

Abstract
Regulation of swelling-activated Cl− current (ICl,swell) is complex, and multiple signaling cascades are implicated. To determine whether protein tyrosine kinase (PTK) modulates ICl,swell and to identify the PTK involved, we studied the effects of a broad-spectrum PTK inhibitor (genistein), selective inhibitors of Src (PP2, a pyrazolopyrimidine) and epidermal growth factor receptor (EGFR) kinase (PD-153035), and a protein tyrosine phosphatase (PTP) inhibitor (orthovanadate). ICl,swell evoked by hyposmotic swelling was increased 181 ± 17% by 100 μM genistein, and the genistein-induced current was blocked by the selective ICl,swell blocker tamoxifen (10 μM). Block of Src with PP2 (10 μM) stimulated tamoxifen-sensitive ICl,swell by 234 ± 27%, mimicking genistein, whereas the inactive analog of PP2, PP3 (10 μM), had no effect. Moreover, block of PTP by orthovanadate (1 mM) inhibited ICl,swell and prevented its stimulation by PP2. In contrast with block of Src, block of EGFR kinase with PD-153035 (20 nM) inhibited ICl,swell. Several lines of evidence argue that the PP2-stimulated current was ICl,swell: 1) the stimulation was volume dependent, 2) the current was blocked by tamoxifen, 3) the current outwardly rectified with both symmetrical and physiological Cl− gradients, and 4) the current reversed near the Cl− equilibrium potential. To rule out contributions of other currents, Cd2+ (0.2 mM) and Ba2+ (1 mM) were added to the bath. Surprisingly, Cd2+ suppressed the decay of Cd2+ plus Ba2+ eliminated time-dependent ICl,swell, and currents between −100 and −100 mV. Nevertheless, these divalent ions did not eliminate ICl,swell or prevent its stimulation by PP2. The results indicate that tyrosine phosphorylation controls ICl,swell, and regulation of ICl,swell by the Src and EGFR kinase families of PTK is antagonistic.
Keywords: volume-sensitive Cl− current, AG 1879, PD-153035, orthovanadate, tamoxifen, genistein, epidermal growth factor receptor
OSMOTIC SWELLING OR HYDROSTATIC inflation of cardiac myocytes and numerous other tissues evokes the volume-sensitive Cl− current ICl,swell. This current is outwardly rectifying, partially inactivated at positive voltages, and blocked by tamoxifen. These biophysical and pharmacological characteristics distinguish ICl,swell from other Cl− currents (for reviews, see Refs. 4 and 28). Under isosmotic conditions, ICl,swell contributes to the background Cl− current (17, 18) and is activated in models of cardiac disease (12) and by stretching β1-integrins (7, 8). Functionally, the activation of ICl,swell influences both cardiac electrical activity (16, 30, 49) and cell volume (11, 12).
The signaling that underlies the activation of ICl,swell is complex, and evidence implicates protein kinases C and A and protein tyrosine kinase (PTK) in its regulation in the heart (4, 28) and other tissues (32). PTK is activated by osmotic swelling of myocytes within 5 s (37, 38) and therefore is well positioned to be an early step in the signaling process. Although substantial evidence indicates that phosphorylation and dephosphorylation of tyrosine residues are involved in the control of ICl,swell, the details remain obscure. Studies with the broad-spectrum PTK inhibitor genistein found that blocking PTK inhibits ICl,swell in dog atrial cells (44), calf pulmonary artery endothelial cells (51), and rabbit ciliary epithelial cells (40). On the other hand, genistein augments ICl,swell in human atrial myocytes (15), and protein tyrosine phosphatase (PTP) inhibitors suppress ICl,swell in bovine chromaffin cells (14) and mouse L-fibroblasts (45). Thus interventions that lead to both phosphorylation and dephosphorylation of tyrosine are capable of inhibiting ICl,swell.
The apparent inconsistency in the relationship between the phosphorylation state of tyrosine residues and the activity of ICl,swell may indicate that regulatory processes are tissue or species specific as previously suggested (32, 33). Another possibility is that ICl,swell is differentially regulated by various families of PTK. Recently, studies on human atrial myocytes revealed that specific inhibition of Src leads to activation of ICl,swell, whereas specific inhibition of epidermal growth factor receptor (EGFR) kinase causes suppression of current (15).
Previous studies on heart focused on the role of PTK in atrial myocytes. The goal of the present study was to evaluate the role of the Src and EGFR kinase families of PTK in the egulation of ICl,swell in ventricular cells. As in human (15) but not canine (44) atrial myocytes, specific inhibition of Src and inhibition of multiple PTKs by genistein stimulated ICl,swell in hyposmotic bathing media, whereas specific inhibition of EGFR kinase suppressed ICl,swell. Moreover, the PTP inhibitor orthovanadate (21) reduced ICl,swell and precluded its activation by Src inhibition. Src activity is not, however, the primary factor that controls the response of ventricular ICl,swell to osmotic stress. Blocking of Src did not alter ICl,swell under isosmotic conditions. Finally, we found that the time-dependent component of ICl,swell at positive voltages could be inhibited without altering the regulation of time-independent ICl,swell by Src. These data suggest that Src- and EGFR kinase-dependent tyrosine phosphorylation and PTP-dependent tyrosine dephosphorylation participate in the regulation of ICl,swell in ventricular myocytes.
METHODS
Ventricular myocyte isolation.
Left ventricular myocytes were freshly isolated from New Zealand White rabbits (~3 kg body wt). Hearts were excised using methods approved by the Institutional Animal Care and Use Committee, mounted on a Langendorff apparatus, and initially perfused with 37°C oxygenated Tyrode solution that contained (in mM) 130 NaCl, 5 KCl, 3 MgCl2, 1.8 CaCl2, 0.4 KH2PO4, 5 HEPES, 15 taurine, 5 creatine, and 10 glucose, pH 7.25. After a 5-min perfusion with Ca2+-free Tyrode solution that contained 0.1 mM Na2-EGTA, the perfusate was switched to Ca2+-free Tyrode solution that contained 0.4–0.5 mg/ml collagenase (type II; Worthington), 0.05 mg/ml pronase (type XIV; Sigma-Aldrich), and 1.5 mg/ml BSA (Sigma-Aldrich). At selected intervals (10–20 min), portions of the left ventricle were excised, cut into strips, placed in test tubes, and gently agitated. After filtration through nylon mesh to remove debris, myocytes were washed twice and stored in modified Kraft-Brühe solution that contained (in mM) 120 K-glutamate, 10 KCl, 10 KH2PO4, 1.8 MgSO4, 0.5 K2-EGTA, 10 taurine, 20 glucose, 10 mannitol, and 10 HEPES (pH 7.2; 295 mosM). Myocytes were used within 8 h of isolation, and only rod-shaped quiescent cells with well-defined regular striations and no evidence of membrane blebbing were selected for study.
Experimental solutions and drugs.
Cells were placed in a poly-l-lysine-coated glass-bottomed chamber (~0.3 ml) mounted on an inverted microscope (Diaphot; Nikon) and were superfused with bathing solution ~22°C) at 2–3 ml/min; solution changes were complete within ~10 s. Anion currents were isolated by replacing Na+ and K+ in the bathing media with equimolar amounts of N-methyl-d-glucamine (NMDG) and adding Cs+ to the bath and pipette solutions. Standard bathing solution contained (in mM) 90 NMDG-Cl, 3 MgCl2, 4.63 CaCl2, 5 Cs2-EGTA, 10 HEPES, 10 glucose, and 0–100 mannitol (pH 7.4). This provides a free Ca2+ concentration of ~1.5 μM (WinMaxC 2.4; www.stanford.edu/~cpatton/maxc.html). In some experiments, 0.2 mM CdCl2 or both 1 mM BaCl2 and 0.2 mM CdCl2 were added to the bath solution. This also necessitated omission of Cs2-EGTA (replaced by 5 mM CsCl), and bath CaCl2 was reduced to 0.1 mM. Bath solutions were designed to allow adjustment of osmolarity with mannitol at a constant ionic strength. Isosmotic (1T) solution was set as 300 mosM, and hypoosmotic (0.7T) solution was ~200 mosM. An osmometer (Osmette S; Precision Systems) was used to routinely verify solution composition.
Tamoxifen (10 mM), genistein (100 mM), the pyrazolopyrimidines PP2 (10 mM, also termed AG 1879) and PP3 (10 mM), and PD-153035 (250 μM) were dissolved in dimethyl sulfoxide (DMSO) at the indicated concentrations and kept frozen (−20°C) in aliquots until use. Cs3VO4 (orthovanadate) was added directly to the bath solution. Tamoxifen, orthovanadate, and DMSO were from Sigma-Aldrich, and the remaining agents were from Calbiochem.
Electrophysiological recordings.
Patch electrodes were made from thin-walled 7740 borosilicate glass (Sutter) and fire polished (initial resistance, 2–3 MΩ). Standard electrode-filling solution contained (in mM) 110 Cs-aspartate, 20 CsCl, 2.5 Mg-ATP, 8 Cs2-EGTA, 0.15 CaCl2, and 10 HEPES, pH 7.1 (liquid junction potential, −11.5 ± 0.7 mV; n = 9). For some experiments, a high- Cl− pipette solution was used; it contained (in mM) 31.7 Cs-aspartate, 98.3 CsCl, 2.5 Mg-ATP, 8 Cs2-EGTA, 0.15 CaCl2, and 10 HEPES, pH 7.1 (liquid junction potential, −6.5 ± 0.5 mV; n = 5). This provided a free Ca2+ concentration of ~60 nM for both pipette solutions. A 3 M KCl agar bridge was used as ground. Seal resistances of 5–30 GΩ were typically achieved, and the measured junction potential was subtracted before seal formation.
Whole cell currents recorded with an Axoclamp 200A or 200B amplifier (Axon) were low pass filtered at 2 kHz (Bessel) and digitized at 5 kHz. Myocytes were dialyzed for 10 min before the recordings commenced. Voltage-clamp protocols and data acquisition were governed by a Digidata 1321A digitizer and pCLAMP 8.0 software (Axon). Successive 500-ms voltage steps were made from a holding potential of −60 mV to test potentials ranging from −100 to +60 or +100 mV in +10-mV increments. Current-voltage (I-V) relationships were plotted from the quasi-steady-state current except for in Figs. 1 and 2, which show currents at 25 ms. Capacitance was calculated with pCLAMP software using a 5-mV step.
Fig. 1.
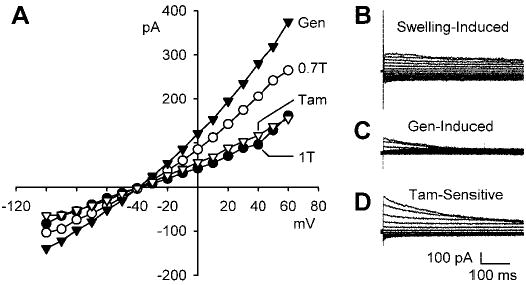
Genistein, a broad-spectrum protein tyrosine kinase (PTK) blocker, augmented swelling-activated Cl− current (ICl,swell). A: current-voltage (I-V) relationship in isosmotic (1T) solution, after swelling in hypoosmotic (0.7T) solution for 10 min, after exposure to 100 μM genistein in 0.7T solution for 10 min (Gen), and after addition of 10 μM tamoxifen (Tam), a selective ICl,swell blocker, to 0.7T plus genistein solution. I-V curves cross near the Cl− equilibrium potential (ECl), which is −42 mV. B: families of swelling-induced difference currents. ICl,swell partially inactivated at positive potentials. C: genistein-induced difference currents in 0.7T solution. D: tamoxifen-sensitive difference currents in 0.7T plus genistein solution. Swelling activated outwardly rectifying ICl,swell that reversed near ECl and was stimulated by genistein. Tamoxifen blocked both the swelling- and genistein-induced components. Calibrations apply to all current records.
Fig. 2.
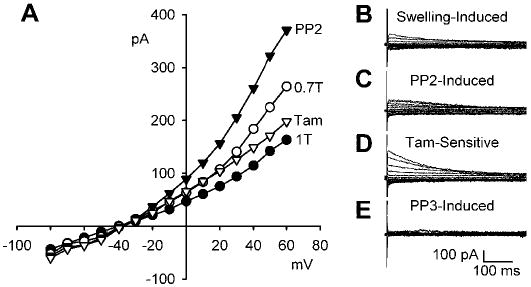
ICl,swell is stimulated by PP2, a selective inhibitor of Src family PTKs, but not by its inactive analog, PP3. A: I-V relationships in 1T solution, after swelling in 0.7T solution for 10 min, after exposure to 10 μM PP2 in 0.7T solution for 10 min (PP2), and after addition of 10 μM tamoxifen to 0.7T solution plus PP2. B: families of swelling-induced difference currents. C: PP2-induced difference currents in 0.7T solution. D: tamoxifen-sensitive difference currents in 0.7T plus PP2 solution. Both the PP2- and swelling-induced currents were sensitive to 10 μM tamoxifen. Selective inhibition of Src by PP2 mimicked the effect of genistein. In contrast, PP3 (an inactive analog of PP2; 10 μM for 10 min) did not alter the magnitude or time course of membrane currents in 0.7T solution. E: PP3-induced difference currents in 0.7T solution. These records were obtained in a different cell than for A-D.
Preliminary studies established that ICl,swell fully activated in <5 min and remained stable for ≥45 min. All interventions were applied for a time sufficient for currents to reach a steady state as judged from I-V curves obtained at selected intervals (typically 1 or 2 min).
Statistics.
Data are expressed as means ± SE, and n refers to the number of cells. Mean currents are presented as current density (in pA/pF) to account for differences in myocyte surface membrane area. Statistical analyses were done using SigmaStat 2.3 or 3.0 software (Systat). For multiple comparisons, a two-way repeated-measures ANOVA was used as appropriate, and the Student-Newman-Keuls test was performed to compare groups. Statistical significance was taken as P < 0.05.
RESULTS
Effects of genistein on ICl,swell.
To assess the effects of PTK on ICl,swell in ventricular myocytes, the broad-spectrum PTK blocker genistein was applied after ICl,swell was activated by osmotic swelling in solutions designed to isolate Cl− currents. Figure 1 illustrates the I-V relationships obtained under each experimental condition and families of difference currents calculated by digital subtraction. As expected, osmotic swelling in 0.7T bath solution induced an outwardly rectifying Cl− current that partially inactivated at positive potentials and reversed at −40.6 ± 2.4 mV, near the calculated Cl− equilibrium potential (ECl) of −42 mV (Fig. 1, A and B). At +60 mV, for example, swelling significantly increased the Cl− current from 1.3 ± 0.2 in 1T to 2.1 ± 0.3 pA/pF after 10 min in 0.7T solution (n = 5; P < 0.001). ICl,swell was further enhanced by a 10-min exposure to 100 μM genistein in 0.7T solution. The genistein-induced difference current outwardly rectified, substantially inactivated at positive potentials, and reversed at the same potential as the swelling-induced current (Fig. 1, A and C). Addition of genistein to 0.7T solution increased the Cl− current at +60 mV from 2.1 ± 0.3 to 2.7 ± 0.3 pA/pF (n = 5; P < 0.001). Thus the swelling-induced current with genistein was 163 ± 17% of the swelling-induced current without genistein in same-cell comparisons (n = 5; P < 0.001). Because genistein caused a much more prominent increase in the outward than the inward current with a physiological Cl− gradient, its stimulation of ICl,swell was not statistically significant at −100 mV.
Tamoxifen blocks ICl,swell but not cAMP-or Ca2+-induced Cl− currents (ICl,cAMP or ICl,Ca, respectively) and can be used to distinguish between these currents under conditions that isolate anionic currents (48, 5). To verify that the genistein-induced current was ICl,swell, myocytes were exposed to 10 μM tamoxifen for 10 min in the continued presence of genistein in 0.7T solution. The tamoxifen-sensitive currents and the I-V relationship after block by tamoxifen are shown (Fig. 1, A and D). Tamoxifen inhibited both the swelling- and genistein-induced currents but did not alter the reversal potential of the I-V curve. At +60 mV in 0.7T bath solution, tamoxifen reduced the Cl− current from 2.7 ± 0.3 after stimulation by genistein to 1.3 ± 0.3 pA/pF (n = 5; P < 0.001), a value indistinguishable from that in 1T solution.
Stimulation of ICl,swell by genistein in 0.7T bath solution was confirmed in an additional seven cells that were not exposed to tamoxifen. On the other hand, partial inhibition of Cl− current by genistein was noted in 3 of 15 cells. In these cells, genistein decreased the current at +60 mV from 1.8 ± 0.1 to 1.3 ± 0.1 pA/pF (n = 3; P < 0.02).
Selective inhibition of Src.
Recently, it was reported that inhibition of Src family PTKs stimulates ICl,swell in human atrial myocytes (15). To test the hypothesis that block of Src is responsible for the stimulation of ICl,swell in rabbit ventricular myocytes, we used PP2, a selective inhibitor of the Src family (3, 23). As before, osmotic swelling in 0.7T solution activated ICl,swell (Fig. 2, A and B). Exposure to 10 μM PP2 for 10 min significantly stimulated ICl,swell in 0.7T solution (Fig. 2C). PP2 augmented the current at +60 mV from 2.1 ± 0.3 to 2.9 ± 0.3 pA/pF (n = 7; P < 0.001); the swelling-activated current in PP2 was 206 ± 10% of the swelling-activated current without PP2. The effect of PP2 at −100 mV was not significant, however, as was the case with genistein. Both the PP2- and swelling-induced currents were sensitive to tamoxifen (Fig. 2D). Exposure to 10 μM tamoxifen for 10 min in the continued presence of PP2 reduced the Cl− current at +60 mV to 1.4 ± 0.2 pA/pF (n = 6; P = 0.001), a value indistinguishable from that in 1T, 1.4 ± 0.4 pA/pF. Thus inhibition of Src by PP2 augmented the tamoxifen-sensitive Cl− current in ventricular myocytes and mimicked the usual effect of genistein.
To exclude the possibility that a nonspecific effect of PP2 was responsible for stimulation of ICl,swell, we applied PP3, an inactive analog of PP2 (3, 46), in separate experiments. Treatment with 10 μM PP3 for 10 min did not alter the magnitude or time dependence of the ICl,swell in 0.7T solution, and the resulting PP3-induced difference current was nil (Fig. 2E). In these cells, swelling increased the current at +60 mV from 1.6 ± 0.2 in 1T to 2.4 ± 0.2 pA/pF in 0.7T solution (n = 6; P < 0.001), but the current was unaffected [2.4 ± 0.3 pA/pF; n = 6; P = not significant (NS)] by addition of PP3 to 0.7T solution. Taken together, these results suggest that Src family PTKs play a critical role in the regulation of swelling-activated Cl− channels in ventricular myocytes.
ICl,swell is thought to contribute to the background Cl− current (17, 18). Therefore, it is important to distinguish whether inhibition of Src augments the response to cell swelling or simply activates ICl,swell independent of cell volume. As shown in Fig. 3, PP2 (10 μM for 10 min) did not alter the I-V relationship for Cl− current under 1T conditions, and the PP2-induced difference current in 1T solution was negligible (Fig. 3, inset). The current at +60 mV was 1.4 ± 0.2 pA/pF in both 1T solution and in 1T solution after treatment with PP2 (n = 7; P = NS). Thus blocking Src enhances activation of ICl,swell in response to swelling but is insufficient to activate ICl,swell or the background Cl− current by itself.
Fig. 3.
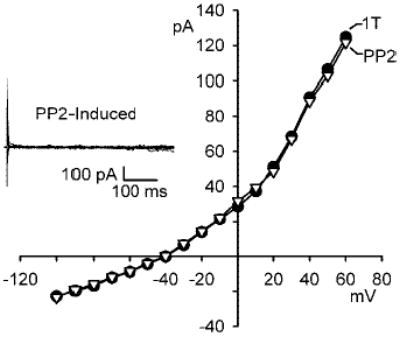
Stimulation of ICl,swell by PP2 requires cell swelling. I-V relationships in 1T and after exposure to 10 μM PP2 in 1T solution for 10 min are shown. Inset: families of PP2-induced difference currents in 1T solution. PP2 did not alter the outwardly rectifying background Cl− current attributed at least in part to ICl,swell under isosmotic conditions. Note the change in scale of I-V relationship compared with previous figures.
Blocking time dependence of ICl,swell.
In several cells, inactivation of ICl,swell at positive potentials appeared to deviate from an exponential decay. This raised the possibility that additional components contributed to the empirically defined ICl,swell. Consequently, we examined the effect of adding 0.2 mM Cd2+ to the bath solution. Figure 4 (A–C and D–F) shows the responses of two of the nine cells studied. As before, osmotic swelling in 0.7T solution evoked an outwardly rectifying Cl− current (Fig. 4, A and D). Cd2+ largely blocked the rapidly inactivating component at positive potentials and had a smaller but variable effect on steady-state current (Fig. 4, B and E). Steady-state current in 0.7T solution with Cd2+ was 109 ± 9% of that without Cd2+ (n = 9; P = NS). The Cd2+-sensitive current (Fig. 4, C and F) exhibited both inactivation at positive potentials and the outward-going rectification that are characteristic of ICl,swell. After suppression of the outward transient by Cd2+, a delayed rectifier appeared to emerge. This is most obvious in Fig. 4E, which illustrates the myocyte with the strongest block of steady-state currents by Cd2+.
Fig. 4.
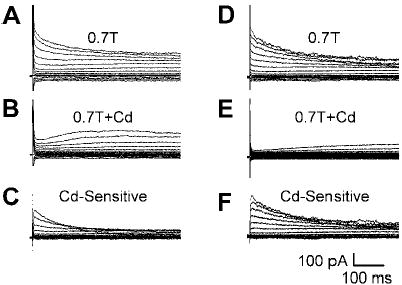
Cd2+ (0.2 mM) suppressed time dependence of ICl,swell at positive potentials and revealed a delayed rectifier current. Examples from two myocytes are shown. A, B, C: data from one myocyte. D, E, F: data from a second myocyte. Families of currents after swelling in 0.7T solution (A and D), after exposure to Cd2+ (B and E), and Cd2+ -sensitive current (C and F) are shown. Cd2+ inhibited the time dependence of ICl,swell at positive potentials and had a variable effect on the steady-state current. After Cd2+ block, a slowly activating outward component of current was apparent. Block of ICl,swell was greatest in the second myocyte (D-F).
The remaining time-dependent component in 0.7T plus Cd2+ solution was Ba2+ sensitive. Figure 5 shows the effect of swelling a myocyte in bathing solutions containing 0.2 mM Cd2+ and 1 mM Ba2+. Under these conditions, the current was essentially time independent over the entire voltage range studied. Nevertheless, osmotic swelling in 0.7T solution increased the Cl− current at +60 mV from 1.5 ± 0.2 to 2.6 ± 0.3 pA/pF (n = 11; P < 0.001). Moreover, block of Src with 10 μM PP2 in 0.7T bath solution caused an additional increase in the outward current to 4.1 ± 0.4 pA/pF (n = 11; P < 0.001). Families of time-independent currents in 1T, 0.7T, and 0.7T plus PP2 solutions are shown in Fig. 5, B-D. Although the addition of Cd2+ and Ba2+ eliminated the time dependence of ICl,swell, the steady-state current densities in 1T and 0.7T solutions with and without these blockers were indistinguishable (cf. Figs. 2 and 5 at +60 mV). In four of these cells, we also verified that the time-independent swelling- and PP2-stimulated currents were blocked by 10 μM tamoxifen after 10 min, as was previously demonstrated for time-dependent currents in the absence of Cd2+ and Ba2+ (see Fig. 2). Addition of tamoxifen (Fig. 5E) significantly reduced the current in 0.7T plus PP2 solution from 3.6 ± 0.4 to 1.3 ± 0.2 pA/pF (n = 4; P < 0.001), a value indistinguishable from that in 1T solution, 1.2 ± 0.3 pA/pF (n = 4; P = NS). As before, I-V curves after activation of ICl,swell, its stimulation, and its inhibition all crossed near ECl.
Fig. 5.
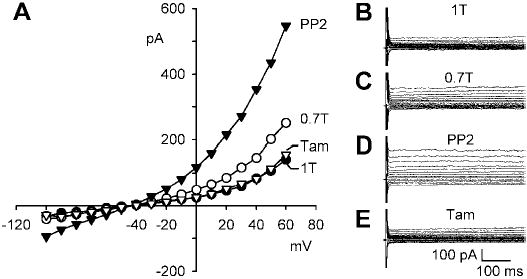
Cd2+ plus Ba2+ eliminated the time dependence of membrane currents but not the stimulation of ICl,swell by PP2. A: I-V relationships in the presence of 0.2 mM Cd2+ and 1 mM Ba2+ in 1T solution, after swelling in 0.7T solution for 10 min, after exposure to 10 μM PP2 for 10 min in 0.7T solution, and after addition of 10 μM tamoxifen for 10 min to block ICl,swell. B, C, D, and E: families of currents in 1T solution, 0.7T solution, 0.7T solution plus PP2, and 0.7T solution plus PP2 and tamoxifen, respectively. PP2 stimulated an outwardly rectifying, tamoxifen-sensitive current that reversed near ECl.
To determine whether Cd2+ and Ba2+ simply shifted the voltage dependence of the currents to more positive potentials (11, 32), the voltage range studied was extended. ICl,swell remained time independent to at least + 100 mV (n = 4) as shown in Fig. 6.
Fig. 6.
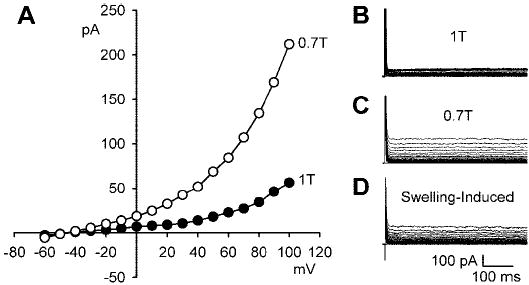
Cd2+ plus Ba2+ eliminated the time dependence of ICl,swell at strongly positive potentials. To test whether Cd2+ (0.2 mM) and Ba2+ (1 mM) shifted the onset of ICl,swell inactivation to more positive voltages, the membrane voltage was stepped from −60 mV to potentials between −60 and +100 mV. A: I-V relationships in 1T solution and after 10 min of swelling in 0.7T solution. B and C: families of currents in 1T and 0.7T solutions, respectively. D: swelling-induced difference current. ICl,swell remained time independent to at least +100 mV.
Symmetrical Cl− gradient.
Both ICl,swell and ICl,cAMP undergo outward rectification with physiological Cl− gradients such as the one used in the experiments described thus far {intracellular Cl− concentration ([Cl−]i) = 20.3 mM; extracellular Cl− concentration ([Cl−]o) = 98.6–105.3 mM}. In contrast, only ICl,swell retains outward rectification in symmetrical high- Cl− solutions (28). Figure 7 shows the effects of osmotic swelling in 0.7T and exposure to PP2 in symmetrical high- Cl− solutions ([Cl−]i = [Cl−]o = 98.6 mM). Swelling induced an outwardly rectifying current that reversed at −2.7 ± 0.3 mV (n = 6) near the expected reversal potential of 0 mV. The current at +60 mV increased from 0.6 ± 0.2 in 1T to 1.6 ± 0.2 pA/pF in 0.7T solution (n = 6; P < 0.005). PP2 additionally augmented the outwardly rectifying current in symmetrical high- Cl− solutions to 2.8 ± 0.4 pA/pF (n = 6; P < 0.001) at +60 mV. In contrast to experiments with a physiological pipette Cl−, a clear PP2-induced stimulation of inward current was detected with elevated pipette Cl− (cf. Fig. 2). At −100 mV, for example, swelling in 0.7T solution increased the Cl− current from −0.4 ± 0.1 to −0.9 ± 0.1 pA/pF (n = 6; P < 0.025), and PP2 additionally increased it to − 1.6 ± 0.2 pA/pF (n = 6; P < 0.003).
Fig. 7.
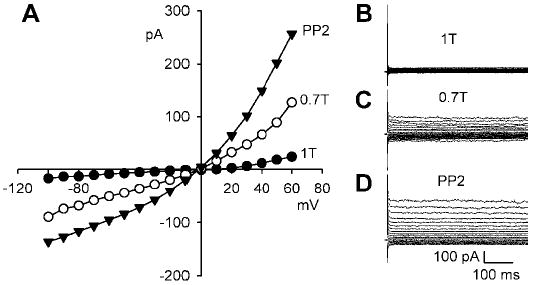
Cl− currents in symmetrical high- Cl− solutions (intracellular and extracellular Cl− concentrations = 98.6 mM). A: I-V relationships for currents in 1T solution, after 10 min of swelling in 0.7T solution, and after 10 min of exposure to 10 μM PP2 in 0.7T solution. B, C, and D: families of currents in 1T, 0.7T, and 0.7T plus PP2 solution, respectively. Swelling and PP2 stimulated outwardly rectifying, time-independent currents that reversed near 0 mV in symmetrical Cl− solutions with 0.2 mM Cd2+ and 1 mM Ba2+ in the bath.
Roles of PTP and EGFR kinase.
If PP2 acts by blocking Src-dependent phosphorylation of a critical tyrosine residue, its action should be opposed by orthovanadate, which inhibits PTP (21) and thereby retains tyrosines in a phosphorylated state. Figure 8 shows a test of this prediction. After activation of ICl,swell in 0.7T solution, myocytes first were exposed to 1 mM orthovanadate for 10 min in 0.7T solution and then were challenged with 10 μM PP2 for 10 min in the continued presence of orthovanadate. Orthovanadate alone reduced the current in 0.7T solution from 3.0 ± 0.2 to 1.8 ± 0.3 pA/pF (n = 4; P < 0.033), an action opposite to that of PP2. Moreover, PP2 failed to significantly stimulate ICl,swell after pretreatment with orthovanadate. The current in PP2 plus orthovanadate was 2.2 ± 0.3 pA/pF, a value not different than that in orthovanadate alone (n = 4; P = NS).
Fig. 8.
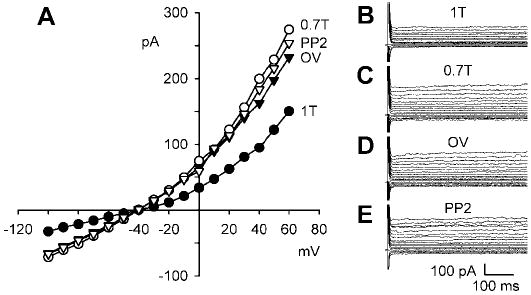
Block of protein tyrosine phosphatase (PTP) by orthovanadate partially inhibits ICl,swell and prevents stimulation of ICl,swell upon block of Src by PP2. A: I-V relationships for currents in 1T solution, after 10 min of swelling in 0.7T solution, after exposure to 1 mM orthovanadate in 0.7T solution for 10 min (OV), and after addition of 10 μM PP2 for 10 min. B, C, D, and E: families of currents in 1T, 0.7T, 0.7T plus orthovanadate, and 0.7T plus orthovanadate and PP2 solution, respectively.
In human atria, ICl,swell is controlled by at least two families of PTKs (Src and EGFR kinase), which have opposite effects on ICl,swell in osmotically swollen myocytes (15). Genistein usually stimulated ICl,swell (see Fig. 1), but inhibition was observed in 20% of myocytes. Therefore, we tested whether EGFR also regulates ICl,swell in the ventricle. Figure 9 illustrates the effects of PD-153035, a highly specific and potent blocker of EGFR kinase (19). As before, ICl,swell was activated by swelling myocytes in 0.7T solution, and then cells were exposed to 20 nM PD-153035 in 0.7T media for 12–15 min. In contrast with the stimulatory effect of blocking Src, blocking EGFR kinase strongly inhibited ICl,swell. Swelling in 0.7T solution increased the current at +60 mV from 1.8 ± 0.4 to 4.0 ± 0.7 pA/pF (n = 4; P = 0.002), and PD-153035 reduced the current to 1.9 ± 0.4 pA/pF, a value indistinguishable from that in 1T solution (n = 4; P = ns). Thus inhibiting EGFR kinase suppressed ~95% of the swelling-induced current.
Fig. 9.
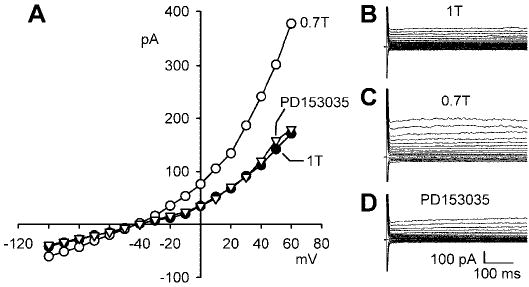
Block of epidermal growth factor receptor (EGFR) kinase by PD-153035 inhibits ICl,swell. A: I-V relationships for currents in 1T solution, after 10 min of swelling in 0.7T solution, and after exposure to 20 nM PD-153035 in 0.7T solution for 12–15 min. B, C, and D: families of currents in 1T, 0.7T, and 0.7T plus PD-153035 solution, respectively.
DISCUSSION
Previous studies on the heart focused on the role of PTK in the regulation of ICl,swell in atria (15, 44). The present study provides the first evidence that ICl,swell in osmotically swollen ventricular myocytes is regulated in an opposing fashion by the Src and EGFR kinase families of PTK and by PTP. PTKs are well placed to be sensors of cell volume and mechanical stretch. These signaling molecules interact with the cytoskeleton, integral membrane proteins, and sarcolemma (6), and tyrosine phosphorylation is among the earliest responses to osmotic swelling in cardiac myocytes and other cells (37, 38).
Antagonistic regulation of ICl,swell by Src and EGFR kinase.
ICl,swell was enhanced by the selective Src family inhibitor PP2. Stimulation of ICl,swell is unlikely to be due to nonspecific effects of PP2, because its inactive analog, PP3, did not alter the magnitude or the time independence of the current. Moreover, as expected for a process that depends on the phosphorylation of tyrosine residues, blocking PTP and thereby tyrosine dephosphorylation with orthovanadate inhibited ICl,swell in 0.7T solution, an effect opposite to that obtained by blocking Src-dependent tyrosine phosphorylation. Ultimately, stimulation of ICl,swell upon blocking Src must result from accumulation of critical tyrosine residues in the dephosphorylated state. Consistent with this idea, suppressing the rate of dephosphorylation by PTP with orthovanadate also precluded ICl,swell stimulation by PP2. Because orthovanadate associates with a variety of phosphate-binding sites, nonspecific effects of this agent cannot be rigorously ruled out.
Regulation of ICl,swell also critically depended on EGFR kinase, a second distinct family of PTK. PD-153035, an extremely potent and selective inhibitor of EGFR kinase (19), completely suppressed ICl,swell in 0.7T solution. AG-1478, the less-potent chloro derivative of PD-153035, also fully inhibited ICl,swell in rabbit ventricular myocytes (unpublished observations). The antagonistic effect of inhibiting Src and EGFR kinase PTK families suggests that at least two distinct tyrosine residues that are phosphorylated by Src and EGFR kinase, respectively, must be involved in the regulation of ICl,swell in rabbit ventricles as we previously proposed for human atria (15). Orthovanadate inhibited ICl,swell, which is expected if the PTP inhibitor primarily opposed the action of Src rather than EGFR kinase. This suggests that the Src family PTK site may be dominant or that orthovanadate differentially modulates the dephosphorylation of the targets of these two PTK families (15).
An antagonistic regulation of ICl,swell by distinct PTK families may in part explain the inconsistent effects of the broad-spectrum PTK inhibitor genistein, which usually stimulated ICl,swell but inhibited the current in 20% of the cells examined. These divergent responses might reflect differences in the activities of Src and EGFR kinase in individual myocytes. The 50% inhibitory concentration of genistein for v-Src and EGFR kinase are quite similar, 26 and 22 μM, respectively, based on in vitro phosphorylation of exogenous substrates (1), and, therefore, 100 μM of genistein should have largely inhibited both PTK families.
Osmotic swelling of neonatal rat ventricular myocytes leads to activation of PTK and tyrosine phosphorylation of target proteins within 5 s, although the PTK involved was not established (37, 38). The present observation that blocking Src stimulates ICl,swell after osmotic swelling but has no effect under isosmotic conditions argues that a swelling-induced activation of Src is unlikely to be responsible for the activation of ICl,swell. On the other hand, blocking EGFR kinase inhibited ICl,swell in 0.7T solution. This raises the possibility that stimulation of EGFR kinase by swelling could contribute to the activation of current seen under these conditions. Consistent with this idea, exogenous EGF activates an outwardly rectifying, tamoxifen-sensitive Cl− current with the characteristics of ICl,swell in rabbit ventricular myocytes (9).
Regulation of swelling-activated Cl− current by PTK appears to be different in rabbit ventricular and canine atrial myocytes. Sorota (44) reported that ICl,swell is inhibited by pretreatment with genistein, whereas acute application of tyrphostin A51, an EGFR kinase inhibitor, and herbimycin A, a Src inhibitor, have no effect. ICl,swell activated by mechanical stretch of rabbit ventricular myocytes is suppressed by acute application of either genistein or PP2 (7). On the other hand, genistein and PP2 stimulate swelling-induced ICl,swell in human atrial myocytes (15), but genistein inhibits ICl,swell in cultured embryonic chick heart (56). Thus the regulation of ICl,swell is likely to depend on the method of stimulation (e.g., swelling vs. stretch) and the particular tissue studied. Interventions that favor tyrosine phosphorylation diminish ICl,swell in bovine chromaffin cells (14) and mouse L-fibroblasts (45), whereas those that suppress tyrosine phosphorylation augment ICl,swell in calf pulmonary artery endothelial cells (51) and rabbit ciliary epithelial cells (40). Moreover, it is apparent that several different molecules act as volume-sensitive anion channels and/or channel regulators and that certain properties of ICl,swell in different tissues are distinct (4). Finally, differences in experimental solutions and conditions (e.g., temperature) may affect the regulation of ICl,swell by signaling pathways.
Time dependence of ICl,swell.
ICl,swell in the heart is usually described as an inactivating current at positive potentials, although in some cases little inactivation is observed (4, 28). The time-dependent genistein- and PP2-stimulated currents recorded here (see Figs. 1 and 2) were consistent with ICl,swell in that they were volume sensitive, outwardly rectifying, and inhibited by tamoxifen. To our surprise, the addition of Cd2+ or Cd2+ and Ba2+ eliminated the time dependence. A 50% higher concentration of Cd2+ (0.3 mM) failed to block the time-dependent, outwardly-rectifying, tamoxifen-sensitive ICl,swell found in human intestinal T84 cells (5), and a time-dependent ICl,swell also was reported in guinea pig myocytes with 0.1– 0.2 mM Cd2+ in the bath solution (43). Nevertheless, in the presence of these divalent ion blockers, PP2 augmented a volume- and tamoxifen-sensitive outwardly rectifying current that reversed at ECl with both physiological and symmetrical Cl− gradients. These characteristics are diagnostic for ICl,swell (4, 28), and thus we attribute both the time-dependent and time-independent components to ICl,swell. One possible mechanism for the block of current decay is a shift in the voltage dependence of ICl,swell to more positive voltages. Extending the range of test voltages to +100 mV did not elicit time dependence in the presence of Cd2+ and Ba2+, whereas current decay was evident at +20 mV in their absence. These findings argue against but do not rigorously exclude a rightward shift in voltage dependence; a shift of >80 mV, which seems unlikely, would be required to explain the data. We also cannot exclude the possibility that distinct Cd2+-sensitive and -insensitive Cl− channels contribute to ICl,swell. If this is the case, both meet the phenomenological definition of ICl,swell.
Although genistein is a popular tool for identifying the involvement of PTK, it previously was found to stimulate ICl,cAMP by a mechanism that is independent of protein phosphorylation (10, 52; cf. 41 and 42), and it also modifies the behavior of gramicidin channels in planar bilayers by altering the energetics of the hydrophobic interaction between the channels and the bilayer (29). Because the cardiac isoform of the cystic fibrosis transmembrane conductance regulator is expressed in ventricular myocytes (27, 58), this raises the possibility that genistein-sensitive ICl,cAMP might contribute to the genistein-sensitive current. Cardiac ICl,cAMP is, however, a time-independent current at all voltages (28, 43); it exhibits a linear I-V relationship in symmetrical high- Cl− solutions (2, 34), and it is insensitive to tamoxifen (50). Moreover, we are unaware of evidence suggesting that PP2 modulates ICl,cAMP. Thus the characteristics of current elicited by genistein and PP2 are inconsistent with ICl,cAMP. It is also unlikely that the current is ICl,Ca. Activation of ICl,Ca requires a Ca2+ transient to elevate the cytoplasmic Ca2+ concentration. In the present studies, cytoplasmic Ca2+ was buffered at ~60 nM with 8 mM EGTA, and bath Ca2+ was either reduced to ~1.5 μM to limit Ca2+ entry, or Ca2+ channels were blocked by Cd2+. These conditions should preclude the occurrence of the Ca2+ transient that is required to elicit a transient outwardcurrent (Ito) due to ICl,Ca (47, 61). In addition, the biophysical characteristics of the genistein- and PP2-induced currents are inconsistent with ICl,Ca. A bell-shaped I-V relationship and inactivation at positive voltages are expected for ICl,Ca with physiological Ca2+ regulation (47, 61), whereas ICl,Ca is a linear and time-independent current when cytoplasmic Ca2+ is fixed at an elevated concentration (60).
We, as others, utilized replacement of K+ and Na+ with internal Cs+ and external NMDG to block cation currents and thereby isolate Cl− currents. It is well known, however, that Cs+ is slightly permeant through a variety of cation channels (22, 24), and the possibility that time-dependent outward currents attributed to Cl− were in fact permeation of Cs+ through cation channels also must be considered. One possibility is Cs+ efflux via Ito, which in rabbit ventricular myocytes is due to Kv1.4 rather than Kv4.x channels (53, 57). Cd2+ block of Ito in rabbit ventricular myocytes and of Kv1.4 expressed in Xenopus oocytes is inconsistent with the present results, however. Cd2+ (10 –500 μM) does not inhibit Kv1.4 currents or Ito; 500 μM Cd2+ shifts the voltage dependence of inactivation rightward by only 12 and 13 mV, respectively, and 100 μM Cd2+ does not significantly shift inactivation of Ito (57). Another possibility is the efflux of Cs+ via L-type Ca2+ channels (ICa-L). This seems unlikely for several reasons. First, osmotic swelling causes partial inhibition of ICa-L in rabbit ventricular myocytes (31), whereas a prolonged swelling-induced stimulation of Ca2+ channels would be required to explain the present data. Second, 50 μM Cd2+ only partially blocked the transient Cl− current (unpublished observations), but should have blocked ~90% of current through L-type Ca2+ channels, which exhibit an apparent dissociation constant (Kd) of 2.14 μM and a Hill coefficient of 0.74 for Cd2+ in rabbit ventricular myocytes (26). Furthermore, previous studies of tail currents recorded during the inactivation of ICl,swell demonstrate reactivation of the underlying conductance on stepping from +80 to −80 mV (43), behavior that is inconsistent with Ca2+ channels.
With Cd2+, a small, slowly activating outward current remained. It is likely that the slow component of delayed rectifier current (IKs) contributes to this time-dependent current. IKs is stimulated by osmotic swelling of myocytes (36, 39, 54, 59), and delayed rectifiers are more permeant to Cs+ than most other K+ channels (the permeability ratio, PCs/PK, is ~0.16; Ref. 22; also see Ref. 24). Moreover, Cd2+ can stimulate both IKs (13) and the rapid component of delayed rectifier current (IKr; Ref. 35) by shifting their voltage dependence. Such a stimulation of delayed rectifier current and suppression of the superimposed ICl,swell transient both are likely to explain why a delayed rectifier emerged after Cd2+ was added to 0.7T bath solution. Extracellular Ba2+ blocks delayed rectifier K+ current in heart (25) as well as currents through KCNQ1 (20), the pore-containing subunit of IKs. The combination of Cd2+ and Ba2+ eliminated the slowly activating outward current observed with Cd2+ alone. Of course, Ba2+ also blocks a number of other K+ channels (24) including the IKr due to human ether-á-go-go-related gene (55), but IKr is not enhanced by cell swelling (36, 54).
In summary, blocking Src family PTKs leads to activation of ICl,swell in rabbit ventricular myocytes after osmotic swelling but not under isosmotic conditions, and blocking PTP suppresses ICl,swell and precludes its activation by inhibition of Src. On the other hand, blocking EGFR kinase PTK inhibited ICl,swell. Thus ICl,swell is regulated antagonistically by two distinct PTK families as well as by other signaling cascades. Moreover, the transient component of ICl,swell is sensitive to Cd2+.
Acknowledgments
The authors thank Steven E. Hutchens for technical assistance.
Footnotes
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grant HL-46764.
References
- 1.Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- 2.Bahinski A, Nairn AC, Greengard P, Gadsby DC. Chloride conductance regulated by cyclic AMP-dependent protein kinase in cardiac myocytes. Nature. 1989;340:718–721. doi: 10.1038/340718a0. [DOI] [PubMed] [Google Scholar]
- 3.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumgarten CM, Clemo HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog Biophys Mol Biol. 2003;82:25–42. doi: 10.1016/s0079-6107(03)00003-8. [DOI] [PubMed] [Google Scholar]
- 5.Bond TD, Ambikapathy S, Mohammad S, Valverde MA. Osmo-sensitive Cl− currents and their relevance to regulatory volume decrease in human intestinal T84 cells: outwardly vs. inwardly rectifying currents. J Physiol (Lond) 1998;511:45–54. doi: 10.1111/j.1469-7793.1998.045bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borg TK, Goldsmith EC, Price R, Carver W, Terracio L, Samarel AM. Specialization at the Z line of cardiac myocytes. Cardiovasc Res. 2000;46:277–285. doi: 10.1016/s0008-6363(99)00433-2. [DOI] [PubMed] [Google Scholar]
- 7.Browe DM, Baumgarten CM. Stretch of β1 integrin activates an outwardly rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J Gen Physiol. 2003;122:689–702. doi: 10.1085/jgp.200308899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by β1-integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Browe DM, Baumgarten CM. Stretch of β1-integrin elicits swelling-activated Cl current via transactivation of EGFR, phosphatidylinositol-3-kinase and NADPH oxidase in rabbit ventricular myocytes (Abstract) Biophys J. 2005;88:289a. [Google Scholar]
- 10.Chiang CE, Chen SA, Chang MS, Lin CI, Luk HN. Genistein directly induces cardiac CFTR chloride current by a tyrosine kinase-independent and protein kinase A-independent pathway in guinea pig ventricular myocytes. Biochem Biophys Res Commun. 1997;235:74–78. doi: 10.1006/bbrc.1997.6739. [DOI] [PubMed] [Google Scholar]
- 11.Clemo HF, Baumgarten CM. Swelling-activated Gd3+-sensitive cation current and cell volume regulation in rabbit ventricular myocytes. J Gen Physiol. 1997;110:297–312. doi: 10.1085/jgp.110.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res. 1999;84:157–165. doi: 10.1161/01.res.84.2.157. [DOI] [PubMed] [Google Scholar]
- 13.Daleau P, Khalifa M, Turgeon J. Effects of cadmium and nisoldipine on the delayed rectifier potassium current in guinea pig ventricular myocytes. J Pharmacol Exp Ther. 1997;281:826–833. [PubMed] [Google Scholar]
- 14.Doroshenko P. Pervanadate inhibits volume-sensitive chloride current in bovine chromaffin cells. Pflügers Arch. 1998;435:303–309. doi: 10.1007/s004240050516. [DOI] [PubMed] [Google Scholar]
- 15.Du XL, Gao Z, Lau CP, Chiu SW, Tse HF, Baumgarten CM, Li GR. Differential effects of tyrosine kinase inhibitors on volume-sensitive chloride current in human atrial myocytes: evidence for dual regulation by Src and EGFR kinases. J Gen Physiol. 2004;123:427–439. doi: 10.1085/jgp.200409013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du XY, Sorota S. Cardiac swelling-induced chloride current depolarizes canine atrial myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H1904–H1916. doi: 10.1152/ajpheart.1997.272.4.H1904. [DOI] [PubMed] [Google Scholar]
- 17.Duan D, Fermini B, Nattel S. α-Adrenergic control of volume-regulated Cl− currents in rabbit atrial myocytes: characterization of a novel ionic regulatory mechanism. Circ Res. 1995;77:379–393. doi: 10.1161/01.res.77.2.379. [DOI] [PubMed] [Google Scholar]
- 18.Duan D, Hume JR, Nattel S. Evidence that outwardly rectifying Cl− channels underlie volume-regulated Cl− currents in heart. Circ Res. 1997;80:103–113. doi: 10.1161/01.res.80.1.103. [DOI] [PubMed] [Google Scholar]
- 19.Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson JM, Leopold WR, Connors RW, Bridges AJ. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science. 1994;265:1093–1095. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]
- 20.Gibor G, Yakubovich D, Peretz A, Attali B. External barium affects the gating of KCNQ1 potassium channels and produces a pore block via two discrete sites. J Gen Physiol. 2004;124:83–102. doi: 10.1085/jgp.200409068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- 22.Hadley RW, Hume JR. Permeability of time-dependent K+ channel in guinea pig ventricular myocytes to Cs+, Na+, NH4+, and Rb+ Am J Physiol Heart Circ Physiol. 1990;259:H1448–H1454. doi: 10.1152/ajpheart.1990.259.5.H1448. [DOI] [PubMed] [Google Scholar]
- 23.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck-and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 24.Hille B.Ion Channels of Excitable Membranes (3rd ed.). Sunderland, MA: Sinauer, 2001.
- 25.Hirano Y, Hiraoka M. Changes in K+ currents induced by Ba2+ in guinea pig ventricular muscles. Am J Physiol Heart Circ Physiol. 1986;251:H24–H33. doi: 10.1152/ajpheart.1986.251.1.H24. [DOI] [PubMed] [Google Scholar]
- 26.Hobai IA, Bates JA, Howarth FC, Levi AJ. Inhibition by external Cd2+ of Na/Ca exchange and L-type Ca channel in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H2164–H2172. doi: 10.1152/ajpheart.1997.272.5.H2164. [DOI] [PubMed] [Google Scholar]
- 27.Horowitz B, Tsung SS, Hart P, Levesque PC, Hume JR. Alternative splicing of CFTR Cl− channels in heart. Am J Physiol Heart Circ Physiol. 1993;264:H2214–H2220. doi: 10.1152/ajpheart.1993.264.6.H2214. [DOI] [PubMed] [Google Scholar]
- 28.Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- 29.Hwang TC, Koeppe RE, Andersen OS. Genistein can modulate channel function by a phosphorylation-independent mechanism: importance of hydrophobic mismatch and bilayer mechanics. Biochemistry. 2003;42:13646–13658. doi: 10.1021/bi034887y. [DOI] [PubMed] [Google Scholar]
- 30.Kocic I, Hirano Y, Hiraoka M. Ionic basis for membrane potential changes induced by hypoosmotic stress in guinea-pig ventricular myocytes. Cardiovasc Res. 2001;51:59–70. doi: 10.1016/s0008-6363(01)00279-6. [DOI] [PubMed] [Google Scholar]
- 31.Li GR, Zhang M, Satin LS, Baumgarten CM. Biphasic effects of cell volume on excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;282:H1270–H1277. doi: 10.1152/ajpheart.00946.2001. [DOI] [PubMed] [Google Scholar]
- 32.Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G. Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol. 1997;68:69–119. doi: 10.1016/s0079-6107(97)00021-7. [DOI] [PubMed] [Google Scholar]
- 33.Okada Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am J Physiol Cell Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- 34.Overholt JL, Hobert ME, Harvey RD. On the mechanism of rectification of the isoproterenol-activated chloride current in guinea-pig ventricular myocytes. J Gen Physiol. 1993;102:871–895. doi: 10.1085/jgp.102.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paquette T, Clay JR, Ogbaghebriel A, Shrier A. Effects of divalent cations on the E-4031-sensitive repolarization current, IKr, in rabbit ventricular myocytes. Biophys J. 1998;74:1278–1285. doi: 10.1016/S0006-3495(98)77841-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rees SA, Vandenberg JI, Wright AR, Yoshida A, Powell T. Cell swelling has differential effects on the rapid and slow components of delayed rectifier potassium current in guinea pig cardiac myocytes. J Gen Physiol. 1995;106:1151–1170. doi: 10.1085/jgp.106.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 38.Sadoshima J, Qiu ZH, Morgan JP, Izumo S. Tyrosine kinase activation is an immediate and essential step in hypotonic cell swelling-induced ERK activation and c-fos gene expression in cardiac myocytes. EMBO J. 1996;15:5535–5546. [PMC free article] [PubMed] [Google Scholar]
- 39.Sasaki N, Mitsuiye T, Noma A. Effects of mechanical stretch on membrane currents of single ventricular myocytes of guinea-pig heart. Jpn J Physiol. 1992;42:957–970. doi: 10.2170/jjphysiol.42.957. [DOI] [PubMed] [Google Scholar]
- 40.Shi C, Barnes S, Coca-Prados M, Kelly ME. Protein tyrosine kinase and protein phosphatase signaling pathways regulate volume-sensitive chloride currents in a nonpigmented ciliary epithelial cell line. Invest Ophthalmol Vis Sci. 2002;43:1525–1532. [PubMed] [Google Scholar]
- 41.Shuba LM, Asai T, Pelzer S, McDonald TF. Activation of cardiac chloride conductance by the tyrosine kinase inhibitor, genistein. Br J Pharmacol. 1996;119:335–345. doi: 10.1111/j.1476-5381.1996.tb15991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shuba LM, McDonald TF. Lack of involvement of G proteins in the activation of cardiac CFTR Cl− current by genistein. Pflügers Arch. 1999;437:796–803. doi: 10.1007/s004240050848. [DOI] [PubMed] [Google Scholar]
- 43.Shuba LM, Ogura T, McDonald TF. Kinetic evidence distinguishing volume-sensitive chloride current from other types in guinea-pig ventricular myocytes. J Physiol (Lond) 1996;491:69–80. doi: 10.1113/jphysiol.1996.sp021197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorota S. Tyrosine protein kinase inhibitors prevent activation of cardiac swelling-induced chloride current. Pflügers Arch. 1995;431:178–185. doi: 10.1007/BF00410189. [DOI] [PubMed] [Google Scholar]
- 45.Thoroed SM, Bryan-Sisneros A, Doroshenko P. Protein phosphotyrosine phosphatase inhibitors suppress regulatory volume decrease and the volume-sensitive Cl− conductance in mouse fibroblasts. Pflügers Arch. 1999;438:133–140. doi: 10.1007/s004240050890. [DOI] [PubMed] [Google Scholar]
- 46.Traxler P, Bold G, Frei J, Lang M, Lydon N, Mett H, Buchdunger E, Meyer T, Mueller M, Furet P. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. J Med Chem. 1997;40:3601–3616. doi: 10.1021/jm970124v. [DOI] [PubMed] [Google Scholar]
- 47.Tseng GN, Hoffman BF. Two components of transient outward current in canine ventricular myocytes. Circ Res. 1989;64:633–647. doi: 10.1161/01.res.64.4.633. [DOI] [PubMed] [Google Scholar]
- 48.Valverde MA, Mintenig GM, Sepulveda FV. Differential effects of tamoxifen and I− on three distinguishable chloride currents activated in T84 intestinal cells. Pflügers Arch. 1993;425:552–554. doi: 10.1007/BF00374885. [DOI] [PubMed] [Google Scholar]
- 49.Vandenberg JI, Bett GC, Powell T. Contribution of a swelling-activated chloride current to changes in the cardiac action potential. Am J Physiol Cell Physiol. 1997;273:C541–C547. doi: 10.1152/ajpcell.1997.273.2.C541. [DOI] [PubMed] [Google Scholar]
- 50.Vandenberg JI, Yoshida A, Kirk K, Powell T. Swelling-activated and isoprenaline-activated chloride currents in guinea pig cardiac myocytes have distinct electrophysiology and pharmacology. J Gen Physiol. 1994;104:997–1017. doi: 10.1085/jgp.104.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voets T, Manolopoulos V, Eggermont J, Ellory C, Droogmans G, Nilius B. Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. J Physiol (Lond) 1998;506:341–352. doi: 10.1111/j.1469-7793.1998.341bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang F, Zeltwanger S, Yang IC, Nairn AC, Hwang TC. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating. Evidence for two binding sites with opposite effects. J Gen Physiol. 1998;111:477–490. doi: 10.1085/jgp.111.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Z, Feng J, Shi H, Pond A, Nerbonne JM, Nattel S. Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circ Res. 1999;84:551–561. doi: 10.1161/01.res.84.5.551. [DOI] [PubMed] [Google Scholar]
- 54.Wang ZR, Mitsuiye T, Noma A. Cell distension-induced increase of the delayed rectifier K+ current in guinea pig ventricular myocytes. Circ Res. 1996;78:466–474. doi: 10.1161/01.res.78.3.466. [DOI] [PubMed] [Google Scholar]
- 55.Weerapura M, Nattel S, Courtemanche M, Doern D, Ethier N, Hebert T. State-dependent barium block of wild-type and inactivation-deficient HERG channels in Xenopus oocytes. J Physiol (Lond) 2000;526:265–278. doi: 10.1111/j.1469-7793.2000.t01-1-00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei H, Mei YA, Sun JT, Zhou HQ, Zhang ZH. Regulation of swelling-activated chloride channels in embryonic chick heart cells. Cell Res. 2003;13:21–28. doi: 10.1038/sj.cr.7290147. [DOI] [PubMed] [Google Scholar]
- 57.Wickenden AD, Tsushima RG, Losito VA, Kaprielian R, Backx PH. Effect of Cd2+ on Kv4.2 and Kv1. 4 expressed in Xenopus oocytes and on the transient outward currents in rat and rabbit ventricular myocytes. Cell Physiol Biochem. 1999;9:11–28. doi: 10.1159/000016299. [DOI] [PubMed] [Google Scholar]
- 58.Wong KR, Trezise AE, Bryant S, Hart G, Vandenberg JI. Molecular and functional distributions of chloride conductances in rabbit ventricle. Am J Physiol Heart Circ Physiol. 1999;277:H1403–H1409. doi: 10.1152/ajpheart.1999.277.4.H1403. [DOI] [PubMed] [Google Scholar]
- 59.Zhou YY, Yao JA, Tseng GN. Role of tyrosine kinase activity in cardiac slow delayed rectifier channel modulation by cell swelling. Pflügers Arch. 1997;433:750–757. doi: 10.1007/s004240050341. [DOI] [PubMed] [Google Scholar]
- 60.Zygmunt AC. Intracellular calcium activates a chloride current in canine ventricular myocytes. Am J Physiol Heart Circ Physiol. 1994;267:H1984–H1995. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- 61.Zygmunt AC, Gibbons WR. Properties of the calcium-activated chloride current in heart. J Gen Physiol. 1992;99:391–414. doi: 10.1085/jgp.99.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]