

Abstract
The first stage in hemostasis is the binding of the platelet membrane receptor glycoprotein (GP) Ib-IX complex to the A1 domain of von Willebrand factor in the subendothelium. A bleeding disorder associated with this interaction is platelet-type von Willebrand disease, which results from gain-of-function (GOF) mutations in amino acid residues 233 or 239 of the GP Ibα subunit of GP Ib-IX. Using optical tweezers and a quadrant photodetector, we investigated the binding of A1 to GOF and loss-of-function mutants of GP Ibα with mutations in the region containing the two known naturally occurring mutations. By dynamically measuring unbinding force profiles at loading rates ranging from 200–20,000 pN/s, we found that the bond strengths between A1 and GP Ibα GOF mutants (233, 235, 237, and 239) were significantly greater than the A1/wild-type GP Ib-IX bond at all loading rates examined (p < 0.05). In addition, mutants 231 and 232 exhibited significantly lower bond strengths with A1 than the wild-type receptors (p < 0.05). We computed unloaded dissociation rate constant (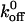 ) values for interactions involving mutant and wild-type GP Ib-IX receptors with A1 and found the A1/wild-type GP Ib-IX
) values for interactions involving mutant and wild-type GP Ib-IX receptors with A1 and found the A1/wild-type GP Ib-IX 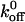 value of 5.47 ± 0.25 s−1 to be significantly greater than the GOF
value of 5.47 ± 0.25 s−1 to be significantly greater than the GOF 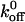 values and significantly less than the loss-of-function
values and significantly less than the loss-of-function 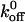 values. Our data illustrate the importance of the bond kinetics associated with the VWF/GP Ib-IX interaction in hemostasis and also demonstrate the drastic changes in binding that can occur when only a single amino acid of GP Ibα is altered.
values. Our data illustrate the importance of the bond kinetics associated with the VWF/GP Ib-IX interaction in hemostasis and also demonstrate the drastic changes in binding that can occur when only a single amino acid of GP Ibα is altered.
INTRODUCTION
To achieve hemostasis, platelets must adhere to the exposed subendothelial matrix in damaged blood vessels. This initial adhesion is mediated by the platelet glycoprotein (GP) Ib-IX-V receptor complex binding to the glycoprotein von Willebrand factor (VWF) present on the exposed subendothelium. The GP Ib-IX-V complex is comprised of four subunits: GP Ibα, GP Ibβ, GP IX, and GP V (Fig. 1). GP Ibα is linked to GP Ibβ through a disulfide bond, whereas GP IX and GP V are noncovalently associated with GP Ib. GP V is not necessary for binding to VWF, but it is a substrate for thrombin (Dong et al., 1997) and appears to function as an ancillary collagen receptor (Moog et al., 2001). GP Ibα contains the binding sites not only for VWF but also for Mac-1, P-selectin, thrombin, high molecular kininogen, and Factor XII (Berndt et al., 2001).
FIGURE 1.
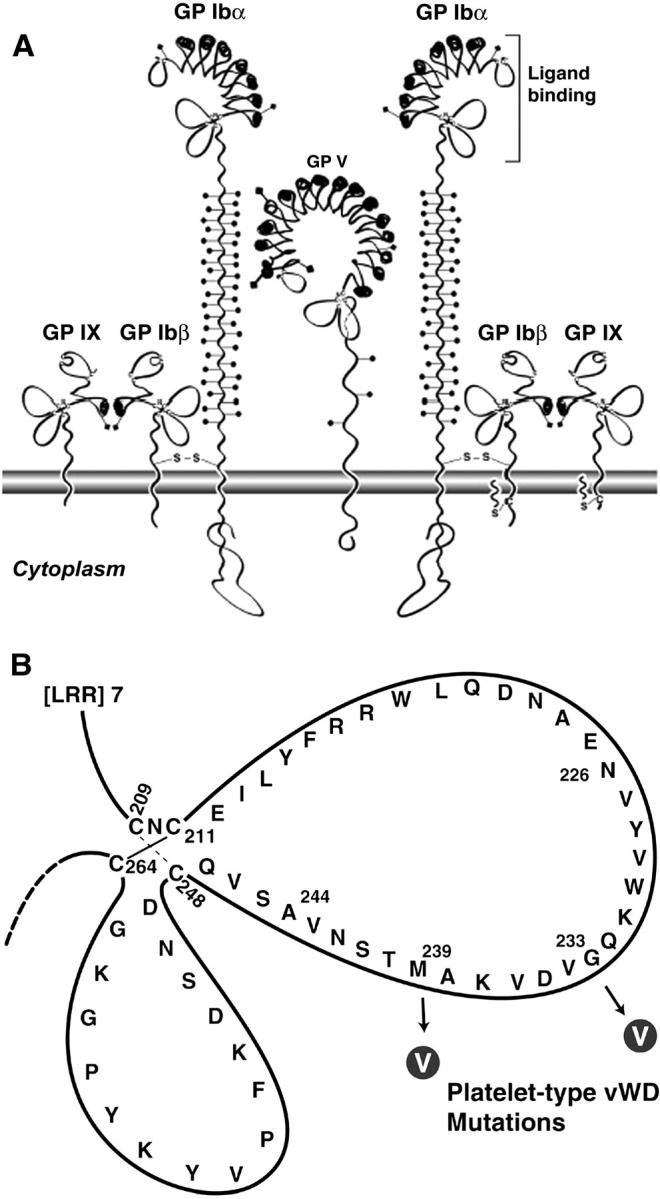
(A) The GP Ib-IX-V complex is composed of four subunits: GP Ibα, GP Ibβ, GP IX, and GP V. GP Ibα is the subunit responsible for binding to VWF A1 domain. (B) Diagram illustrating the GP Ibα C-terminal flank disulfide loops. The two mutations associated with ptVWD involve the conversion of nonvaline residues to valines at amino acid residues 233 and 239.
VWF is a large glycoprotein, the mature subunit consisting of 2050 amino acid residues, which is primarily stored in the α-granules of platelets and the Weibel-Palade bodies of endothelial cells, from which it is released upon cell activation. VWF contains three consecutive, homologous A domains, with the A1 domain containing the GP Ib-IX binding site (Sadler, 1998). After GP Ib-IX-V has bound to VWF, a cascade of interactions ensues that results in platelet activation and subsequent aggregation, ultimately leading to the formation of a platelet-rich thrombus. These interactions include secretion of agonists (e.g., ADP) by platelets, elevation of cytosolic Ca2+, and Ca2+-dependent activation of GP IIb-IIIa (Kroll et al., 1996; Ruggeri, 1997; Goto et al., 1998).
When unusually large VWF multimers are released from endothelial cells, they are processed into smaller and less adhesive plasma forms by the ADAMTS-13 enzyme (Gerritsen et al., 2001;Fujikawa et al., 2001). Normally the plasma form of VWF does not interact with GP Ib-IX-V; however, plasma VWF and GP Ib-IX-V will interact with each other when they are exposed to pathologic shear stress such as those in arteries occluded by atherosclerotic plaque. The presence of various exogenous modulators can also induce binding in vitro when high shear stress is absent (Berndt et al., 1992).
The crystal structure of the GP Ibα/VWF complex reveals two main contact areas bridged by an area of solvated charge interaction (Huizinga et al., 2002). The first and largest contact area, between VWF and GP Ibα, involves leucine-rich repeats 5–8 and the COOH-terminal flank interacting with the top portion of the A1 domain. The second contact area, between a part of the NH2-terminal domain and the leucine-rich repeat 1 of GP Ibα, involves three portions of the bottom face of the A1 domain. When GP Ibα interacts with VWF, the loop from amino acid residue 227 to residue 241 in the COOH-terminal flank of GP Ibα (Fig. 1 B) is hypothesized to change conformation. Gain-of-function (GOF) mutations in this loop can lead to platelet-type von Willebrand disease (ptVWD), where afflicted individuals are thrombocytopenic and have a propensity to bleed easily. Specifically, mutations of glycine (Miller et al., 1991) or methionine (Russell and Roth, 1993) at residues 233 and 239, respectively, to valine (Fig. 1 B) cause the platelet receptor to bind VWF more readily (Tait et al., 2001). Although mutations most likely modulate the affinity of GP Ibα for VWF in ptVWD platelets, avidity modulation that results in an increased number of bonds could also be playing a role in the excessive VWF binding. The increased propensity to bleed in individuals with ptVWD is thought to result from rapid clearance from the plasma of the highest molecular weight multimers of VWF, which are particularly effective in mediating platelet adhesion (Arya et al., 2002). In vitro, the platelets of patients with ptVWD bind VWF at much lower modulator concentrations than are required to induce VWF binding to normal platelets (Miller and Castella, 1982; Weiss et al., 1982).
Several previous studies have investigated VWF adhesion to GP Ibα containing the ptVWD mutations expressed in heterologous cell lines. Dong et al. (2000) observed that transfected cells expressing the GOF mutants rolled significantly more slowly than wild-type cells over immobilized VWF under flow, required lower modulator concentrations to bind VWF, and supported cell adhesion at much lower surface-coating densities of VWF. Tait et al. (2001) examined the VWF-binding ability of similar mutants using microcapillary tubes and found that ristocetin-mediated VWF/mutant GP Ib-IX adhesion was greater than VWF/wild-type GP Ib-IX adhesion in terms of cell tethering to the VWF-coated substrate, slower cell rolling velocity, and decreased cell detachment with increasing shear rate. In addition, Doggett et al. (2003) have investigated the interaction between ptVWD platelets and A1 using a flow chamber system, and found that the GP Ibα-233 mutation promoted and stabilized platelet adhesion at shear rates that do not support adhesion between wild-type GP Ib-IX-V and VWF. Although the Doggett et al. study was useful in characterizing the interaction between ptVWD GP Ib-IX-V receptors and VWF at various shear rates, it employed bulk-flow measurement techniques as opposed to single-molecule methods. In this study, we used optical tweezers to investigate the effect of substituting valine for other amino acids in GP Ibα on binding A1 at a single-bond level over a wide range of loading conditions. With single-molecule measurements, one can begin to understand important physiological functional fluctuations that may not be apparent by observing average values of physical quantities. For example, one is able to distinguish whether the affinity or avidity of a particular bond is modified by an external stimulus (Arya et al., 2003).
Using a Chinese hamster ovary cell model that expresses several ptVWD mutant receptors as part of recombinant GP Ib-IX complexes, we aim to explore the intricacies of VWF binding-affinity modulation that occur when mutations are introduced into the region of GP Ibα associated with ptVWD. Measurements of forces to break the bonds between A1 and GP Ibα at various loading rates allows subsequent investigation of molecular unbinding mechanics through computation of the dissociation constants for the interactions between A1 and mutant or wild-type GP Ib-IX receptor complexes.
MATERIALS AND METHODS
Cells expressing GP Ib-IX complexes
The method for preparing the cells that express mutant receptors used in this study has been previously described (Dong et al., 2000). Briefly, six mutations were created in which each codon for nonvaline residues within a region spanning Lys231–Met239 was converted to a valine codon. The mutated GP Ibα cDNAs were then transfected into Chinese hamster ovary (CHO) βIX cells (which stably express GP Ibβ and GP IX) using Lipofectamine (Invitrogen, Carlsbad, CA). Since the GP V subunit is not required for the VWF/GP Ib-IX interaction, it was not included in our transfection system. The mutant cells used in the study expressed GP Ib-IX complexes with GP Ibα mutations at residues 231, 232, 233, 235, 237, and 239. CHO αβIX cells, which expressed wild-type GP Ibα, were also prepared and used to investigate A1 binding. CHO βIX cells were used as negative controls since they do not bind VWF. The diameter of the CHO cells was typically 10 μm.
Preparation of A1 and protein-coated beads
The interactions of the A1 domain with the different GP Ib-IX mutants were investigated. Recombinant VWF A1 domain was produced using Escherichia coli by a previously described method (Cruz et al., 1993). It is important to note that the recombinant VWF A1 domain used in our study was not glycosylated. Glycosylation, in the isolated domain and in the intact protein, appears to negatively modulate the binding of VWF to GP Ibα. For example, Cruz et al. (1993) have shown that glycosylation of the VWF A1 domain increases its solubility but reduces its affinity for GP Ib-IX. These investigators showed that the affinity of glycosylated A1, produced in CHO cells, for platelet GP Ib-IX was threefold lower when compared to the nonglycosylated version. Our own optical tweezers-based study (Arya et al., 2002) showed that isolated, nonglycosylated A1 and unusually large VWF, which presumably contained glycosylated A1, adhered to GP Ib-IX with similar bond strengths. These studies all suggest that in the isolated A1 domain, the nonglycosylated form assumes the active conformation present in unusually large VWF, or becomes induced when VWF binds collagen or is activated by ristocetin. Consistent with this finding, early studies showed that desialylation of intact plasma VWF produced a form capable of binding and activating platelets spontaneously (De Marco and Shapiro, 1981). Activation of VWF by either shear stress, collagen binding, desialylation, or the nonphysiological modulators ristocetin and botrocetin, is necessary for its binding to GP Ibα and therefore for studies of that interaction. The nonglycosylated form of A1 thus provides a useful reagent for the studies of this interaction, and numerous studies have used A1 produced from E. coli to investigate structural and functional attributes of VWF. For example, Emsley et al. (1998) determined the crystal structure of A1 domain using nonglycosylated A1 from E. coli. In addition, Miyata and Ruggeri (1999) studied A1/GP Ibα adhesion under flow using nonglycosylated A1 from E. coli.
Polystyrene beads (Polysciences, Warrington, PA) with a diameter of 2.0 μm were coated with A1 according to the following procedure. Single drops of bead suspensions were added to tubes containing A1 at a concentration of either ∼10 or 100 μg/ml in citrate phosphate buffer. The beads and A1 domain were then incubated for 1 h at room temperature with gentle mixing on an aliquot shaker. The suspension was subsequently centrifuged for 2 min in an Eppendorf microcentrifuge at 15,000 rpm. The supernatant was removed, and the beads were resuspended by vortex in Dulbecco's phosphate-buffered saline solution (pH 7.3) containing 10 mg/ml bovine serum albumin, 1 mg/ml sodium azide, and 5% glycerol. The beads were then washed in 1 ml Dulbecco's phosphate-buffered saline solution, incubated for 30 min, centrifuged for 2 min, and resuspended in 0.5 ml of Dulbecco's solution.
Optical tweezers setup and measurements of time-resolved unbinding forces
We used a titanium-sapphire laser (Ti:sapph) (3900 S, Spectra-Physics, Santa Clara, CA) tuned to 830 nm in our optical tweezers setup (Fig. 2). Light-induced damage to cells varies with the incident wavelength, but little or no damage has been demonstrated at 830 nm in an optical trap (Neuman et al., 1999;Leitz et al., 2002).
FIGURE 2.

Optical tweezers setup. Light from a titanium-sapphire laser (λ = 830 nm) is reflected by a dichroic mirror and focused by a high-numerical-aperture (1.3) objective lens of an inverted microscope to form an optical trap. A charge-coupled device camera collects 10% of the light for image visualization, and a quadrant photodetector uses the remaining light for dynamically monitoring the displacement of the bead from the trapping center during the bond rupture process. The CHO cell and A1-coated bead are in a solution chamber mounted onto a piezoelectrically driven translational stage used to position the cell at an appropriate distance from the trapped bead.
The Ti:sapph laser was pumped by a solid-state, frequency-doubled neodymium yttrium vanadate (Nd:YVO4) laser operating at a wavelength of 532 nm (Millennia V, Spectra-Physics). The laser light then passed through an attenuator (925B, Newport Electronics, Irvine, CA) used to control the beam intensity. An external dichroic mirror placed just before the bottom entrance port allowed transmission of the laser light into an inverted microscope (Axiovert S100TV, Carl Zeiss, Jena, Germany). The laser light was focused by a high numerical aperture (1.3) objective lens onto a diffraction-limited spot to form an optical trap (Ashkin et al., 1986).
A glass-bottom culture dish (P35GC-0-10-C, MatTek, Ashland, MA) was used to hold the cells and beads. A 10-mm-diameter German glass no. 0 coverslip was located in the center of the 35-mm dish. Using a micropipette, we added 100 μL of cells and 5 μL of beads to the dish, which was left exposed to the air to prevent heating of the sample by the laser. The cell/bead suspension could be left uncovered for 1 h before evaporation became significant. The cell chamber was illuminated from the top with white light for visualizing the specimens. The external dichroic mirror reflected the light <650 nm toward a beam splitter, which transmitted 10% of the light to a charged-coupled device (CCD) camera (CCD 100, DAGE-MTI, Michigan City, IN) for imaging purposes. Due to slight leakage of the laser beam into the imaging path, an infrared filter was placed before the beam splitter to pass all light <700 nm. The remaining 90% of the light was sent to a quadrant photodetector (Hamamatsu, S4939, Somerville, NJ), mounted on a micrometer-controlled stage, used for measurements of dynamic unbinding force between the receptor and its ligand.
We use the dynamic displacement of the trapped bead from the trapping center in response to an external force application (e.g., molecular forces acting upon the trapped bead) as a technique to obtain time-resolved force measurements. Specifically, we project the change in the shadow of the trapped 2-μm bead by a 15× eyepiece in response to external force application onto the surface of the quadrant photodetector, generating a voltage signal on each of its four quadrants (V1–V4), and relate the voltage to bond loading and, hence, to the unbinding force. The solution chamber containing cells was mounted onto a piezoelectrically driven translational stage (P-527.3CL, Physik Instrumente, Waldbronn, Germany). Movement of the piezoelectric stage was controlled by a function generator (Stanford Research Systems, DS345, Sunnyvale, CA). The resolution of the piezoelectric stage was 10 nm in the x and y directions and 2 nm in the z direction (laser-beam propagation direction).
The analog photodetector output voltage resulting from the movement of a trapped bead during bond rupture were amplified by a circuit (QP50-6SD, Pacific Silicon Sensor, Westlake Village, CA) and digitized with an A-D converter (Wavebook512, Iotech, Cleveland, OH). The sampling frequency for all experiments was 50 kHz. The bandwidth of the detection system was 257 kHz and the signal/noise ratio (SNR) was 120. The SNR was determined experimentally by analyzing the variation in the photodetector voltage signal of a bead responding to movement of the piezoelectric stage driven by a triangular waveform of 1 Hz. The SNR was then computed as the background subtracted mean of the signal divided by the mean of the standard deviation of the signal. The drift performance of the detection system was found to be 15 nm/min. We analyzed the digital signals by a LabView virtual instrument to compute the dynamic unbinding forces.
Calibration procedure
During the force calibration process, a bead was trapped and different fluid velocities were applied through the use of the piezoelectric stage to induce a viscous drag force on the trapped bead. A triangular waveform was used to drive the stage, and the frequencies applied ranged from 1 to 6 Hz (Ermilov and Anvari, 2004). The forces induced using these frequencies were in the same range as the rupture forces measured in our experiments. The continuous change in the shadow of the trapped bead was projected onto the surface of the quadrant photodetector generating a voltage signal on each of its four quadrants (V1–V4). Bead displacement from the trapping center in the x direction (corresponding to the direction of fluid flow) in response to the viscous drag force was characterized by the voltage difference Vx = (V1 − V4) + (V2 − V3). For a given laser power, Vx was recorded at different fluid velocities, which corresponded to different trapping forces. The forces were computed by using a modified version of Stokes law,
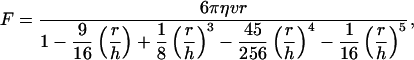 |
(1) |
in which η is the solution viscosity, v is the stage velocity, r is the bead radius, and h is the distance between the center of the bead and the coverslip. The denominator represents the correction factor that must be used to correct the drag force due to the sphere's proximity to the coverslip (Svoboda and Block, 1994).
In this study, we performed the calibration when the pumping laser was set at a maximal output of 5 W, which resulted in Ti:sapph laser power of 310 mW after the microscope objective. Calibration of the bead displacement and trapping force as a function of Vx for a 2-μm-diameter polystyrene bead trapped at h = 10 μm from the coverslip are shown in Fig. 3. The height was determined by focusing the objective on surface-bound debris and then moving the objective up 10 μm using the fine-focus adjustment of the microscope. The light level during bond rupture experiments was kept at the same level as in the calibration procedure. The relationship between photodetector voltage and bead displacement was determined to be ∼0.03 V/μm by immobilizing the bead on the coverslip and moving the stage to a distance equal to the diameter of the bead (2 μm). Since our experiments required the bead to be 10 μm above the coverslip, during the voltage-displacement calibration procedure we made sure to keep the bead in the same focal plane that was used in the trials to determine rupture force. The value of 0.03 V/μm was multiplied by the slope of the line in Fig. 3 B to compute the trap stiffness (∼200 pN/μm). In Fig. 3 B, the force values were obtained by moving the piezoelectric stage at a known velocity, and applying Stokes law.
FIGURE 3.

Calibration of bead displacement and force as a function of photodetector differential output voltage, Vx. (A) There was a linear relationship between bead displacment and Vx. (B) The linear relationship between force and Vx yielded a slope of ∼6.2 pN/mV.
Interaction of A1 and GP Ib-IX
The A1/GP Ib-IX time-resolved rupture force was measured using the quadrant photodetector, which recorded the change in shadow of the trapped bead projected on its quadrants during unbinding experiments. The CHO cell was placed in the solution chamber 10 min before an experiment to allow firm adhesion to the coverslip. We optically trapped a bead coated with A1 at a distance of 4 μm from the cell, and moved the transfected cell toward the trapped bead at a constant velocity by using the piezoelectrically driven stage. The cell was then immediately retracted from the trapped bead at the same velocity. As the piezoelectric stage moved the cell, the photodetector measured the amount of the change in the shadow of the trapped bead, which was induced by displacement of the bead from the trap center. As bonds were breaking and displacing the bead, a continuous curve of Vx against time was obtained. The bead was recentered at the beginning of each experiment.
To account for the cell's shadow on the photodetector, we subtracted the retraction voltage measurements from the approach voltage measurements. This procedure had to be performed since the cell's contribution to the photodetector voltage signal was many times that of the bead, depending on the distance between the bead and the cell. Using the curve depicting the photodetector voltage difference in the approach and retraction phases together with the trapping force calibration curve (Fig. 2 B), the dynamic bond rupture force was measured.
To confirm that the subtraction process was valid, we used 2.0-μm-diameter fluorescent beads (F-8826, Molecular Probes, Eugene, OR) with peak excitation and emission wavelengths of 580 and 605 nm, respectively, coated with A1. For fluorescence excitation (XBO xenon short-arc lamp, 75 W), only the image of the trapped fluorescent bead was projected on the quadrant photodetector. In this way, the image of the cell did not influence our force measurements. Fluorescence illumination passes through an excitation filter (D535/50, Chroma Technology, Rockingham, VT) into the objective to illuminate the trapped fluorescent bead. The fluorescent light emitted from the bead was recollected by the objective, and separated from the excitation light by the dichroic mirror. A filter (D606/55, Chroma Technology) was placed in front of the quadrant photodetector to specifically select the fluorescent light from the bead. The forces for bond rupture using the subtraction method and fluorescent method were found to be comparable.
Velocities of 0.5, 1, 5, 10, 50, and 100 μm/s were investigated, and the laser power remained constant regardless of pulling velocity. The loading rates were computed by multiplying the pulling rate by the trap stiffness and ranged from 200 to 20,000 pN/s. No exogenous modulator was required as we have previously shown that immobilized A1 spontaneously binds to GP Ib-IX (Arya et al., 2002).
Model of bond dissociation
The Bell model is used frequently to evaluate protein-protein interactions under an applied load (Bell, 1978). Under the Bell model, an applied force F distorts the energy landscape of a receptor-ligand complex. When a bond is loaded, the activation barrier is lowered, and the bond will tend to break faster than its unloaded counterpart. The lowering of the activation barrier causes the dissociation rate constant, koff, to increase when the bond is loaded. In the Bell model, the dissociation constant is exponentially related to the applied force F by the equation
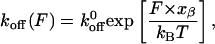 |
(2) |
where 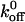 is the unloaded (spontaneous) dissociation rate, kB is Boltzmann's constant (1.3807 × 10−23 J/K), T is absolute temperature, and xβ is the distance between the bound state and transition state along the reaction coordinate. Since the force on the bond is not applied instantaneously, one can consider the load-dependent dissociation by incorporating the loading rate into Eq. 2 and deriving the following equation,
is the unloaded (spontaneous) dissociation rate, kB is Boltzmann's constant (1.3807 × 10−23 J/K), T is absolute temperature, and xβ is the distance between the bound state and transition state along the reaction coordinate. Since the force on the bond is not applied instantaneously, one can consider the load-dependent dissociation by incorporating the loading rate into Eq. 2 and deriving the following equation,
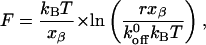 |
(3) |
where r is a constant loading rate (Evans and Ritchie, 1997). This expression predicts a linear relationship between rupture force and the logarithm of the loading rate (Merkel et al., 1999; Tees et al., 2001).
Statistics
Each result is reported as the mean ± SD. The error bars in all graphs indicate standard deviation. The Student's t-test was used to determine if there were statistically significant differences among the computed 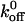 values and rupture force values.
values and rupture force values.
RESULTS
Time-resolved measurements of unbinding forces in wild-type and mutant and GP Ib-IX complexes
Time-resolved unbinding profiles for the A1/wild-type and mutant GP Ib-IX interactions were recorded at loading rates of 100, 200, 1,000, 2,000, 10,000, and 20,000 pN/s. Only the forces in the x direction were analyzed since negligible forces were observed in the y direction during the bond detachment process. In Fig. 4 A, the wild-type CHO αβIX cell was moved away from the trapped A1-coated bead at a pulling rate of 5 μm/s corresponding to an applied loading rate of 1000 pN/s. The minimum value represents the rupture force and corresponds to ∼21 pN. The force profile for a trial in which no adhesion occurred is shown in Fig. 4 B. One should note the positive forces in both graphs that correspond to impingement of the cell upon the trapped bead. We also computed loading rates from the slope before the point of rupture in the force-time graphs and found them to be within 12% of applied values based on the product of stage velocity and trapping stiffness (data not shown). Our force profiles did not show multiple peaks characteristic of sequential breakage (Chen and Moy, 2000), but rather one large peak suggesting that the bonds broke simultaneously.
FIGURE 4.
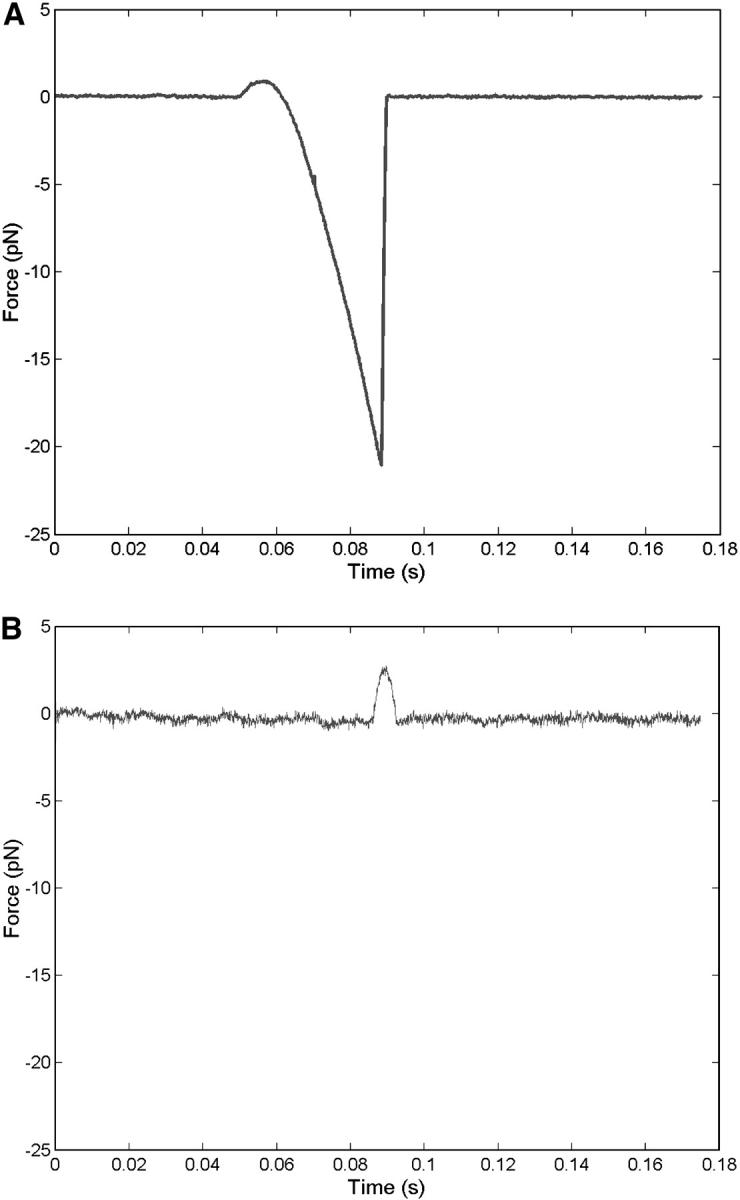
Sample traces of A1/wild-type unbinding force profile acquired using quadrant photodetector. Graphs depict experiments in which adhesion occurred (A) and did not occur (B). Profiles were obtained at 1000 pN/s.
No adhesion was observed between the CHO βIX cells (expressing GP Ibβ and GP IX, but not GP Ibα) and the beads coated with A1 domain, confirming that the A1 domain binds exclusively to the GP Ibα. It is highly improbable that the measured forces were associated with the removal of adsorbed A1 from the polystyrene beads. Gergely et al. (2000) have studied the process of adsorption and found that, within milliseconds, the force required to rupture the nonspecific adhesion between fibronectin and hydrophobic silica was ∼200 pN. In addition, the adhesive forces between proteins and polystyrene after long-term adsorption are several orders of magnitude greater than the forces we have measured (Sagvolden, 1999).
Although passive adsorption has frequently been used to immobilize ligands onto surfaces in previous studies (Gergely et al., 2000), potential problems are associated with this method. When ligands are passively immobilized, the molecules can attach to the surface in many different orientations. If the orientation is not correlated with the specific receptor-ligand interaction, the functionality of the ligand could be compromised (Davies et al., 1994). Instead of passive adsorption, others have used antibodies to attach ligands to surfaces. For example, in their study of selectin-leukocyte interactions, Hanley et al. (2004) used anti-human IgG-Fc monoclonal antibodies to functionalize molecular force probe cantilevers. The cantilevers were subsequently incubated with L- or E-selectin/IgG-Fc chimera protein to ensure proper functional orientation of the selectin. In our study, at least some of the immobilized A1 molecules were clearly functional, since significant A1/GP Ib-IX adhesion was observed.
The plasma membrane of the CHO cells formed visible tethers in 5% of our experiments. In the rare cases where tethers did form, the forces were an order of magnitude greater than the typical rupture forces. We do not include results from experiments in which tethers formed.
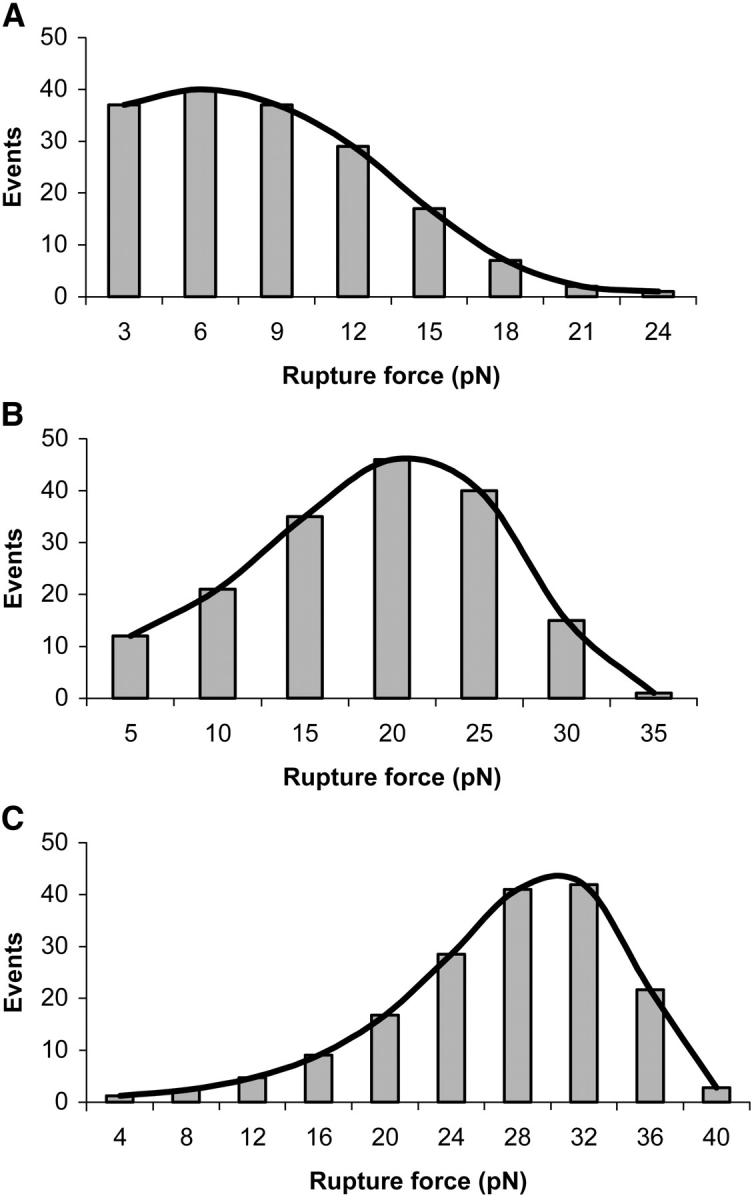
Histograms of rupture forces for A1/wild-type GP Ib-IX bonds obtained at loading rates ranging from 100 to 10,000 pN/s are shown in Fig. 5. We observed that the bond rupture force for the interaction between the A1 domain and both wild-type and mutant GP Ib-IX complexes increased logarithmically with loading rate (Fig. 6). Using Eq. 3 in conjunction with the force-loading rate data, we computed 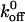 and xβ values for the interactions between A1 and the mutant and wild-type receptors (Table 1). Differences between the
and xβ values for the interactions between A1 and the mutant and wild-type receptors (Table 1). Differences between the 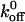 and xβ values for the A1/wild-type GP Ib-IX and A1/mutant GP Ib-IX interactions were all statistically significant (p < 0.05).
and xβ values for the A1/wild-type GP Ib-IX and A1/mutant GP Ib-IX interactions were all statistically significant (p < 0.05).
FIGURE 5.
Histograms depicting distributions of A1/wild-type GP Ib-IX rupture forces. Superposed on the histograms are best-fit curves used to determine the most frequent rupture force. The loading rates were 100 pN/s (A), 1000 pN/s (B), and 10,000 pN/s (C).
FIGURE 6.
Rupture force as a function of loading rate plot for the A1/wild-type GP Ib-IX and mutant GP Ib-IX interactions. The loading rates ranged between 100 and 20,000 pN/s. Mutants 231 and 232 were found to bind less strongly to A1 than mutants 233, 235, 237, 239, and the wild-type cells.
TABLE 1.
Dissociation rates and xβ values of A1 bonds with wild-type and mutated GP Ib-IX receptors
| Cell type |
 (s−1) (s−1) |
xβ (nm) |
|---|---|---|
| Wild-type | 5.47 ± 0.25 | 0.71 ± 0.03 |
| 231 mutant | 7.99 ± 0.53 | 0.97 ± 0.06 |
| 232 mutant | 10.8 ± 0.34 | 1.4 ± 0.04 |
| 233 mutant | 4.07 ± 0.31 | 0.47 ± 0.03 |
| 235 mutant | 3.73 ± 0.29 | 0.44 ± 0.03 |
| 237 mutant | 3.27 ± 0.22 | 0.45 ± 0.03 |
| 239 mutant | 4.21 ± 0.38 | 0.55 ± 0.05 |
| Wild-type (Doggett, 2002) | 3.45 ± 0.37 | 0.016 ± 0.002 |
| Wild-type (Kumar, 2003) | 5.66 | 0.0058 |
| Wild-type (Miura, 2000) | 0.0038 | — |
| 233 (Doggett, 2003) | 0.67 ± 0.11 | 0.031 ± 0.005 |
| 237 (Kumar, 2003) | 2.56 | 0.0089 |
For each cell type in our experiments, there were 170 A1/GP Ib-IX adhesive events and statistically significant differences among the 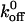 values (p < 0.05).
values (p < 0.05).
DISCUSSION
Platelets must adhere to subendothelial matrix proteins and other activated platelets at sites of vascular injury to achieve hemostasis and participate in subsequent vascular repair. The first step in hemostasis involves bond formation between the platelet GP Ib-IX-V receptor complex and VWF. In addition to mediating platelet adhesion, the GP Ib-IX-V complex on the surface of the platelet also transduces signals required for platelet activation (Kroll et al., 1991). One consequence of these signals is to induce a fibrinogen-binding conformation of αIIbβ3, which then mediates platelet aggregation. There is also evidence that the binding of VWF to GP Ib-IX can induce cytoskeletal reorganization in platelets under physiological flow conditions (Yuan et al., 1999). Although the A1/GP Ib-IX interaction is critical in the process of hemostasis formation, fundamental data regarding the mechanics of this particular bond is lacking.
Under equilibrium conditions, all molecules in solution will bond and dissociate without any applied force. However, an applied force on a bond reduces the rupture time to less than the characteristic bond lifetime, toff, required for spontaneous dissociation (Evans, 2001). The values for toff vary widely for different types of bonds, but many biological interactions have characteristic toff values ranging from 0.1–1 s (Hanley et al., 2003; Chesla et al., 2000). It has been well established that energetic barriers along the pathways for dissociation determine bond lifetime, and these barriers will be lowered if an external force is applied. Under the Bell model, the rate of unbinding will increase exponentially with force.
From our experimental data using A1 and wild-type CHO αβIX cells, we used the slope of the line relating force and loading rate and determined 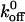 to be 5.47 s−1. Although we are using the model associated with a single sharp energy barrier (Evans and Ritchie, 1997), we are not excluding the possibility that the A1/GP Ib-IX energy landscape has multiple transition states. For example, Zhang et al. (2002) observed two distinct linear regimes in their LFA-1/ICAM-1 unbinding data, thereby indicating two activation barriers for the interaction. Dissociation constants were also computed for the interactions between A1 and the mutant GP Ib-IX complexes. The GOF mutants (233, 235, 237, and 239) all exhibited
to be 5.47 s−1. Although we are using the model associated with a single sharp energy barrier (Evans and Ritchie, 1997), we are not excluding the possibility that the A1/GP Ib-IX energy landscape has multiple transition states. For example, Zhang et al. (2002) observed two distinct linear regimes in their LFA-1/ICAM-1 unbinding data, thereby indicating two activation barriers for the interaction. Dissociation constants were also computed for the interactions between A1 and the mutant GP Ib-IX complexes. The GOF mutants (233, 235, 237, and 239) all exhibited 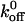 values <5.47 s−1 whereas the two loss-of-function mutants (231 and 232) both had
values <5.47 s−1 whereas the two loss-of-function mutants (231 and 232) both had 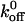 values >5.47 s−1.
values >5.47 s−1.
Several previous studies have estimated 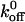 values for the A1/GP Ibα interaction using conventional binding assays. From saturation binding experiments involving radiolabeled VWF A1 domain, Miura et al. (2000) suggested that slow intrinsic binding kinetics were responsible for rapid GP Ibα adhesion to VWF. Miura et al. reported a
values for the A1/GP Ibα interaction using conventional binding assays. From saturation binding experiments involving radiolabeled VWF A1 domain, Miura et al. (2000) suggested that slow intrinsic binding kinetics were responsible for rapid GP Ibα adhesion to VWF. Miura et al. reported a 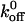 value of 0.0038 s−1 for the interaction between radiolabeled A1 and GP Ibα present on formalin-fixed platelets. The
value of 0.0038 s−1 for the interaction between radiolabeled A1 and GP Ibα present on formalin-fixed platelets. The 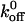 value estimated by Miura et al. was three orders of magnitude less than the value reported by us and the value of 3.45 s−1 reported by Doggett et al. (2002), who studied the A1/GP Ibα interaction using a parallel-plate flow chamber. It is important to note that Miura et al. measured the kinetics of binding between the receptor and ligand using gamma radiation counting without any force applied to the bond. It is clear that the presence of an applied force on the bond greatly influences the kinetics. In a subsequent study, Doggett et al. examined the 233 valine mutation associated with ptVWD and computed
value estimated by Miura et al. was three orders of magnitude less than the value reported by us and the value of 3.45 s−1 reported by Doggett et al. (2002), who studied the A1/GP Ibα interaction using a parallel-plate flow chamber. It is important to note that Miura et al. measured the kinetics of binding between the receptor and ligand using gamma radiation counting without any force applied to the bond. It is clear that the presence of an applied force on the bond greatly influences the kinetics. In a subsequent study, Doggett et al. examined the 233 valine mutation associated with ptVWD and computed 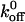 for the A1/mutant GP Ib-IX interaction to be 0.67 s−1 (Doggett et al., 2003). In addition, Kumar et al. (2003) estimated the
for the A1/mutant GP Ib-IX interaction to be 0.67 s−1 (Doggett et al., 2003). In addition, Kumar et al. (2003) estimated the 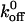 values for the A1/wild-type GP Ib-IX and A1/mutant 237 interactions using a parallel-plate flow chamber to be 5.66 s−1 and 2.56 s−1, respectively. As in the aforementioned studies, our results indicate a decrease in
values for the A1/wild-type GP Ib-IX and A1/mutant 237 interactions using a parallel-plate flow chamber to be 5.66 s−1 and 2.56 s−1, respectively. As in the aforementioned studies, our results indicate a decrease in 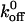 values for the A1/mutant GP Ib-IX interactions in comparison to the A1/wild-type GP Ib-IX interactions. It is important to note Doggett et al. (2002) used platelets in their experiments, whereas we used transfected CHO cells. In addition, Kumar et al. (2003) independently estimated
values for the A1/mutant GP Ib-IX interactions in comparison to the A1/wild-type GP Ib-IX interactions. It is important to note Doggett et al. (2002) used platelets in their experiments, whereas we used transfected CHO cells. In addition, Kumar et al. (2003) independently estimated 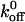 based on the duration of transient tethering events between GP Ibα and the A1-coated substrate under conditions of hydrodynamic flow. In contrast, our optical tweezers-based assay was performed without any hydrodynamic flow to examine the kinetics of bond rupture at the single-molecule level. It is important to note that both flow chambers and optical tweezers impose forces on the bonds but not necessarily the same forces. These forces are clearly important in many biological phenomena, including the immune response and hemostasis, and from an evolutionary standpoint, have most likely influenced the structures and functions of the proteins associated with these processes.
based on the duration of transient tethering events between GP Ibα and the A1-coated substrate under conditions of hydrodynamic flow. In contrast, our optical tweezers-based assay was performed without any hydrodynamic flow to examine the kinetics of bond rupture at the single-molecule level. It is important to note that both flow chambers and optical tweezers impose forces on the bonds but not necessarily the same forces. These forces are clearly important in many biological phenomena, including the immune response and hemostasis, and from an evolutionary standpoint, have most likely influenced the structures and functions of the proteins associated with these processes.
We computed the Bell model parameter of xβ for A1 binding to wild-type and mutated GP Ib-IX receptors. Although our xβ values are larger than those found for selectins (Chen and Springer, 2001) and the xβ values from the Kumar and Doggett studies, we are confident that we are not extracting the receptor from the plasma membrane. In a previous study, we showed that the bond strength between plasma VWF and GP Ib-IX was dependent upon the modulator, either botrocetin or ristocetin, that was used to facilitate the interaction (Arya et al., 2002). If we were actually extracting the receptor, the force observed would be the same regardless of which modulator was used. It is important to note that our 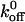 values are close to the previously reported
values are close to the previously reported 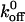 values for the A1/ wild-type and mutant GP Ib-IX interactions; however, our xβ values are an order of magnitude greater than the values reported by Kumar and Doggett. Like the other studies, we are also using the model employing a single sharp energy barrier. However it is quite possible that two or more energy barriers exist for the A1/GP Ib-IX interaction. We surmise that the other studies are probing the high-strength regime of potentially two energy barriers. This higher energy barrier corresponds to the smaller of two possible xβ values. Using a two barrier system, our
values for the A1/ wild-type and mutant GP Ib-IX interactions; however, our xβ values are an order of magnitude greater than the values reported by Kumar and Doggett. Like the other studies, we are also using the model employing a single sharp energy barrier. However it is quite possible that two or more energy barriers exist for the A1/GP Ib-IX interaction. We surmise that the other studies are probing the high-strength regime of potentially two energy barriers. This higher energy barrier corresponds to the smaller of two possible xβ values. Using a two barrier system, our 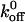 values are similar for surpassing both energy barriers, and thus, we have the same
values are similar for surpassing both energy barriers, and thus, we have the same 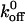 values as the others. However, it is generally accepted that the slope associated with a high-strength regime is greater than the slope associated with a low-strength regime (Evans, 1998). As a result, the xβ value (i.e., the inverse slope) related to the low-strength regime is greater than the xβ value associated with the high-strength regime. Thus, our xβ values, which correspond to the lower-energy barrier, will be larger than the xβ values of others, which may indeed correspond to the higher-energy barrier. In addition, one should notice that there appears to be a consistent pattern associated with the deviations of the measured force values from the fitted values in Fig. 6. Although our group and others investigating the VWF/GP Ib-IX interaction have all used the single sharp energy barrier model, this consistency suggests a more complicated energy landscape with multiple transition states. Using this logic, one can see why our
values as the others. However, it is generally accepted that the slope associated with a high-strength regime is greater than the slope associated with a low-strength regime (Evans, 1998). As a result, the xβ value (i.e., the inverse slope) related to the low-strength regime is greater than the xβ value associated with the high-strength regime. Thus, our xβ values, which correspond to the lower-energy barrier, will be larger than the xβ values of others, which may indeed correspond to the higher-energy barrier. In addition, one should notice that there appears to be a consistent pattern associated with the deviations of the measured force values from the fitted values in Fig. 6. Although our group and others investigating the VWF/GP Ib-IX interaction have all used the single sharp energy barrier model, this consistency suggests a more complicated energy landscape with multiple transition states. Using this logic, one can see why our 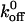 values are similar to those reported by others, but our xβ values are greater than those previously reported.
values are similar to those reported by others, but our xβ values are greater than those previously reported.
Many similarities exist between the trafficking of cells into tissues during the immune response and platelet adhesion to the vessel wall during hemostasis. For example, the three stages that govern the conversion of an unbound blood cell to an adherent one under flow conditions in both the immune response and hemostasis are initial tethering, transient rolling adhesion, and firm adhesion. The selectins are critical in mediating the initial tethering events of leukocytes to the blood vessel wall during inflammation. As suggested by Doggett et al. (2002), we also believe that the A1/GP Ib-IX bond does indeed possess kinetic properties similar to the bonds formed by selectins, and this crucial interaction helps to promote rapid platelet adhesion to the VWF matrix under flow conditions. A previous study using surface plasmon resonance techniques found the unloaded dissociation constant for the P-selectin/P-selectin glycoprotein ligand-1 interaction to be ∼1.4 s−1(Mehta et al., 1998), which is on the same order as the 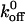 value for the A1/GP Ib-IX interaction. This is reasonable since the selectins are involved in the process of inflammation, similar to the process of hemostasis associated with adhesion of platelets to vascular surfaces.
value for the A1/GP Ib-IX interaction. This is reasonable since the selectins are involved in the process of inflammation, similar to the process of hemostasis associated with adhesion of platelets to vascular surfaces.
One should note that the physiologic shear rates (several hundred s−1) imposed on platelets during the initial phase of hemostasis are on the same order of magnitude as the higher loading rates (10,000–20,000 pN/s) used in this study (Litvinov et al., 2002). It is also important to realize that the A1/GP Ib-IX rupture values were slightly below 40 pN at a loading rate as high as 20,000 pN/s. This force value is much lower than bond strengths as determined by atomic force microscopy between various integrins and extracellular matrix proteins (∼100 pN), interactions required for irreversible cell adhesion (Lehenkari and Horton, 1999). Thus this difference in bond strengths is reasonable since the VWF/GP Ib-IX interaction is not associated with irreversible platelet adhesion but rather is responsible for the tethering and rolling of platelets within the vessel.
Since the pulling rate in some of the trials was 100 μm/s, it is important to address the importance of inertia and drag on our observed rupture forces. Ermilov and Anvari have analyzed the significance of inertia and hydrodynamic coupling on observed forces at different pulling rates (Ermilov and Anvari, 2004). Their theoretical calculations clearly indicate that the inertia of a 2-μm polystyrene microsphere is negligible during an exponential increase of microsphere velocity from 0 to 1000 μm/s. The inertia is <5 pN even when the time constant is 1 μs. Since we are only accelerating the bead to a maximum velocity of 100 μm/s, the inertia is certainly negligible. Also, the time constant for our system exceeds 1 μs. In addition, the calculations of Ermilov and Anvari indicate that the drag contributes significantly to the rupture force when the pulling rate is ∼1000 μm/s. This velocity is an order of magnitude greater than the highest pulling rate used in our study.
One of the primary goals of force spectroscopy studies is to correlate the structure of a protein to its various biological functions. The crystal structure of GP Ibα indicates the presence of a β-loop comprising residues 227–241 (Huizinga et al., 2002). Extending over the interior of the GP Ibα leucine-rich repeat concave face, the loop forms a projection that appears to hinder the binding of the VWF A1 domain. Thus, a conformational change in the loop may be necessary for A1/GP Ibα binding to occur. Uff et al. (2002) believe the projection could either fold back and actively promote binding or simply be removed. Based on our adhesion studies, individual residues in the region of the loop play a critical role in binding to A1. We found that valine mutations in residues 233, 235, 237, and 239 increased the A1/GP Ibα single-bond strength with respect to the wild-type receptor regardless of loading rate. These GOF mutations lie on the outer tips of the loop and are most likely promoting a more open conformation of the glycoprotein (Uff et al., 2002). It is possible that residues 231 and 232 substantially decreased the A1/GP Ibα bond strength due to steric hindrance. We surmise that the projection of the loop did not extend out to a sufficient length in the LOF mutants. Thus, our adhesion studies indicate that individual amino acid mutations in GP Ibα can dramatically affect the ability of GP Ib-IX to bind A1, and alter the bond kinetics associated with the VWF/GP Ib-IX interaction in hemostasis. Future studies should focus on analysis of the VWF/GP Ib-IX interaction under hydrodynamic flow on the single-bond level to further delineate the molecular aspects of ptVWD.
CONCLUSION
The use of optical tweezers in conjunction with optical detection of the trapped bead displacement using a quadrant photodetector allowed us to dynamically monitor unbinding forces between VWF A1 domain and wild-type or mutant GP Ib-IX receptor complexes. Based on these measurements performed at loading rates ranging between 200 and 20,000 pN/s, we observed that single point mutations in the GP Ibα subunit of GP Ib-IX could induce significant changes in the receptor's ability to bind A1. The unbinding studies demonstrated a logarithmic increase in mutant and wild-type GP Ib-IX/A1 rupture force as the loading rate increased. Although the detailed structure of the mutant complexes has yet to be determined, we surmise that changes in the β-loop of GP Ibα are responsible for the increase or decrease in binding between A1 and the various GP Ib-IX receptors across all loading rates examined.
References
- Arya, M., B. Anvari, G. M. Romo, M. A. Cruz, J. F. Dong, L. V. McIntire, J. L. Moake, and J. A. Lopez. 2002. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 99:3971–3977. [DOI] [PubMed] [Google Scholar]
- Arya, M., J. A. Lopez, G. M. Romo, M. A. Cruz, A. Kasirer-Friede, S. J. Shattil, and B. Anvari. 2003. Glycoprotein Ib-IX-mediated activation of integrin α(IIb)β(3): effects of receptor clustering and von Willebrand factor adhesion. J Thromb. Haemost. 1:1150–1157. [DOI] [PubMed] [Google Scholar]
- Ashkin, A., J. M. Dziedzic, J. E. Bjorkholm, and S. Chu. 1986. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 11:288–290. [DOI] [PubMed] [Google Scholar]
- Bell, G. I. 1978. Models for the specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- Berndt, M. C., Y. Shen, S. M. Dopheide, E. E. Gardiner, and R. K. Andrews. 2001. The vascular biology of the glycoprotein Ib-IX-V complex. Thromb. Haemost. 86:178–188. [PubMed] [Google Scholar]
- Berndt, M. C., C. M. Ward, W. J. Booth, P. A. Castaldi, A. V. Mazurov, and R. K. Andrews. 1992. Identification of aspartic acid 514 through glutamic acid 542 as a glycoprotein Ib-IX complex receptor recognition sequence in von Willebrand factor. Mechanism of modulation of von Willebrand factor by ristocetin and botrocetin. Biochemistry. 31:11144–11151. [DOI] [PubMed] [Google Scholar]
- Chen, A., and V. T. Moy. 2000. Cross-linking of cell surface receptors enhances cooperativity of molecular adhesion. Biophys. J. 78:2814–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., and T. A. Springer. 2001. Selectin receptor-ligand bonds: Formation limited by shear rate and dissociation governed by the Bell model. Proc. Natl. Acad. Sci. USA. 98:950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesla, S. E., P. Li, S. Nagarajan, P. Selvaraj, and C. Zhu. 2000. The membrane anchor influences ligand binding two-dimensional kinetic rates and three-dimensional affinity of FcγRIII (CD16). J. Biol. Chem. 275:10235–10246. [DOI] [PubMed] [Google Scholar]
- Cruz, M. A., R. I. Handin, and R. J. Wise. 1993. The interaction of the von Willebrand factor-A1 domain with platelet glycoprotein Ib/IX. The role of glycosylation and disulfide bonding in a monomeric recombinant A1 domain protein. J. Biol. Chem. 268:21238–21245. [PubMed] [Google Scholar]
- Davies, J., A. C. Dawkes, A. G. Haymes, C. J. Roberts, R. F. Sunderland, M. J. Wilkins, M. C. Davies, S. J. Tendler, D. E. Jackson, and J. C. Edwards. 1994. A scanning tunnelling microscopy comparison of passive antibody adsorption and biotinylated antibody linkage to streptavidin on microtiter wells. J. Immunol. Methods. 167:263–269. [DOI] [PubMed] [Google Scholar]
- De Marco, L., and S. S. Shapiro. 1981. Properties of human asialo-factor VIII. A ristocetin-independent platelet-aggregating agent. J. Clin. Invest. 68:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett, T. A., G. Girdhar, A. Lawshe, J. L. Miller, I. J. Laurenzi, S. L. Diamond, and T. G. Diacovo. 2003. Alterations in the intrinsic properties of the GPIbα-VWF tether bond define the kinetics of the platelet-type von Willebrand disease mutation, Gly233Val. Blood. 102:152–160. [DOI] [PubMed] [Google Scholar]
- Doggett, T. A., G. Girdhar, A. Lawshe, D. W. Schmidtke, I. J. Laurenzi, S. L. Diamond, and T. G. Diacovo. 2002. Selectin-like kinetics and biomechanics promote rapid platelet adhesion in flow: the GPIbα-vWF tether bond. Biophys. J. 83:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, J., A. J. Schade, G. M. Romo, R. K. Andrews, S. Gao, L. V. McIntire, and J. A. Lopez. 2000. Novel gain-of-function mutations of platelet glycoprotein IBα by valine mutagenesis in the Cys209-Cys248 disulfide loop. Functional analysis under statis and dynamic conditions. J. Biol. Chem. 275:27663–27670. [DOI] [PubMed] [Google Scholar]
- Dong, J. F., G. Sae-Tung, and J. A. Lopez. 1997. Role of glycoprotein V in the formation of the platelet high-affinity thrombin-binding site. Blood. 89:4355–4363. [PubMed] [Google Scholar]
- Emsley, J., M. Cruz, R. Handin, and R. Liddington. 1998. Crystal structure of the von Willebrand Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 273:10396–10401. [DOI] [PubMed] [Google Scholar]
- Ermilov, S., and B. Anvari. 2004. Dynamic measurements of transverse optical trapping force in biological applications. Ann. Biomed. Eng. 32:1016–1026. [DOI] [PubMed] [Google Scholar]
- Evans, E. 1998. Energy landscapes of biomolecular adhesion and receptor anchoring at interfaces explored with dynamic force spectroscopy. Faraday Discuss. 1:1–16. [DOI] [PubMed] [Google Scholar]
- Evans, E. 2001. Probing the relation between force—lifetime—and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 30:105–128. [DOI] [PubMed] [Google Scholar]
- Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa, K., H. Suzuki, B. McMullen, and D. Chung. 2001. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 98:1662–1666. [DOI] [PubMed] [Google Scholar]
- Gergely, C., J. Voegel, P. Schaaf, B. Senger, M. Maaloum, J. K. Horber, and J. Hemmerle. 2000. Unbinding process of adsorbed proteins under external stress studied by atomic force microscopy spectroscopy. Proc. Natl. Acad. Sci. USA. 97:10802–10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen, H. E., R. Robles, B. Lammle, and M. Furlan. 2001. Partial amino acid sequence of purified von Willebrand factor-cleaving protease. Blood. 98:1654–1661. [DOI] [PubMed] [Google Scholar]
- Goto, S., Y. Ikeda, E. Saldivar, and Z. M. Ruggeri. 1998. Distinct mechanisms of platelet aggregation as a consequence of different shearing flow conditions. J. Clin. Invest. 101:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley, W., O. McCarty, S. Jadhav, Y. Tseng, D. Wirtz, and K. Konstantopoulos. 2003. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 278:10556–10561. [DOI] [PubMed] [Google Scholar]
- Hanley, W. D., D. Wirtz, and K. Konstantopoulos. 2004. Distinct kinetic and mechanical properties govern selectin-leukocyte interactions. J. Cell Sci. 117:2503–2511. [DOI] [PubMed] [Google Scholar]
- Huizinga, E. G., S. Tsuji, R. A. Romijn, M. E. Schiphorst, P. G. de Groot, J. J. Sixma, and P. Gros. 2002. Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science. 297:1176–1179. [DOI] [PubMed] [Google Scholar]
- Kroll, M. H., T. S. Harris, J. L. Moake, R. I. Handin, and A. I. Schafer. 1991. von Willebrand factor binding to platelet GpIb initiates signals for platelet activation. J. Clin. Invest. 88:1568–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll, M. H., J. D. Hellums, L. V. McIntire, A. I. Schafer, and J. L. Moake. 1996. Platelets and shear stress. Blood. 88:1525–1541. [PubMed] [Google Scholar]
- Kumar, R. A., J. F. Dong, J. A. Thaggard, M. A. Cruz, J. A. Lopez, and L. V. McIntire. 2003. Kinetics of GPIbα-vWF-A1 tether bond under flow: effect of GPIbα mutations on the association and dissociation rates. Biophys. J. 85:4099–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehenkari, P. P., and M. A. Horton. 1999. Single integrin molecule adhesion forces in intact cells measured by atomic force microscopy. Biochem. Biophys. Res. Commun. 259:645–650. [DOI] [PubMed] [Google Scholar]
- Leitz, G., E. Fallman, S. Tuck, and O. Axner. 2002. Stress response in Caenorhabditis elegans caused by optical tweezers: wavelength, power, and time dependence. Biophys. J. 82:2224–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinov, R. I., H. Shuman, J. S. Bennett, and J. W. Weisel. 2002. Binding strength and activation state of single fibrinogen-integrin pairs on living cells. Proc. Natl. Acad. Sci. USA. 99:7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, P., R. D. Cummings, and R. P. McEver. 1998. Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J. Biol. Chem. 273:32506–32513. [DOI] [PubMed] [Google Scholar]
- Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- Miller, J. L., and A. Castella. 1982. Platelet-type von Willebrand's disease: characterization of a new bleeding disorder. Blood. 60:790–794. [PubMed] [Google Scholar]
- Miller, J. L., D. Cunningham, V. A. Lyle, and C. N. Finch. 1991. Mutation in the gene encoding the alpha chain of platelet glycoprotein Ib in platelet-type von Willebrand disease. Proc. Natl. Acad. Sci. USA. 88:4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, S., C. Q. Li, Z. Cao, H. Wang, M. R. Wardell, and J. E. Sadler. 2000. Interaction of von Willebrand factor domain A1 with platelet glycoprotein Ibα-(1–289). Slow intrinsic binding kinetics mediate rapid platelet adhesion. J. Biol. Chem. 275:7539–7546. [DOI] [PubMed] [Google Scholar]
- Miyata, S., and Z. M. Ruggeri. 1999. Distinct structural attributes regulating von Willebrand factor A1 domain interaction with platelet glycoprotein Ibα under flow. J. Biol. Chem. 274:6586–6593. [DOI] [PubMed] [Google Scholar]
- Moog, S., P. Mangin, N. Lenain, C. Strassel, C. Ravanat, S. Schuhler, M. Freund, M. Santer, M. Kahn, B. Nieswandt, C. Gachet, J. P. Cazenave, and F. Lanza. 2001. Platelet glycoprotein V binds to collagen and participates in platelet adhesion and aggregation. Blood. 98:1038–1046. [DOI] [PubMed] [Google Scholar]
- Neuman, K. C., E. H. Chadd, G. F. Liou, K. Bergman, and S. M. Block. 1999. Characterization of photodamage to Escherichia coli in optical traps. Biophys. J. 77:2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri, Z. M. 1997. Mechanisms initiating platelet thrombus formation. Thromb. Haemost. 78:611–616. [PubMed] [Google Scholar]
- Russell, S. D., and G. J. Roth. 1993. Pseudo-von Willebrand disease: a mutation in the platelet glycoprotein Ib α gene associated with a hyperactive surface receptor. Blood. 81:1787–1791. [PubMed] [Google Scholar]
- Sadler, J. E. 1998. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 67:395–424. [DOI] [PubMed] [Google Scholar]
- Sagvolden, G. 1999. Protein adhesion force dynamics and single adhesion events. Biophys. J. 77:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda, K., and S. M. Block. 1994. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 23:247–285. [DOI] [PubMed] [Google Scholar]
- Tait, A. S., S. L. Cranmer, S. P. Jackson, I. W. Dawes, and B. H. Chong. 2001. Phenotype changes resulting in high-affinity binding of von Willebrand factor to recombinant glycoprotein Ib-IX: analysis of the platelet-type von Willebrand disease mutations. Blood. 98:1812–1818. [DOI] [PubMed] [Google Scholar]
- Tees, D. F., R. E. Waugh, and D. A. Hammer. 2001. A microcantilever device to assess the effect of force on the lifetime of selectin-carbohydrate bonds. Biophys. J. 80:668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uff, S., J. M. Clemetson, T. Harrison, K. J. Clemetson, and J. Emsley. 2002. Crystal structure of the platelet glycoprotein Ib(alpha) N-terminal domain reveals an unmasking mechanism for receptor activation. J. Biol. Chem. 277:35657–35663. [DOI] [PubMed] [Google Scholar]
- Weiss, H. J., D. Meyer, R. Rabinowitz, G. Pietu, J. P. Girma, W. J. Vicic, and J. Rogers. 1982. Pseudo-von Willebrand's disease. An intrinsic platelet defect with aggregation by unmodified human factor VIII/von Willebrand factor and enhanced adsorption of its high-molecular-weight multimers. N. Engl. J. Med. 306:326–333. [DOI] [PubMed] [Google Scholar]
- Yuan, Y., S. Kulkarni, P. Ulsemer, S. L. Cranmer, C. L. Yap, W. S. Nesbitt, I. Harper, N. Mistry, S. M. Dopheide, S. C. Hughan, D. Williamson, C. de la Salle, H. H. Salem, F. Lanza, and S. P. Jackson. 1999. The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. J. Biol. Chem. 274:36241–36251. [DOI] [PubMed] [Google Scholar]
- Zhang, X., E. Wojcikiewicz, and V. T. Moy. 2002. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys. J. 83:2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]