rate of a fluorophore. Its value depends on instrumental parameters, such as the excitation wavelength and the quantum yield of the detector. If instrumental parameters are kept constant, molecular brightness characterizes a photophysical property of the fluorescent molecule.
Molecular brightness is a useful marker for monitoring protein association. If a fluorescently labeled protein diffuses through the observation volume, it will produce a burst of detected photons. The average photon count rate of these bursts determines the molecular brightness of the labeled protein. If such a protein associates to form a homodimer, the new complex will carry two fluorescent labels. Diffusion of the complex through the observation volume will produce, on average, twice as many photons than is the case for the monomeric protein, because two independently fluorescing molecules are participating. Consequently, the molecular brightness of the dimer is twice that of the monomer (Müller et al., 2000). To measure over wide concentration ranges as frequently encountered in cells, a new PCH model was developed that takes nonideal detector effects into account (Hillesheim and Muller, 2003). Using this new theory, we recently demonstrated that brightness analysis provides a quantitative approach to study homodimer formation in cells (Chen et al., 2003).
Here, we expand brightness analysis to the case of binary protein mixtures to quantify hetero- and homointeractions between them. The relative ratio of expressed proteins varies from cell to cell, and a method that determines the coexpression ratio is needed. Each protein species is marked with its own fluorescent marker. We use the fluorescent proteins CFP and YFP in this study. The spectral difference in their fluorescence emission is used to determine the coexpression ratio of both proteins from a dual-color intensity ratio measurement and the independently determined brightness of the fluorescent markers. In addition, we have to account for energy transfer between CFP and YFP. We use fluorescence lifetime measurements of the donor to characterize an apparent fluorescence resonance energy transfer (FRET) efficiency, which is used to connect the experimentally measured intensity ratio with the coexpression ratio.
Once the coexpression ratio of a cell is established, we switch from the dual-channel setup to a single-channel brightness experiment and perform PCH analysis to determine the molecular brightness of protein complexes. We developed a theory to describe the influence of FRET on brightness analysis. By choosing the right excitation conditions we are able to eliminate the influence of FRET on the brightness of a heterodimer that contains a donor-acceptor pair. We choose excitation conditions where the brightness of CFP and YFP are virtually the same. Thus, a heterodimer will appear twice as bright as the corresponding monomers. This allows us to take advantage of our existing single-channel PCH theory to probe the interactions between the proteins. In other words, we do not separate species by color, but solely by their brightness difference. To distinguish homo- from heterodimers, the excitation wavelength is changed to make CFP “dark,” so that we monitor the molecular brightness of YFP fusion proteins only.
We choose the ligand-binding domain of the two nuclear receptors, retinoid X receptor (RXR) and retinoic acid receptor (RAR), as our model system. We previously examined the behavior of each of these proteins in cells with brightness analysis (Chen et al., 2003). Because RXRLBD is able to form a heterodimer with RARLBD and also a homodimer with itself, it is a suitable system to test our technique. Homodimer formation depends on the presence of ligand and offers an additional mechanism to control the composition of the mixture. We present two-photon fluorescence fluctuation experiments of cells expressing both proteins and demonstrate that RXRLBD forms a very tight heterodimer with RARLBD. Homodimer formation of RXRLBD requires the presence of ligand and is less tight than the heterodimer. Consequently, we only observed homodimers when the concentration of RXRLBD exceeds the concentration of RARLBD. Our results demonstrate the potential of brightness analysis for quantitative characterization of complex protein interactions in cells.
THEORY
The average fluorescence intensity 〈F〉 is given by
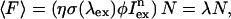 |
(1) |
with the absorption cross-section σ(λex) at the excitation wavelength λex, the fluorescence quantum yield φ, the excitation intensity Iex at the sample, and the number of molecules N in the observation volume. The factor η takes the detection efficiency of the optics and the detector into account. The factor n equals 1 for one-photon excitation and is 2 for two-photon excitation. The product of the factors within the bracket describes the photon count rate of a single molecule (Müller, 2004). The molecular brightness ɛ is given by the product of the photon count rate and the sampling time at the detector,
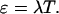 |
(2) |
Equation 2 is valid if the diffusion time is much shorter than the sampling time T.
Our dual-channel detection path contains a dichroic that splits the fluorescence into two detection channels according to color. We label the detection channel as red and green. The subscripts (r) and (g) are used to denote properties of the red and green channel, respectively. If no photons are absorbed by the dichroic mirror the single-channel photon count rate is simply the sum of the photon count rates in each detection channel,
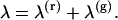 |
(3) |
Measuring the intensity ratio of a fluorescent dye determines its brightness and photon count ratio,
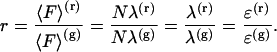 |
(4) |
This allows us to determine the brightness ratio rCFP of CFP and the brightness ratio rYFP of YFP, which characterizes the relative brightness of the proteins with the two-channels setup.
Now, let us consider a mixture of two proteins, A and B, which are noninteracting. Protein A and B are labeled with CFP and YFP, respectively. If the two proteins do not interact, then the intensity ratio of the two detection channels rF = 〈F〉(r)/〈F〉(g) is given by
 |
(5) |
where the parameter rλ = λYFP/λCFP is the brightness ratio between YFP and CFP, and rN = NA,total/NB,total is the total number of protein A over protein B, which is also the ratio of the total number of YFP over CFP molecules. Experimentally, rλ, rF, rYFP, and rCFP can be determined independently, and rN is calculated from Eq. 5.
For interacting proteins one has to take fluorescence resonance energy transfer (FRET) into account. If FRET is present, the donor fluorescence will quench, and the acceptor fluorescence will enhance. Consequently, the intensity ratio, rF, will be influenced by the presence of energy transfer. Let 〈FD〉 be the fluorescence intensity of a donor in the absence of FRET. Energy transfer to an acceptor molecule leads to a reduction of the fluorescence  of the donor,
of the donor,
 |
(6) |
where E is the efficiency of the energy transfer. Consequently, the photon count rate of a single donor molecule 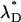 in the presence of FRET is related to the photon count rate of the donor molecule λD in the absence of FRET by
in the presence of FRET is related to the photon count rate of the donor molecule λD in the absence of FRET by
 |
(7) |
The fluorescence of the acceptor molecule 〈FA〉 without FRET is given by
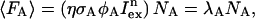 |
(8) |
with λA as the photon count rate of the acceptor.
The presence of FRET in an acceptor-donor complex leads to an enhancement of the fluorescence 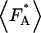 of the acceptor molecule,
of the acceptor molecule,
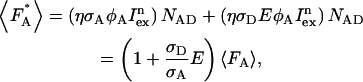 |
(9) |
where NAD equals the number of acceptor-donor complexes. Because the fluorescence intensity is related to the photon count rate of a single molecule by 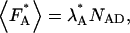 the photon count rate of the acceptor molecule in the presence of FRET is given by
the photon count rate of the acceptor molecule in the presence of FRET is given by
 |
(10) |
In addition to the efficiency E of energy transfer one needs to consider the ratio of the excitation cross sections of the donor and acceptor molecule at the excitation wavelength. Equations 7 and 10 describe the influence of FRET on the photon count rate of acceptor and donor molecules. Multiplying both equations with the sampling time T describes, according to Eq. 2, the influence of FRET on the brightness of the donor and acceptor. Usually we will detect fluorescence from both the acceptor and the donor in the detection channel, and we have to determine the brightness of the protein complex. The photon count rate of a heterodimer containing an acceptor-donor pair is thus given by
 |
(11) |
For example, if the donor and acceptor molecule are identical, such as is the case for a homodimer, the brightness of the dimer is twice the brightness of the monomer,  independently of homo-energy transfer occurring between the identical fluorophores.
independently of homo-energy transfer occurring between the identical fluorophores.
We now specifically consider the CFP-YFP fluorescence pair. CFP serves as the donor and YFP is the acceptor. The two-photon cross sections of both fluorescent proteins are at an excitation wavelength of 905 nm identical (Zipfel et al., 2003), σD(905 nm)/σA(905 nm) ≈ 1. In addition, the photon count rate of CFP and YFP measured at this excitation wavelength in our single-channel setup is identical within 10%, λA ≈ λD. For this special case FRET is not affecting the brightness of protein complexes containing both CFP and YFP. For example, a heterodimer that carries a CFP and YFP label leads to a photon count rate of 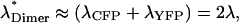 which is independent of FRET. We use λ to indicate the photon count rate of a single fluorescent protein.
which is independent of FRET. We use λ to indicate the photon count rate of a single fluorescent protein.
Let us consider a mixture of two monomers (A and B) and their dimer AB. Each monomer of A carries a CFP molecule and each monomer B carries a YFP label. The FRET efficiency between the donor and acceptor of the heterodimer is E. The fluorescence intensity in detection channel (i) of such a mixture is given by
 |
(12) |
Using photon count rates, Eq. 12 becomes
 |
(13) |
where we used the fact that σCFP/σYFP ≈ 1 for excitation at 905 nm. The total number of proteins A is NA,total = NA + NAB, and the total number of proteins B is similarly given by NB,total = NB + NAB. We define the degree of dimerization by
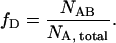 |
(14) |
We now rewrite the intensity ratio rF = 〈F〉(r)/〈F〉(g), which was introduced in Eq. 5 for noninteracting proteins, for the case of interacting proteins,
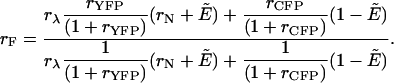 |
(15) |
We introduced the apparent FRET efficiency  which is related to the FRET efficiency E of the dimer by
which is related to the FRET efficiency E of the dimer by 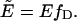 Solving Eq. 15 for rN yields
Solving Eq. 15 for rN yields
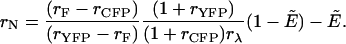 |
(16) |
The only parameter unknown in the above equation is the apparent FRET efficiency, which will be determined from fluorescence lifetime measurements.
The fluorescence decay of the donor in the absence of FRET, FD(t) = αD exp(−t/τD), changes to FD*(t) = αD exp(−t/τD*) in the presence of FRET. For a multi-exponential decay process, 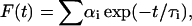 we use the average lifetime 〈τ〉 defined by
we use the average lifetime 〈τ〉 defined by
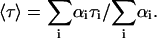 |
(17) |
This definition of the average lifetime is proportional to the average fluorescence intensity 〈F〉. The FRET efficiency is usually determined from measurements of the donor fluorescence or from its lifetime in the presence and absence of FRET (Lakowicz, 1999),
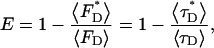 |
(18) |
where  is the fluorescence lifetime in the presence of FRET and 〈τD〉 is the lifetime in the absence of FRET. We observe fluorescence from a mixture of monomers A without FRET and dimers AB that undergo FRET. The fluorescence intensity is given by
is the fluorescence lifetime in the presence of FRET and 〈τD〉 is the lifetime in the absence of FRET. We observe fluorescence from a mixture of monomers A without FRET and dimers AB that undergo FRET. The fluorescence intensity is given by  which leads to an apparent FRET efficiency of
which leads to an apparent FRET efficiency of
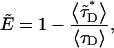 |
(19) |
where 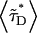 is the average lifetime of the mixture,
is the average lifetime of the mixture,
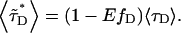 |
(20) |
We previously developed an improved PCH theory to examine protein interactions in living cells over a wide concentration range (Chen et al., 2003). The low laser power used in cellular applications and the high concentration of proteins typically encountered leads to a signal/noise ratio that does not allow the direct resolution of a monomer/dimer equilibrium. Instead, we measure an apparent brightness that contains contributions of the brightnesses of the monomer and the dimer. The apparent molecular brightness was determined by analyzing data using PCH and moment analysis (Chen et al., 1999; Qian and Elson, 1990). For a mixture of species the apparent brightness is given by a nonlinear combination of the brightness ɛi and the occupation number Ni of each species (Chen et al., 2003),
 |
(21) |
The apparent brightness of a monomer/dimer mixture will lie between the brightness values of the monomer and dimer,
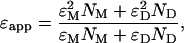 |
(22) |
where ɛM and ɛD are the brightness values, and NM and ND are the occupation numbers of the monomer and dimer, respectively. Because both CFP and YFP have virtually identical brightness values at an excitation wavelength of 905 nm in our single-channel setup, all monomeric proteins carry the brightness ɛM and all dimers have a brightness of ɛD = 2ɛM. This means that we cannot distinguish between proteins labeled with CFP and YFP by brightness analysis, nor can we distinguish between a homodimer and a heterodimer. We perform an additional measurement at a different excitation wavelength where only the acceptor YFP is excited in order to distinguish homo- from heterodimers.
METHODS
Experimental setup
A mode-locked Ti:Sapphire laser (Tsunami, Spectra Physics, Mountain View, CA) pumped by an intracavity doubled Nd:YVO4 laser (Spectra Physics) serves as source for two-photon excitation. The laser produces 100-fs pulses with a repetition frequency of 80 MHz (tunable between 700 and 1000 nm). The experiments were carried out using a Zeiss Axiovert 200 microscope (Carl Zeiss, Thornwood, NY) with a 63× Plan Apochromat oil immersion objective (NA = 1.4). The power at the sample was determined by measuring the laser power directly after the objective. We used a power of 0.5 mW at the sample for the cell measurements at all wavelengths. A modified microscope's turret was used for all cellular measurements. In addition to the filter cube used for the fluorescent light path, another filter cube with a dichroic (Chroma Technology, Brattleboro, VT) for two-photon excitation was added, rotated by 90° with respect to the original cube. This arrangement allows the laser beam to enter the microscope turret from the side, while at the same time preserving the fluorescence microscope capabilities of the instrument. We used the fluorescence path of the microscope to locate and position cells and subsequently switched to two-photon microscopy for fluctuation experiments. Cells are mounted on an electronic stage (MS-2000 XYZ, ASI, Eugene, OR). The location of each measurement within cells is recorded from the electronic readout of the x,y position of the stage.
The fluorescence is sent to the bottom port for intensity ratiometric measurements. A dichroic mirror with a center wavelength of 525 nm (525DCXRU, Chroma Technology) splits the fluorescence into two detection channels and the intensities of the dual-color experiment are recorded by two avalanche photo diodes (APD) (Model SPCM-AQR-14, Perkin-Elmer, Vaudreuil, Canada). Single-color brightness measurements are performed by sending the fluorescence to the side port. The fluorescence is recorded by another APD (Model SPCM-AQR-14). The data acquisition time for measurements of the intensity ratio is on the order of 5 s, whereas brightness measurements require on the order of 50 s. The TTL-output of each APD unit is connected to a PCI data acquisition card (ISS, Champaign, IL), which stores the complete sequence of photon counts using a sampling frequency of 20 kHz. The recorded photon counts were stored and analyzed with programs written for IDL 5.4 (Research Systems, Boulder, CO).
A time-correlated single photon counting module was used to measure fluorescence lifetimes (TimeHarp 200, Picoquant, Germany). Monitoring the laser pulses with a photodiode (DET210, Thorlabs, Newton, NJ) provides the synchronization signal. The fluorescence signal was monitored with a PMT (H7421-40, Hamamatsu, Japan) under magic-angle conditions. A bandpass filter (FF495-EX01-25, Semrock, Rochester, NY) was placed in front of the PMT to block the light emitted from YFP. The fluorescence fluctuation lifetime data are analyzed with GLOBALS Unlimited (Urbana, IL).
Expression vectors and cell measurements
RXRLBD-YFP, RARLBD-CFP, and RARLBD-YFP plasmids are subcloned from existing RXRLBD-EGFP and RARLBD-EGFP plasmids (Chen et al., 2003) using pECFP-C1 and pEYFP-C1 (Clontech, Palo Alto, CA). CFP-RARLBD-YFP was constructed by inserting a PCR-amplified YFP fragment into RARLBD YFP SacII and BamHI sites. All sequences were verified by automatic sequencing. CV-1 cells were obtained from ATCC (Manassas, VA) and maintained in 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and EMEM media. Transfections were carried out by using transfectin (Bio-Rad, Hercules, CA) according to manufacturer's instructions. The RXR agonist used in this study is AGN 194209 (Farooqui et al., 2003).
Cells were subcultured into eight-well coverglass chamber slides (Naglenunc International, Rochester, NY). Before conducting measurements, the growth media was exchanged to Dulbecco's phosphate-buffered saline with calcium and magnesium (Biowhittaker, Walkersville, MD). RXR agonist was added to the media at 3-μM concentration. Fluorescence fluctuation spectroscopy measurements were performed 10 min after the addition of ligand. To estimate the uncertainty in measuring the brightness in cells, we divided the data set of a single measurement into shorter segments, calculated the brightness of each segment, and determined the standard deviation in brightness of one segment. We estimate the experimental error in brightness for the complete data set by dividing the standard deviation by the square-root of the number of segments. This approach assumes statistical independence between individual segments. We checked many of our cell measurements and consistently recovered errors between 4% and 8%.
Control experiments
Photobleaching and saturation of the fluorophores would complicate data analysis of fluorescence fluctuation experiments. We confirmed that, for the power range used in this study, photobleaching and saturation are absent by performing control experiments as previously described (Chen et al., 2003). Before starting the actual experiments, we always perform a brightness calibration by measuring cells that express RARLBD-YFP, RARLBD-CFP, and CFP-RARLBD-YFP, because we previously showed that RARLBD remains monomeric in cells. Measurements of RARLBD-YFP and RARLBD-CFP serve to establish the brightness ɛMonomer of a monomer, and measurements on CFP-RARLBD-YFP were performed to check that the total brightness of the two fluorophores is additive. For experiments with an excitation wavelength of 965 nm, where CFP is dark, we used RARLBD-YFP and CFP-RARLBD-YFP to calibrate the brightness of the monomer. The total concentration of the expressed proteins in cells was determined using the brightness of a monomer and the total measured intensity as previously described (Chen et al., 2003). At 905 nm we determine the total protein concentration of RXRLBD-YFP and RARLBD-CFP. At 965 nm we only determine the protein concentration of RXRLBD-YFP, because CFP is dark.
RESULTS
Protein coexpression ratio from dual-color intensity measurements
The absolute concentration of an expressed protein varies widely from cell to cell after transient transfection. Since the expression level of proteins is not directly under our experimental control, it is necessary to determine the expressed protein ratio in each cell from experiment. This is especially important for single-channel brightness analysis, where knowledge of the expressed protein ratio provides an important constraint for the analysis and interpretation of data. In this study CFP and YFP serve as fluorescent tags to uniquely mark each protein species. Dual-color detection is used to determine the expression ratio of the two fusion proteins from the intensity ratio of the two detection channels. For the moment we restrict ourselves to the case of two noninteracting protein species. In other words, there is no energy transfer between the fluorescent proteins. We excite CFP and YFP at 905 nm and split the fluorescence emission into two channels with a dichroic filter. The emission spectra of CFP and YFP strongly overlap, and each channel contains signal contributions from both proteins. We therefore first determine the intensity ratio (see Eq. 4) for each protein by measuring cells transfected with either CFP or YFP fusion protein. We determined intensity ratios of rCFP = 0.4 and rYFP = 2.3 for CFP and YFP, respectively. These intensity ratios are not sufficient to determine the coexpression ratio. The intensity ratio of a mixture depends also on the brightness of the individual proteins. A brighter protein will contribute more to the intensity ratio of the mixture than a dimmer protein. We measured the brightness of CFP and YFP in 10 cells each using our single-color setup and calculated an averaged brightness ratio of rλ = 1.1 between YFP and CFP.
If protein A is labeled with CFP and protein B is labeled with YFP, then the coexpression ratio rN = NA,total/NB,total of the two proteins equals the ratio rN = NYFP,total/NCFP,total of the total number of CFP and YFP proteins. The solid line in Fig. 1 is a theoretical curve of the intensity ratio as a function of protein coexpression ratio calculated from Eq. 5 based on the experimental parameters determined for CFP and YFP. Thus, an intensity ratio of rF = 1.0 corresponds to a 1:1 coexpression of both proteins in the absence of FRET.
FIGURE 1.

Intensity ratio rF as a function of the coexpression ratio rN = NYFP/NCFP. The solid line represents the relationship between the intensity ratio and the coexpression ratio in the absence of FRET. The dashed line was calculated assuming a stoichiometric binding model and a FRET efficiency of 27%. An intensity ratio of 1.25 (dotted arrow) leads to a coexpression ratio of ∼2 (solid arrow) in the absence of FRET, while representing equimolar expression in the presence of FRET (dashed arrow). Each shaded bar indicates the range of intensity ratios selected for further brightness analysis in our experiments.
Protein coexpression ratio in the presence of FRET
We now determine the coexpression ratio of the fusion proteins RARLBD-CFP and RXRLBD-YFP. But first we perform control experiments on cells either expressing RARLBD-CFP or RXRLBD-YFP. The measured intensity ratios of the two proteins are identical to the ratios obtained earlier for CFP and YFP. Their molecular brightness values are also within experimental error identical to the brightness values of CFP and YFP (data not shown).
It is known that RARLBD and RXRLBD form a tight heterodimer in vitro. Because CFP and YFP are a good FRET pair, we suspect that dimer formation of RARLBD-CFP and RXRLBD-YFP in vivo leads to energy transfer between the fluorescent proteins. The presence of FRET quenches the intensity of CFP and raises the intensity of YFP, which affects the value of the intensity ratio measurements. Because the intensity ratio is needed to determine the coexpression ratio, we developed an expression (Eq. 15) that takes the influence of FRET quantitatively into account. Independent measurements are needed to determine the exact amount of the apparent FRET efficiency between RARLBD-CFP and RXRLBD-YFP. We measure the fluorescence lifetime of the donor CFP with a narrow bandpass filter that completely cuts off the YFP emission. The apparent FRET efficiency is determined form Eq. 19 by comparing the fluorescence lifetime of the protein mixture 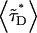 with the fluorescence lifetime of RARLBD-CFP alone 〈τD〉. We performed a series of measurements on cells that coexpress RARLBD-CFP and RXRLBD-YFP. For each cell, we first measure the dual-color intensity ratio by directing the fluorescence emission to the emission port with the dual-channel setup. Then we redirect the fluorescence emission to another microscope port to perform fluorescence lifetime measurements. Fig. 2 graphs the fluorescence intensity ratio of RARLBD-CFP and RXRLBD-YFP against the average fluorescence lifetime
with the fluorescence lifetime of RARLBD-CFP alone 〈τD〉. We performed a series of measurements on cells that coexpress RARLBD-CFP and RXRLBD-YFP. For each cell, we first measure the dual-color intensity ratio by directing the fluorescence emission to the emission port with the dual-channel setup. Then we redirect the fluorescence emission to another microscope port to perform fluorescence lifetime measurements. Fig. 2 graphs the fluorescence intensity ratio of RARLBD-CFP and RXRLBD-YFP against the average fluorescence lifetime 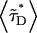 of the donor CFP. There are two striking features to be noted. First, the average fluorescence lifetime of the donor changes as a function of intensity ratio, indicating the presence of FRET. Second, there is a unique lifetime for each intensity ratio. Because each intensity ratio encodes a particular coexpression ratio, the lifetime depends only on the coexpression ratio.
of the donor CFP. There are two striking features to be noted. First, the average fluorescence lifetime of the donor changes as a function of intensity ratio, indicating the presence of FRET. Second, there is a unique lifetime for each intensity ratio. Because each intensity ratio encodes a particular coexpression ratio, the lifetime depends only on the coexpression ratio.
FIGURE 2.

Fluorescence lifetime of the donor RAR-CFP as a function of the intensity ratio of RXR-YFP and RAR-CFP coexpressed in CV-1 cells. The symbols are the experimentally determined fluorescence lifetime values of RAR-CFP, and the solid line is the theoretical predication assuming stoichiometric binding with an apparent FRET efficiency of 27%. The fluorescence lifetime of RAR-CFP is 2.38 ns in the absence of RXR-YFP. The lifetime of RAR-CFP decreases as the concentration of RXR-YFP is raised and reaches a limiting value once all RAR-CFP are bound to RXR-YFP.
The degree of dimerization of a reaction depends on the absolute concentration of both proteins and their dissociation coefficient KD. Concentrations below the value of the KD lead to a dimer fraction that is lower than the maximum possible. Concentrations much larger than the KD value lead to complete dimerization of every available protein pair. This limiting case is also called the stoichiometric binding regime, because the effect of entropy is negligible. We expect to see, for a 1:1 coexpression ratio with increasing absolute protein concentration, a steady increase in the dimer fraction until it reaches a limiting value of 1. Because the average lifetime of the donor depends on the dimer fraction according to Eq. 20, we also expect to see, for a 1:1 coexpression ratio, that the lifetime changes as a function of protein concentration. However, no changes in lifetime are observed, although the protein concentrations measured span more than one order-of-magnitude for a given intensity ratio. The only explanation consistent with the experimental observation is that all our measurements are taking place under conditions of stoichiometric binding. Thus, the KD of the heterodimer must be much lower than 500 nM, which corresponds to the lowest protein concentration shown in Fig. 3. An in vitro study determined a KD of ∼400 pM for the heterodimer of RXRLBD and RARLBD (Dong and Noy, 1998). Thus our result is consistent with the in vitro study, since we expect similar binding properties in vivo and in vitro. Stoichiometric binding leads to a simple relationship between the dimer fraction fD and the coexpression ratio rN = NRXRLBD-YFP/NRARLBD-CFP,
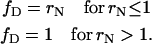 |
(23) |
In other words, the dimer fraction depends only on the coexpression ratio and is independent of the absolute value of the protein concentrations.
FIGURE 3.

Relative brightness of RARLBD-CFP and RXRLBD-YFP in CV-1 cells with intensity ratios between 1.15 and 1.35. This range of intensity ratios corresponds to coexpression ratios NRXRLBD-YFP/NRARLBD-CFP from 0.9 to 1.2. (A) At an excitation wavelength of 905 nm CFP and YFP are equally bright, and the relative brightness of the heterodimer is ∼2. (B) At 965 nm, only YFP is excited, and the apparent brightness of the heterodimer is reduced to the YFP brightness. The relative brightness of the heterodimer is virtually identical in the absence (▪) and in the presence (▵) of RXR agonist, which indicates that RXRLBD is unable to form homodimers in the presence of equal concentrations of RARLBD. The experimental error in brightness is shown for selected cells.
Because of energy transfer between RARLBD-CFP and RXRLBD-YFP, the lifetime of the donor RARLBD-CFP varies as a function of the intensity ratio. When RARLBD-CFP is in great excess, we expect that the average fluorescence lifetime is similar to that of RARLBD-CFP alone. As the RXRLBD-YFP concentration increases, the fluorescence lifetime of RARLBD-CFP decreases, because of the presence of FRET in the heterodimers, and will level off once all RARLBD-CFP form heterodimers. We indeed observe, in Fig. 2, a decrease in fluorescence lifetime as a function of intensity ratio. At an intensity ratio of 0.4, only RARLBD-CFP is present, and the fluorescence lifetime 〈τD〉 of the donor is 2.38 ns. This lifetime decreases with increasing intensity ratio until the dimer fraction fD equals 1. The limiting lifetime value of 1.75 ns corresponds to a FRET efficiency of E = 0.27. The effective FRET efficiency is given by 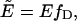 which allows us to calculate the intensity ratio and the fluorescence lifetime for stoichiometric binding using Eqs. 15 and 20. The solid line in Fig. 2 was calculated using the measured intensity ratios of the fluorescent proteins, rCFP and rYFP, their brightness ratio rλ, and a FRET efficiency of E = 0.27. We used the same parameters to calculate the relationship between the coexpression ratio rN and the intensity ratio rF for stoichiometric binding from Eq. 15, which is shown in Fig. 1 as a dashed line. The solid line represents the case without FRET. The differences between the two curves clearly highlight the importance of including FRET into the model. For example, a 1:1 coexpression is characterized by an intensity ratio of 1.0 in the absence of FRET, but increases to a value of 1.25 in the presence of 27% FRET. Note that the intensity ratio of 1.25 corresponds to a 1:2 CFP/YFP coexpression ratio in the absence of FRET. In summary, we established a relationship between the intensity ratio and the coexpression ratio for the brightness experiments that follow.
which allows us to calculate the intensity ratio and the fluorescence lifetime for stoichiometric binding using Eqs. 15 and 20. The solid line in Fig. 2 was calculated using the measured intensity ratios of the fluorescent proteins, rCFP and rYFP, their brightness ratio rλ, and a FRET efficiency of E = 0.27. We used the same parameters to calculate the relationship between the coexpression ratio rN and the intensity ratio rF for stoichiometric binding from Eq. 15, which is shown in Fig. 1 as a dashed line. The solid line represents the case without FRET. The differences between the two curves clearly highlight the importance of including FRET into the model. For example, a 1:1 coexpression is characterized by an intensity ratio of 1.0 in the absence of FRET, but increases to a value of 1.25 in the presence of 27% FRET. Note that the intensity ratio of 1.25 corresponds to a 1:2 CFP/YFP coexpression ratio in the absence of FRET. In summary, we established a relationship between the intensity ratio and the coexpression ratio for the brightness experiments that follow.
Single channel molecular brightness measurements on heterocomplexes
Moment or PCH analysis of fluorescence fluctuation experiments provides information about the brightness of proteins. We have used brightness analysis to detect homointeractions between proteins in cells (Chen et al., 2003). This type of analysis can also be applied to detect heteroprotein interactions using single-color fluorescence fluctuation spectroscopy. Let us illustrate the concept using a special case. Two proteins A and B are coexpressed with a 1:1 concentration ratio. Protein A is labeled with YFP and protein B is labeled with CFP. At an excitation wavelength of 905 nm, CFP fusion protein has almost the same brightness as YFP fusion protein. Thus, from a molecular brightness point of view, these two species are indistinguishable. Let us further assume that protein A exists in three different states, as monomer A, as homodimer A2, and as heterodimer AB. Protein B only exists in two states, as monomer B and as heterodimer AB. Table 1 summarizes the apparent brightness values that result from a number of different combinations of these protein states. If the brightness ɛM of the monomers of A and B is ɛ, then the brightness ɛD of the heterodimer AB and the homodimer A2 is 2ɛ.
TABLE 1.
Apparent brightness of binary protein mixtures

Each protein, A and B, carries a fluorophore with a brightness of ɛ. The apparent brightness of the monomeric proteins is ɛ. The other panels show the apparent brightness for different protein mixtures. To facilitate experimental distinction between these different mixtures, we also consider the case where only A carries a fluorescent label and B is nonfluorescent.
For convenience, we introduce the relative brightness ρɛ, which is the ratio of the apparent brightness to the brightness of a monomer, ρɛ = ɛapp/ɛM. A mixture of noninteracting monomers (A+B) results in a brightness of ɛ, which is equivalent to a relative brightness of 1. For a mixture of monomers in equilibrium with the heterodimer (A+B+AB) the relative brightness will lie between 1 and 2. The same situation applies for a mixture of homodimers A2 and monomeric B (A2+B). The relative brightness of this mixture has a value between 1 and 2. The exact value depends on the composition of the mixture and is determined by
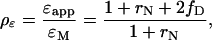 |
(24) |
which we derived from Eq. 22 for the special case of a dimer brightness ɛD = 2ɛ. As is apparent from Table 1, although it is very easy to distinguish a pure heterodimer from a monomer, it is much more tricky to differentiate a heterodimer-monomer mixture (AB+A+B) from a homodimer-monomer mixture (A2+B), because of the overlapping apparent brightness values in both cases. Thus, in general it will not be possible to identify the sample composition by measurement of the apparent brightness alone. To address this problem, we render CFP nonfluorescent by moving the two-photon excitation wavelength to 965 nm. Consequently, the brightness of protein B drops to 0 and the brightness of the heterodimer AB reduces from 2ɛ to the brightness ɛ of protein A (Table 1). The apparent brightness of the heterodimer-monomer mixture (AB+A+B) reduces to ɛ, whereas the brightness of the homodimer-monomer mixture (A2+B) increases to 2ɛ. In other words, the composition of protein mixtures can be identified by selectively turning on and off the brightness of fluorescent proteins by changing the excitation wavelength.
The molecular brightness of RARLBD-CFP and RXRLBD-YFP in cells
We previously demonstrated that the brightness of RARLBD in cells is equal to that of a monomer in the presence and absence of its agonist, all-trans-retinoic acid. In other words, RARLBD does not form homocomplexes. RXRLBD is mostly monomeric based on brightness in the absence of 9-cis-retinoic acid, but exhibits an increase in brightness at high protein concentrations, which indicates the presence of homocomplexes. Upon ligand addition, the apparent brightness of RXRLBD increases for all concentrations measured and reaches the brightness of a dimer at high protein concentrations. This result is consistent with a shift of the homodimer-monomer equilibrium to a higher binding affinity.
In this study, we wish to distinguish homo- and heterodimer formation of RXRLBD and RARLBD by applying molecular brightness analysis to cells transfected with RARLBD-CFP and RXRLBD-YFP. For each cell we perform a dual-channel intensity measurement to determine its protein expression ratio, followed by a single-channel brightness measurement at 905 nm to establish the brightness of the average protein complex. We also record the position of each cell using the electronic controlled stage of the microscope. This procedure allows us to revisit cells and measure brightness with an excitation wavelength of 965 nm where only YFP is excited, which is essential for distinguishing between heterodimers and homodimers of RXRLBD. The same sequence of measurements is carried out after addition of ligand. Fig. 3 shows the brightness ratio ρɛ of cells with intensity ratios from 1.15 to 1.35, which corresponds to approximately equimolar expression of both proteins. At an excitation of 905 nm, the relative brightness values of the RARLBD-CFP/RXRLBD-YFP mixture is 2 at all concentrations measured, thus indicating the presence of a purely dimeric species (Fig. 3 A). The relative brightness values are virtually unchanged upon adding RXR agonist. Measuring the same cells at 965 nm reveals a relative brightness of RXRLBD-YFP, which is 1 (Fig. 3 B). This means that RXRLBD-YFP is not able to form homodimers in the presence of equal amount of RARLBD, but must be present as a heterodimer. In other words, our study confirms in vitro results that demonstrate that RARLBD forms a very tight heterodimer with RXRLBD, and that RXRLBD cannot form a homodimer in the presence of equimolar concentrations of RARLBD (Dong and Noy, 1998).
Next, we examine the power of brightness analysis by studying situations where one of the protein components is in excess. We first choose cells that express more RARLBD-CFP than RXRLBD-YFP. Specifically, we picked cells with intensity ratios from 0.7 to 0.9, which corresponds to concentration ratios NRXRLBD-YFP/NRARLBD-CFP = 0.35–0.6. The relative brightness measured at 905 nm as a function of protein concentration is shown in Fig. 4 A. The relative brightness is concentration-independent, and its value is lower than that of a dimer. This reduced relative brightness indicates the presence of a monomeric species. This is expected, because the fraction of RARLBD-CFP, which is in excess over the RXRLBD-YFP concentration, cannot find a heterobinding partner and has to be in monomeric form. Upon adding RXR agonist, the relative brightness stays the same, which indicates that the excess fraction of RARLBD-YFP remains monomeric. Calculating the expected relative brightness from Eq. 24 for concentration ratios of 0.35–0.6 predicts values between 1.5 and 1.75. This range is marked as dotted lines in Fig. 4 A, and our data fall well between these limits. To check our model we also measured the brightness at 965 nm, where only YFP is excited (Fig. 5, solid squares). The relative brightness of RXRLBD-YFP is equivalent to that of a monomer. Thus, no homocomplexes of RXRLBD-YFP are found. RXRLBD-YFP forms a tight heterodimer with its partner RARLBD-CFP.
FIGURE 4.

Relative brightness of RARLBD-CFP and RXRLBD-YFP in CV-1 cells at an excitation wavelength of 905 nm. (A) Cells with intensity ratios from 0.7 to 0.9 (NRXRLBD-YFP/NRARLBD-CFP from 0.36 to 0.6). Excess of RARLBD leads to an apparent brightness (▪), which is less than that expected for a full dimer. Adding ligand leaves the apparent brightness (▵) unchanged. (B) Cells with intensity ratios from 1.5 to 1.7 (NRXRLBD-YFP/NRARLBD-CFP from 1.8 to 2.8). Excess of RXRLBD leads to an apparent brightness (▪), which is less than that expected for a full dimer. Adding ligand restores the apparent molecular brightness (▵) of the mixture to that of a pure dimer population. The dashed lines correspond to the range of relative brightness values in the absence of ligand and are calculated from the simple binding model discussed in the text.
FIGURE 5.

Relative brightness of RARLBD-CFP and RXRLBD-YFP in CV-1 cells in the presence of RXR agonist at an excitation wavelength of 965 nm. Cells with an excess of RARLBD (NRXRLBD-YFP/NRARLBD-CFP from 0.36 to 0.6) have an apparent brightness equal to monomeric RXRLBD-YFP. Cells with an excess of RXRLBD (NRXRLBD-YFP/NRARLBD-CFP from 1.8 to 2.8) have an apparent brightness, which is larger than that of a monomer, but less than that of a dimer. The dashed lines indicate the relative brightness range expected from the binding model described in the text.
We also measured cells that express more RXRLBD-YFP than RARLBD-CFP. Cells with intensity ratios of 1.5–1.7 were selected, which corresponds to concentration ratios NRXRLBD-YFP/NRARLBD-CFP = 1.8–2.8. The relative brightness values measured at 905 nm as a function of concentration are shown in Fig. 4 B. Again, the relative brightness of these cells is lower than the value required for a pure dimer. We expect that both proteins form a tight heterodimer. In addition, based on our previous study we anticipate that RXRLBD-YFP remains essentially monomeric in the absence of ligand. This model allows us to calculate the expected relative brightness from Eq. 24, which gives values from 1.5 to 1.75 (shown as dashed lines in Fig. 4 B). These brightness predictions are in good agreement with our experimental data.
Adding RXR agonist increases the relative brightness (open triangles in Fig. 4 B) to that expected for a purely dimeric species. To better understand this behavior, we also measured the brightness at 965 nm, where only YFP is excited (open triangles in Fig. 5). The relative brightness in the presence of RXR agonist is concentration-independent and larger than that of YFP alone, but less than that of dimeric YFP. This clearly demonstrates the presence of homocomplexes of RXRLBD-YFP. We introduce a simple model where RXRLBD-YFP forms a tight heterodimer with RARLBD-CFP, and any excess concentration of RXRLBD-YFP forms a homodimer in the presence of RXR agonist. This simple model fits our data very well. Both the homodimer and the heterodimer have a brightness of 2ɛ at an excitation wavelength of 905 nm. Thus we expect a relative brightness of 2 in the presence of agonist as experimentally observed (see triangles in Fig. 4 B). At an excitation wavelength of 965 nm the relative brightness of the heterodimer is reduced to 1, whereas the relative brightness of the homodimer remains 2. Calculating the relative brightness for this model gives values between 1.4 and 1.6, which are in good agreement with the experimental data (see Fig. 5).
DISCUSSION
Determining the protein coexpression ratio is crucial for a quantitative analysis of protein heterointeractions in cells. We use a dual-color fluorescence setup to determine the coexpression ratio. Both CFP and YFP are conveniently coexcited at 905 nm and have virtually the same molecular brightness. Knowledge of this brightness and the measured dual-color intensity ratio determine the coexpression ratio in the absence of FRET. Calculating the coexpression ratio in the presence of FRET requires an additional parameter, the apparent FRET efficiency 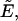 which is experimentally determined from Eq. 19 by lifetime measurements of the donor. Ignoring FRET leads to a systematic bias when selecting cells on the basis of dual-color intensity ratio measurements as illustrated in Fig. 1. For example, an intensity ratio of 1.25 would be interpreted as a 1:2 coexpression ratio, although actually reflecting a 1:1 expression ratio.
which is experimentally determined from Eq. 19 by lifetime measurements of the donor. Ignoring FRET leads to a systematic bias when selecting cells on the basis of dual-color intensity ratio measurements as illustrated in Fig. 1. For example, an intensity ratio of 1.25 would be interpreted as a 1:2 coexpression ratio, although actually reflecting a 1:1 expression ratio.
The term “apparent FRET” is used to indicate that not all donor molecules are experiencing energy transfer. For example, CFP in monomeric form will not contribute to the FRET signal. The average lifetime of the donor 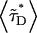 is given by a superposition of the lifetimes in the presence and absence of FRET,
is given by a superposition of the lifetimes in the presence and absence of FRET,  where f− and f+ represent the normalized fraction of donor molecules, with and without energy transfer, respectively. The apparent FRET efficiency is defined by
where f− and f+ represent the normalized fraction of donor molecules, with and without energy transfer, respectively. The apparent FRET efficiency is defined by 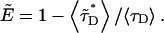 As we have shown, our model proteins RARLBD and RXRLBD form a very tight heterodimer in vivo, and all our measurements are performed at protein concentrations above the KD of the heterodimer. In other words, we encounter stoichiometric binding, where the degree of binding is solely determined by the ratio of the protein concentrations. The simplicity of this binding model allows us to perform an experimental test of our theory. The average lifetime of the donor depends only on the intensity ratio and is determined from Eqs. 23 and 16, as shown in Fig. 2. We observe a FRET efficiency of 27% for the heterodimer of RARLBD-CFP and RXRLBD-YFP.
As we have shown, our model proteins RARLBD and RXRLBD form a very tight heterodimer in vivo, and all our measurements are performed at protein concentrations above the KD of the heterodimer. In other words, we encounter stoichiometric binding, where the degree of binding is solely determined by the ratio of the protein concentrations. The simplicity of this binding model allows us to perform an experimental test of our theory. The average lifetime of the donor depends only on the intensity ratio and is determined from Eqs. 23 and 16, as shown in Fig. 2. We observe a FRET efficiency of 27% for the heterodimer of RARLBD-CFP and RXRLBD-YFP.
We would like to emphasize that measuring the average donor lifetime, and thereby the apparent FRET efficiency, is needed only for the accurate determination of the protein coexpression ratio. The examination of protein-protein interactions by brightness analysis does not rely on FRET. This differs from traditional FRET spectroscopy, where a positive FRET signal is needed to detect protein-protein interactions (Day et al., 2001). The absence of a FRET signal, however, cannot rule out protein association. Brightness analysis, on the other hand, detects protein association by an increase of the brightness, which occurs both in the presence and absence of FRET. We choose experimental conditions where the brightness increase is independent of FRET. This simplifies the interpretation of the experimentally measured brightness. Brightness analysis also characterizes the formation of homocomplexes, where FRET spectroscopy is unsuitable, and provides quantitative information about the degree of binding.
We introduced a theory that describes the influence of energy transfer on the brightness of the donor and acceptor. The brightness of the donor increases and the brightness of the acceptor decreases as shown in Eqs. 7 and 10. The brightness of a dimer that carries a donor-acceptor pair is given by the sum of their brightnesses. It is important to note that judicious selection of the excitation wavelength and optical filters allows the brightness of the dimer to be independent of energy transfer. For example, an excitation wavelength where the two-photon cross-sections of the donor and acceptor are identical, together with optical filters that give identical brightness for donor and acceptor, are ensuring a brightness of the dimer that is twice the brightness of the monomer (see Eq. 11). These conditions are approximately fulfilled for exciting the CFP/YFP pair at 905 nm. The ratio of their two-photon cross sections is 1 (Zipfel et al., 2003), and their brightnesses differ by only 10%. This difference in brightness is acceptable, given the experimental uncertainties of cellular measurements.
Another condition required for a quantitative interpretation of brightness is that the fluorescence of the label is not quenched by protein-protein interactions. We verified this previously for the fluorescent protein EGFP (Chen et al., 2003). The same conclusion holds for the fluorescent proteins CFP and YFP (data not shown). We found that the molecular brightness of RARLBD-CFP and RXRLBD-YFP is the same as either CFP or YFP alone within the experimental uncertainty of brightness experiments in cells. We also observed doubling of the brightness when expressing both proteins in a 1:1 ratio, as expected for a heterodimer in the absence of quenching (shown in Fig. 3 A).
Endogenous RXR and RAR could interact with the fluorescently labeled RXRLBD and RARLBD, thereby introducing a nonfluorescent background species. The consequence of such a background species is the reduction of the measured apparent brightness. We previously demonstrated that endogenous RXR and RAR are undetectable in COS-1 cells (Chen et al., 2003). Here we use CV-1 cells, which are closely related to COS-1. COS-1 is the SV40-transformed CV-1 cell line. Thus, we expect that endogenous RXR and RAR are absent in the CV-1 cell line. In addition, if endogenous protein were present, it would compete with labeled protein in forming binding complexes. Complexes formed between a labeled and an endogenous protein have a lower brightness than complexes between labeled proteins, which would result in a reduction of the apparent brightness to values less than twice the monomer brightness. Our results in Fig. 3 show that equimolar concentrations of RXRLBD and RARLBD lead to a brightness of twice the monomer brightness, indicating a pure species of labeled heterodimers. In other words, the influence of endogenous protein is negligible over the concentration range studied.
RXR is a unique nuclear receptor because it serves as a heterodimer partner for many other nuclear receptors, such as RAR, T3R, and VDR (Mangelsdorf et al., 1995). Previously, we have shown that RXRLBD is capable of forming homodimers after ligand activation in cells. In this study we examined RXRLBD in the presence of RARLBD. We found that RXRLBD is not capable of forming homodimers in the presence of an equal or higher concentration of RARLBD. This demonstrates that the affinity for the heterodimer is much higher than the affinity for the homodimer. Our finding is in agreement with other studies using purified proteins that conclude RAR and RXR form a very tight heterodimer in vitro (Dong and Noy, 1998; Poujol et al., 2003).
To clearly distinguish between hetero- and homodimers the excitation wavelength was changed from 905 nm to 965 nm to selectively excite the acceptor YFP. The two-photon cross-section of YFP at this wavelength exceeds the cross-section of CFP by ∼20 times. In other words, the brightness of CFP is approximately a factor-of-20 smaller than the brightness of YFP, and can be safely ignored in most circumstances. Measuring the brightness at both wavelengths provides a convenient method to distinguish between homodimers and heterodimers as illustrated in Table 1. Our experimental brightness values are in good agreement with a simple binding model, where RXRLBD and RARLBD form a tight heterodimer. Any excess population of RXRLBD is a monomer in the absence of ligand and forms homodimers in the presence of ligand.
Labeling of individual protein species with differently colored dyes is an elegant approach for studying protein-protein interactions. Analysis of such experiments with dual-color fluorescence fluctuation spectroscopy is an obvious choice, and has been primarily the focus of fluorescence cross-correlation analysis (Bacia et al., 2002). However, dual-color cross-correlation analysis in living cells with fluorescent proteins is challenging. There are only a limited number of fluorescent proteins to choose from. For example, the CFP and YFP pair, which is widely used for FRET studies, exhibits strong spectral overlap. The spectral overlap leads to cross talk between the detection channels and severely complicates dual-channel cross-correlation analysis.
Dual-color PCH analysis offers another approach to analyze fluctuation experiments. We recently demonstrated that it is possible to resolve a diluted CFP/YFP mixture in vitro using dual-color PCH (Chen et al., 2004). However, cellular experiments require measurements over a wide concentration range, and a dual-color PCH theory that takes deadtime and afterpulsing of the photo detector into account still needs to be developed. In addition, a quantitative treatment of FRET in dual-color PCH is still missing. Thus, we decided to focus on detecting homo- and heterointeractions, not by differences in color, but solely based on brightness in a single-channel setup. By exciting CFP and YFP at 905 nm we eliminate brightness differences between the fluorophores and eliminate the influence of FRET on the brightness of the heterodimer.
Our method of brightness analysis is applicable to other protein systems as well. In our particular case, we take advantage of the tight binding between RXRLBD and RARLBD. This results in a unique relationship between the average fluorescence lifetime of the donor and the coexpression ratio as shown in Fig. 2. In general, the degree of binding not only depends on the coexpression ratio but on the absolute concentrations of the reactants as well. Measuring the fluorescence lifetime of the donor, the dual-color intensity ratio, and the single-color brightness of each cell determines the apparent FRET efficiency, the coexpression ratio, and the brightness of the sample. The absolute protein concentration is determined from the fluorescence intensity. Different excitation wavelengths are used to distinguish between homo- and heterointeractions. Collecting brightness data in this manner allows our distinguishing different binding models by comparing the experimental data with model-dependent predictions.
CONCLUSIONS
Molecular brightness is inherently sensitive to the stoichiometry of a protein complex. In the past, we demonstrated that molecular brightness is a robust method for characterizing the oligomerization of a homocomplex. Here, we show that molecular brightness is capable to determine the composition of a heterocomplex as well. We demonstrate that a lifetime and a dual-color intensity ratio measurement are sufficient to characterize the coexpression ratio. A theory that describes the presence of energy transfer on brightness was introduced and used to find excitation conditions for CFP and YFP that eliminate the influence of energy transfer on the brightness of heterodimers. Brightness analysis was applied to demonstrate heterodimer formation between RXRLBD and RARLBD. In addition, by exciting at different wavelengths we were able to distinguish homodimer and heterodimer complexes. Our theory allows a quantitative interpretation of the brightness in terms of protein populations. We like to stress that this is the first quantitative study of both homo- and heterodimer formation in cells. In summary, brightness analysis is a promising tool for quantitative analysis of protein-protein interactions in living cells.
Acknowledgments
This work was supported by grants from the National Institutes of Health (No. GM64589) and the National Science Foundation (No. PHY-0346782). Y.C. acknowledges support by a postdoctoral fellowship from the National Institutes of Health (GM020853). L.N.W. acknowledges support from the National Institutes of Health (Nos. DK54733, DK60521, and K02 DA13926).
References
- Bacia, K., I. V. Majoul, and P. Schwille. 2002. Probing the endocytic pathway in live cells using dual-color fluorescence cross-correlation analysis. Biophys. J. 83:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berland, K. M. 1995. Two-Photon Fluctuation Correlation Spectroscopy: Method and Applications to Protein Aggregation and Intracellular Diffusion. University of Illinois at Urbana-Champaign, Urbana, IL.
- Brock, R., M. A. Hink, and T. M. Jovin. 1998. Fluorescence correlation microscopy of cells in the presence of autofluorescence. Biophys. J. 75:2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., J. D. Müller, Q. Ruan, and E. Gratton. 2002. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., J. D. Müller, P. T. So, and E. Gratton. 1999. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 77:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., M. Tekmen, L. Hillesheim, J. Skinner, B. Wu, and J. Mueller. 2004. Dual-color photon counting histogram. Biophys J. 88:2177–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., L. N. Wei, and J. D. Müller. 2003. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc. Natl. Acad. Sci. USA. 100:15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, R. N., A. Periasamy, and F. Schaufele. 2001. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 25:4–18. [DOI] [PubMed] [Google Scholar]
- Denk, W., J. H. Strickler, and W. W. Webb. 1990. Two-photon laser scanning fluorescence microscopy. Science. 248:73–76. [DOI] [PubMed] [Google Scholar]
- Dong, D., and N. Noy. 1998. Heterodimer formation by retinoid X receptor: regulation by ligands and by the receptor's self-association properties. Biochemistry. 37:10691–10700. [DOI] [PubMed] [Google Scholar]
- Farooqui, M., P. J. Franco, J. Thompson, H. Kagechika, R. A. Chandraratna, L. Banaszak, and L. N. Wei. 2003. Effects of retinoid ligands on RIP140: molecular interaction with retinoid receptors and biological activity. Biochemistry. 42:971–979. [DOI] [PubMed] [Google Scholar]
- Hillesheim, L. N., and J. D. Muller. 2003. The photon counting histogram in fluorescence fluctuation spectroscopy with non-ideal photodetectors. Biophys. J. 85:1948–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- Mangelsdorf, D. J., C. Thummel, M. Beato, P. Herrlich, G. Schutz, K. Umesono, B. Blumberg, P. Kastner, M. Mark, P. Chambon, et al.,. 1995. The nuclear receptor superfamily: the second decade. Cell. 83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, J. D. 2004. Cumulant analysis in fluorescence fluctuation spectroscopy. Biophys. J. 86:3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, J. D., Y. Chen, and E. Gratton. 2000. Resolving heterogeneity on the single molecular level with the photon counting histogram. Biophys. J. 78:474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politz, J. C., E. S. Browne, D. E. Wolf, and T. Pederson. 1998. Intranuclear diffusion and hybridization state of oligonucleotides measured by fluorescence correlation spectroscopy in living cells. Proc. Natl. Acad. Sci. USA. 95:6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poujol, N., E. Margeat, S. Baud, and C. A. Royer. 2003. RAR antagonists diminish the level of DNA binding by the RAR/RXR heterodimer. Biochemistry. 42:4918–4925. [DOI] [PubMed] [Google Scholar]
- Qian, H., and E. L. Elson. 1990. Distribution of molecular aggregation by analysis of fluctuation moments. Proc. Natl. Acad. Sci. USA. 87:5479–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel, W. R., R. M. Williams, and W. W. Webb. 2003. Nonlinear magic: multiphoton microscopy in the biosciences. Nat. Biotechnol. 21:1369–1377. [DOI] [PubMed] [Google Scholar]