

Abstract
Manganese(II) porphyrins are isoelectronic with iron(III) porphyrins, and previously reported work suggests that manganese nitrosyl porphyrins are good structural models for their kinetically unstable and biologically relevant ferric–NO analogues. We have prepared a new set of six-coordinate manganese nitrosyl porphyrins of the general form (por)Mn(NO)(L) (por = TTP, T(p-OCH3)PP; L = piperidine, methanol, 1-methylimidazole) in moderate to high yields. The (por)Mn(NO)(pip) complexes were prepared from the reductive nitrosylation of the (por)MnCl compounds with NO in the presence of piperidine. The IR spectra of the (por)Mn(NO)(pip) compounds as KBr pellets show new strong bands at 1746 cm−1 (for TTP) and 1748 cm−1 (for (T(p-OCH3)PP) due to the NO ligands. Attempted crystallization of one of these compounds (por = TTP) from dichloromethane–methanol resulted in the generation of the methanol complex (TTP)Mn(NO)(CH3OH). Reaction of the (por)Mn(NO)(pip) compounds with excess 1-methylimidazole gave the (por)Mn(NO)(1-MeIm) derivatives in good yields. The IR spectra of these compounds show νNO bands that are ~12 cm−1 lower than those of the (por)Mn(NO)(pip) precursors, indicative of greater Mn→NO π-backdonation in the 1-MeIm derivatives. X-Ray crystal structures of three of these compounds, namely (TTP)Mn(NO)(CH3OH), (TTP)Mn(NO)(1-MeIm) and (T(p-OCH3)PP)Mn(NO)(1-MeIm) were obtained, and reveal that the NO ligands in these complexes are linear.
Introduction
The biological role of nitric oxide (NO) is tied in with its interactions with iron porphyrins in heme biomolecules.1 For example, the receptor for NO is the heme-containing enzyme soluble guanylyl cyclase (sGC), and this enzyme contains histidine as an axial ligand to iron. NO binds to the ferrous heme site in sGC to give a six-coordinate (por)Fe(NO)(His) complex that eventually converts to a five-coordinate (por)Fe(NO) species.2–5 The binding of NO to the heme iron in sGC has been correlated with activation of this enzyme, resulting eventually in vasodilation. Other histidine-liganded hemes also form adducts with NO, and these include hemoglobin, myoglobin, cytochrome oxidase, nitrophorins, FixL and nitrite reductase.1 In many cases, the binding of NO has been shown to be biologically relevant.
NO binds to both ferric and ferrous hemes, although NO binding to ferric hemes is weaker than that for ferrous hemes.6 The biological relevance of NO binding to ferric heme is best exemplified by ferric nitrophorins from Rhodnius prolixus.7 The nitrophorins are ferric heme proteins contained in the saliva of the organism, and they bind NO. When the insect bites the host, NO is released from the ferric nitrosyl nitrophorins, resulting in local vasodilation which ensures that the insect obtains a sufficient blood meal.
Efforts to study well-characterized six-coordinate ferric nitrosyl porphyrins have been hampered by the fact that they are difficult to obtain pure, and only a few have been obtained in sufficient quantities for detailed spectroscopic and crystallographic studies.8 Such ferric nitrosyl porphyrins belong to the {FeNO}6 class as defined by Enemark and Feltham.9–11
We are interested in the study of manganese nitrosyl porphyrins belonging to the {MnNO}6 classification. These are isoelectronic with their ferric nitrosyl {FeNO}6 and d6 ferrous carbonyl FeCO counterparts. Manganese-substituted hemoproteins containing NO continue to provide very valuable information to complement that obtained from their isoelectronic and biologically relevant FeCO analogues. Examples of such Mn-substituted nitrosyl complexes include those of hemoglobin (Hb), myoglobin (Mb), sGC, neuronal nitric oxide synthase, cytochrome c and peroxidases.4,12–15 MnHbNO and MnMbNO have been prepared as spectroscopic models for their EPR-silent Fe–CO analogues.16–18 The MnHbNO derivative was shown to behave similarly to native HbCO in terms of ligand rebinding kinetics and cooperativity.19 In the case of Mn-substituted sGC, binding of NO generated a six-coordinate (porphyrinato)Mn(NO)(His) species that did not activate the enzyme.4 Other manganese hemes have been prepared for NO-sensing20–22 and for peroxynitrite decomposition.23,24
To date, there are only two X-ray crystal structures of manganese nitrosyl porphyrins, namely those of (TTP)Mn(NO) and (TPP)Mn(NO)(4-Mepip).25 In order to better understand the nature of binding of NO to manganese porphyrins and to determine the nature of the trans effect of the bound NO ligand in these complexes, we have prepared a new set of synthetic manganese nitrosyl porphyrin complexes containing methanol and N-donor ligands. In this article, we report their syntheses and structural characterization by spectroscopy and high-resolution X-ray crystallography.
Experimental
All reactions were performed under an atmosphere of pre-purified nitrogen using standard Schlenk glassware and/or in an Innovative Technology Labmaster 100 Dry Box. Solutions for spectral studies were also prepared under a nitrogen atmosphere. Solvents were distilled from appropriate drying agents under nitrogen just prior to use: CH2Cl2 (CaH2).
Chemicals
(TTP)MnCl and (T(p-OCH3)PP)MnCl were prepared by literature methods.26,27 Piperidine (99.5+%), 1-methylimidazole (1-MeIm, 99+%) and anhydrous CH3OH (99.8%) were purchased from Aldrich Chemical Co. and used as received. Chloroform-d (99.8%) was obtained from Cambridge Isotope Laboratories. Nitric oxide (98%, Matheson Gas) for the synthesis work was passed through KOH pellets and two cold traps (dry-ice/acetone, −78 °C) to remove higher nitrogen oxides.
Instrumentation
Infrared spectra were recorded on a Bio-Rad FT-155 FTIR spectrometer. Proton NMR spectra were obtained on Varian VXR-S 500 MHz or Varian Mercury VX 300 MHz spectrometers for low-temperature and room-temperature measurements, respectively, and the signals referenced to the residual signal of the solvent employed (CHCl3 at 7.24 ppm). All coupling constants are in Hz. ESI mass spectra were obtained on a Micromass Q-TOF mass spectrometer. Elemental analyses were performed by Atlantic Microlab, Norcross, Georgia.
Preparation of (por)Mn(NO)(pip) compounds (por = TTP, T(p-OCH3)PP).
A Schlenk flask was charged with (TTP)MnCl (0.035 g, 0.046 mmol), CH2Cl2 (8 mL) and piperidine (0.7 mL). The mixture was stirred to generate a green solution, and NO gas was bubbled through the solution for 20 min. [Note: during this time, an uncharacterized white gaseous product was generated from solution.] The color of the reaction mixture turned bright red. Nitrogen was bubbled through the solution for 5 min to remove any unreacted NO and other gaseous products. Anhydrous methanol (15 mL) was added to the solution, and the volume of the solution was reduced in vacuo until a solid precipitated. The supernatant solution was discarded, and the red–purple solid was dried in vacuo to give (TTP)Mn(NO)(pip)·0.65CH2Cl2 (0.026 g, 0.029 mmol, 63% isolated yield). Anal. Calc. for C53H47N6OMn·0.65CH2Cl2: C, 72.07; H, 5.44; N, 9.40; Cl, 5.15. Found: C, 71.78; H, 5.66; N, 9.53; Cl, 4.99%. IR (KBr, cm−1): νNO = 1746s; also 3022w, 2935w, 2920w, 2859w, 1533m, 1503m, 1450m, 1404vw, 1346m, 1305w, 1268vw, 1209w, 1181m, 1108w, 1070m, 1027vw, 1015w, 1001s, 871w, 847vw, 796s, 718m, 631w, 552vw, 522m. 1H NMR (CDCl3, −50 °C): δ 8.74 (s, 8H, pyrrole-H of TTP), 8.03 (d, J = 7, 4H, o-H of TTP), 7.99 (d, J = 7, 4H, o′-H of TTP), 7.52 (app t (overlapping d’s), 8H, m/m′-H of TTP), 5.29 (s, CH2Cl2), 2.67 (s, 12H, CH3 of TTP), 0.15 (br d, 1H of pip), −0.40 (br d, 2H of pip), −0.78 (br d, 1H of pip), −1.34 (br d, 2H of pip), −3.42 (app q, J = 13, 2H of pip), −3.76 (br d, 2H of pip), −5.48 (app t, J = 13, 1H of pip). The peaks of the piperidine ligand were not observed in the 1H NMR spectrum of the complex at room temperature. ESI mass spectrum: m/z 723 [(TTP)Mn]+ (100%).
Attempts to obtain suitable crystals of this sample for a single-crystal X-ray structural determination have so far not been successful. Unexpectedly, however, crystals grown by slow solvent evaporation of a CH2Cl2–CH3OH (2 : 1) solution mixture of (TTP)Mn(NO)(pip) under inert atmosphere were identified as (TTP)Mn(NO)(CH3OH) by X-ray crystallography. The IR spectrum of (TTP)Mn(NO)(CH3OH) (as a KBr pellet) showed a strong band at 1743 cm−1 assigned to νNO.
The (T(p-OCH3)PP)Mn(NO)(pip) compound was prepared similarly from (T(p-OCH3)PP)MnCl and NO gas in the presence of piperidine. The red-purple product was obtained in 81% isolated yield. IR (KBr, cm−1): νNO = 1748s; also 3032vw, 2997vw, 2935w, 2834w, 1608m, 1574vw, 1533m, 1512s, 1502s, 1464m, 1439m, 1410vw, 1347m, 1303w, 1287m, 1248s, 1206w, 1175s, 1107w, 1071w, 1039w, 1026m, 1002s, 872vw, 848w, 803s, 719m, 637vw, 632vw, 607m, 575vw, 555w, 538m. 1H NMR (CDCl3, −50 °C): δ 8.73 (s, 8H, pyrrole-H of T(p-OCH3)PP), 8.04 (d, J = 8, 4H, o-H of T(p-OCH3)PP), 8.01 (d, J = 8, 4H, o′-H of T(p-OCH3)PP), 7.22 (br (overlapping with CHCl3 peak), 8H, m/m′-H of T(p-OCH3)PP), 5.29 (s, CH2Cl2), 4.07 (s, 12H, OCH3 of T(p-OCH3)PP), 0.12 (br d, 1H of pip), −0.43 (br d, 2H of pip), −0.80 (br d, 1H of pip), −1.36 (br d, 2H of pip), −3.46 (app q, J = 13, 2H of pip), −3.79 (br d, 2H of pip), −5.50 (app t, J = 13, 1H of pip). The peaks of the piperidine ligand were not observed in the 1H NMR spectrum of the complex at room temperature. ESI mass spectrum: m/z 787 [(T(p-OCH3)-PP)Mn]+ (100%).
Preparation of (por)Mn(NO)(1-MeIm) compounds (por = TTP, T(p-OCH3)PP).
To a stirred CH2Cl2 solution (8 mL) of (TTP)Mn(NO)(pip)·0.65CH2Cl2 (0.020 g, 0.022 mmol) was added 1-MeIm (0.01 mL). The mixture was stirred for 5 h. The solvent was removed in vacuo, and the residue was redissolved in a CH2Cl2–CH3OH (1 : 1, 10 mL) mixture. To this solution was added 1-MeIm (0.1 mL). Slow evaporation of this solution at room temperature under inert atmosphere gave spectroscopically pure (TTP)Mn(NO)(1-MeIm) (0.012 g, 0.014 mmol, 64% isolated yield) as purple crystals. IR (KBr, cm−1): νNO = 1738s, 1732s; also 3128vw, 3019vw, 2952vw, 2918vw, 2866vw, 1532w, 1506m, 1457vw, 1347m, 1304vw, 1284vw, 1233w, 1209w, 1181m, 1108m, 1081w, 1071m, 1001s, 943vw, 908vw, 846vw, 796s, 731w, 719m, 670vw, 660vw, 628vw, 615vw, 523m. 1H NMR (CDCl3, −40 °C): δ 8.69 (s, 8H, pyrrole-H of TTP), 8.04 (d, J = 7, 4H, o-H of TTP), 7.91 (d, J = 7, 4H, o′-H of TTP), 7.50 (d, J = 7, 4H, m-H of TTP), 7.45 (d, J = 7, 4H, m′-H of TTP), 4.60 (s, 1H of 1-MeIm), 2.66 (s, 12H, CH3 of TTP), 2.06 (s, 3H, CH3 of 1-MeIm), 1.17 (s, 1H of 1-MeIm), 0.73 (s, 1H of 1-MeIm). The peaks of the 1-MeIm ligand were not observed in the 1H NMR spectrum of the complex at room temperature. ESI mass spectrum: m/z 805 [(TTP)Mn(1-MeIm)]+ (57%), 723 [(TTP)Mn]+ (100%).
The purple (T(p-OCH3)PP)Mn(NO)(1-MeIm) compound was obtained in 77% isolated yield after recrystallization from CH2Cl2–CH3OH (2 : 1) in the presence of excess 1-MeIm at room temperature under inert atmosphere. IR (KBr, cm−1): νNO = 1736s; also 3128vw, 3033vw, 2998vw, 2958vw, 2934vw, 2906vw, 2833vw, 1606m, 1533w, 1501s, 1466w, 1460w, 1439w, 1346m, 1303vw, 1285m, 1247s, 1205vw, 1177s, 1108w, 1089vw, 1073vw, 1037vw, 1024w, 1001s, 849vw, 808m, 799m, 735w, 719w, 670vw, 662vw, 607w, 582vw, 540w. 1H NMR (CDCl3, −40 °C): δ 8.70 (s, 8H, pyrrole-H of T(p-OCH3)PP), 8.07 (dd, J = 8/2, 4H, o-H of T(p-OCH3)PP), 7.93 (dd, J = 8/2, 4H, o′-H of T(p-OCH3)PP), 7.22 (dd (overlapping with CHCl3 peak), 4H, m-H of T(p-OCH3)PP), 7.18 (dd, J = 8/2, 4H, m′-H of T(p-OCH3)PP), 4.60 (s, 1H of 1-MeIm), 4.06 (s, 12H, OCH3 of T(p-OCH3)PP), 2.07 (s, 3H, CH3 of 1-MeIm), 1.15 (s, 1H of 1-MeIm), 0.72 (s, 1H of 1-MeIm). The peaks of the 1-MeIm ligand were not observed in the 1H NMR spectrum of the complex at room temperature. ESI mass spectrum: m/z 787 [(T(p-OCH3)PP)Mn]+ (100%).
Structural determinations by X-ray crystallography
X-Ray data were collected on a Bruker Apex diffractometer using Mo-Kα (λ = 0.71073 Å) radiation. The structures were solved by the direct method using the SHELXTL system (Version 6.12; 1997) and refined by full-matrix least squares on F2 using all reflections. All the non-hydrogen atoms were refined anisotropically. All the hydrogen atoms were included with idealized parameters. Displacement ellipsoids in Figs. 1 and 2 are drawn at the 35% probability level. Details of the crystal data and refinement are given in Table 1.
Fig. 1.
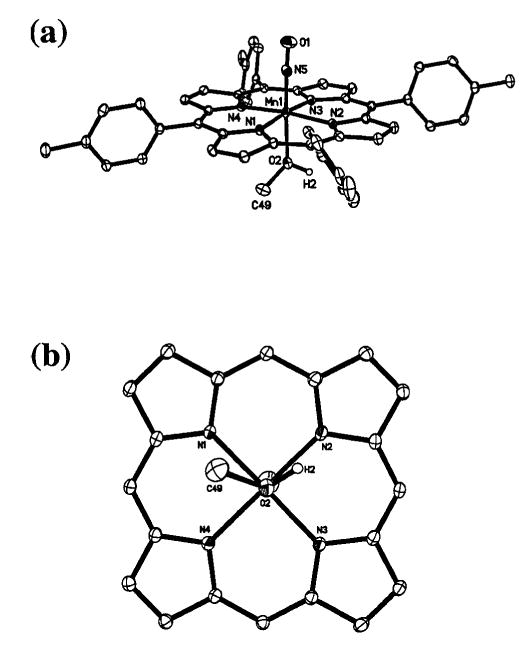
(a) Molecular structure of (TTP)Mn(NO)(CH3OH). Only the crystallographically ordered molecule is shown. (b) View of the orientation of the methanol ligand relative to the porphyrin skeleton. The porphyrin tolyl substituents have been omitted for clarity.
Fig. 2.
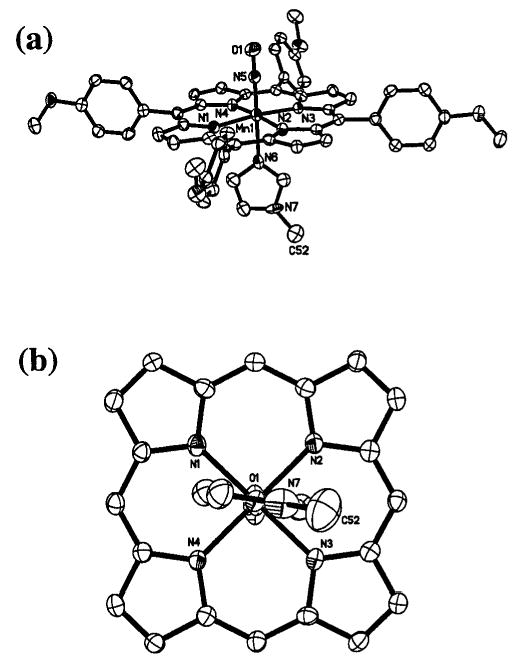
(a) Molecular structure of (T(p-OCH3)PP)Mn(NO)(1-MeIm). Hydrogen atoms have been omitted for clarity. (b) View of the orientation of the 1-MeIm ligand relative to the porphyrin skeleton. The porphyrin p-methoxyphenyl substituents have been omitted for clarity.
Table 1.
Crystal data and structure refinement
| (TTP)Mn(NO)(CH3OH)·solva | (TTP)Mn(NO)(1-MeIm)·0.28CH2Cl2 | (T(p-OCH3)PP)Mn(NO)(1-MeIm)·CH2Cl2 | ||||
| Formula | C49.90H43.33Cl0.27N5O2.77Mn | C52.28H42.56Cl0.56N7OMn | C53H44Cl2N7O5Mn | |||
| Mr | 821.69 | 859.65 | 984.79 | |||
| T/K | 96(2) | 120(2) | 178(2) | |||
| Crystal system | Triclinic | Triclinic | Triclinic | |||
| Space group |
P
|
P
|
P
|
|||
| a/Å | 11.3170(6) | 14.9067(8) | 12.1992(16) | |||
| b/Å | 12.3768(7) | 15.0365(8) | 12.7458(17) | |||
| c/Å | 23.0336(12) | 15.8656(9) | 15.151(2) | |||
| agr;/° | 97.9110(10) | 104.7090(10) | 90.136(2) | |||
| β/° | 102.9260(10) | 90.8320(10) | 90.287(2) | |||
| γ/° | 97.0580(10) | 97.3400(10) | 99.228(2) | |||
| V, Z | 3074.9(3) Å3, 3 | 3407.4(3) Å3, 3 | 2325.3(5) Å3, 2 | |||
| Dc/g cm−3 | 1.331 | 1.257 | 1.407 | |||
| μ/mm−1 | 0.389 | 0.370 | 0.457 | |||
| F(000) | 1288 | 1343 | 1020 | |||
| Crystal size/mm | 0.22 × 0.18 × 0.16 | 0.42 × 0.38 × 0.28 | 0.18 × 0.04 × 0.04 | |||
| θ Range for data collection/° | 1.68–27.00 | 1.84–26.50 | 1.62–25.00 | |||
| Index ranges, hkl | −14 to 14, −15 to 15, −29 to 29 | −18 to 18, −18 to 18, −19 to 19 | −14 to 14, −14 to 15, −17 to 17 | |||
| Reflections collected | 37158 | 36783 | 23420 | |||
| Independent reflns (Rint) | 13303 (0.0162) | 13983 (0.0198) | 8054 (0.0715) | |||
| Max., min. transmission | 0.9403, 0.9193 | 0.9036, 0.8602 | 0.9820, 0.9223 | |||
| Data/restraints/parameters | 13303/124/916 | 13983/69/948 | 8054/63/672 | |||
| Goodness-of-fit on F2 | 1.127 | 1.193 | 1.004 | |||
| Final R indices [I > 2σ(I)] | R1 = 0.0670, wR2 = 0.1769 | R1 = 0.0780, wR2 = 0.2205 | R1 = 0.0750, wR2 = 0.1721 | |||
| R Indices (all data) | R1 = 0.0685, wR2 = 0.1778 | R1 = 0.0811, wR2 = 0.2220 | R1 = 0.1437, wR2 = 0.1896 | |||
| Largest diff. peak, hole/e Å−3 | 1.311, −0.776 | 1.257, −0.592 | 1.190, −0.458 |
Crystal contains both CH3OH and CH2Cl2 in fractional occupancy.
(i) (TTP)Mn(NO)(CH3OH). Crystals for X-ray crystallography were grown during an attempt at crystallizing (TTP)-Mn(NO)(pip) using CH2Cl2–CH3OH. X-Ray diffraction intensity data, which approximately covered the full sphere of the reciprocal space, were measured as a series of ω oscillation frames each 0.3° for 21 s frame−1. The detector was operated in 512 × 512 mode and was positioned 6.00 cm from the crystal. Coverage of unique data was 99.0% complete to 54° (2θ). Cell parameters were determined from a non-linear least squares fit of 7582 reflections in the range of 2.8 < θ < 25.9°. A total of 37158 reflections were measured. The data were corrected for absorption by multi-scan method from equivalent reflections giving the minimum and maximum transmissions of 0.9193 and 0.9403. The asymmetric unit contains one and a half units of the C49H40N5O2Mn complex (with the Mn(2) atom situated very near the inversion center) and fractional amounts of dichloromethane (0.2) and methanol (1.15) solvent molecules. SHELXTL restrains of DFIX and ISOR were applied to the atoms belonging to the disordered axial group and the solvent molecules to achieve convergence during least squares refinement. The final R1 = 0.067 is based on 12836 “observed reflections” [I > 2σ(I )], and wR2 = 0.178 is based on all reflections (13303 reflections).
(ii) (TTP)Mn(NO)(1-MeIm)·0.28CH2Cl2. Suitable crystals for X-ray crystallography were grown by slow evaporation of a CH2Cl2–CH3OH (1 : 1) solution of the compound containing 1-MeIm at room temperature under inert atmosphere. Coverage of unique data was 99.1% complete to 53° (2θ). Cell parameters were determined from a non-linear least squares fit of 8729 reflections in the range of 3.6 < θ < 26.2°. A total of 13983 reflections were measured. The data were corrected for absorption by multi-scan method from equivalent reflections giving the minimum and maximum transmissions of 0.8602 and 0.9036. The asymmetric unit contains one and a half units of the C52H42N7OMn complex (with the Mn(2) atom situated very near the inversion center) and fractional dichloromethane solvent molecules. The unit of the C52H42N7OMn complex that lies at the inversion center is disordered at the axial sites. The N(10) atom from the NO group lies very close to the N(11) atom of the 1-methylimidazole ligand. As a result, the refinement of the positional and thermal parameters is compromised. The Mn–N–O angles and N–O bond distances in the two molecules are significantly different. However, in view of the axial disorder in the disordered molecule, it is not possible to ascertain whether these differences in bond angles and bond lengths are real or an artifact of the poor refinement due to the disorder. SHELXTL restrains of DFIX, ISOR and EADP were applied to achieve convergence during least squares refinement (the disordered NO group in the molecule #2 was allowed to refine without any restraints). The final R1 = 0.078 is based on 13129 “observed reflections” [I > 2σ(I)], and wR2 = 0.222 is based on all reflections (13983 reflections).
(iii) (T(p-OCH3)PP)Mn(NO)(1-MeIm)·CH2Cl2. Suitable crystals for X-ray crystallography were grown by slow evaporation of a CH2Cl2–CH3OH (2 : 1) solution of the compound containing 1-MeIm at room temperature under inert atmosphere. Coverage of unique data was 98.4% complete to 50° (2θ). Cell parameters were determined from a non-linear least squares fit of 7823 reflections in the range of 3.2 < θ < 24.4°. A total of 23420 reflections were measured. The data were corrected for absorption by the multi-scan method from equivalent reflections giving minimum and maximum transmissions of 0.9223 and 0.9820.
An initial data set was collected at 100(2) K and yielded poor refinement. Better data were obtained at 178(2) K, showing much sharper spots on the frames. There is a highly disordered CH2Cl2 solvent molecule and some minor disorder involving the nitrosyl O(1) atom, the imidazole N(7) atom and the p-methoxy C(27), C(34), C(41) and C(48) atoms of the porphyrin aryl substituents. The non-solvent atoms were refined without resolving into their disordered components due to the very close proximity of these components. The overall geometry of the molecule of interest is sound. SHELXTL restrains of DFIX, ISOR and SADI were applied to the atoms belonging to the disordered solvent molecule to achieve convergence during least squares refinement. The final R1 = 0.075 is based on 4461 “observed reflections” [I > 2σ(I )], and wR2 = 0.1896 is based on all reflections (8054 unique reflections). A reviewer suggested that we consider the possibility of a monoclinic crystal system, since two angles are close to 90°. We re-examined this possibility, and better fits of the data were obtained for a triclinic cell (Rint = 0.07) than for a monoclinic cell (Rint = 0.57); the correct Laue symmetry is rather than 2/m. We thank the reviewer for this suggestion.
CCDC reference numbers 215499–215501.
See http://www.rsc.org/suppdata/dt/b3/b308143p/ for crystallographic data in CIF or other electronic format.
Results and discussion
The synthesis and chemistry of manganese- and other metalloporphyrin nitrosyl complexes has been reviewed recently.1 Scheidt and coworkers showed, almost thirty years ago, that reductive nitrosylation of manganese(III) porphyrins took place in the presence of aliphatic amines to generate manganese nitrosyl porphyrins.28,29 We have used similar methodology for the preparation of (TTP)Mn(NO)(pip) and (T(p-OCH3)PP)-Mn(NO)(pip) in 63 and 81% isolated yields, respectively (eqn. (1); por = TTP or T(p-OCH3)PP; pip = piperidine).
| (1) |
These red–purple complexes are soluble in CH2Cl2, CHCl3 and acetone, but are only slightly soluble in hexane and methanol. The complexes are moderately stable in air in the solid state, showing no noticeable signs of decomposition over a one month period as judged by IR and 1H NMR spectroscopy. Their solutions, however, are air-sensitive.
The IR spectra of the (por)Mn(NO)(pip) complexes (as KBr pellets) show new strong bands at 1746 cm−1 (for TTP) and 1748 cm−1 (for (T(p-OCH3)PP), respectively, which are attributed to the terminal NO ligands. These bands are similar to that reported for the crystallographically characterized (TPP)-Mn(NO)(4-Mepip) compound (νNO 1740 cm−1) that exhibits a linear NO geometry.28,29 Although the (por)Mn(NO)(pip) compounds are isoelectronic with their ferric–NO counterparts, the νNO bands of the manganese compounds are significantly lower than those of the isolable ferric [(por)Fe(NO)(N-base)]+ complexes (νNO 1894–1921 cm−1) reported by Scheidt and Ellison.30 Parthasarathi and Spiro have suggested, based on resonance Raman studies, that there is reduced metal→NO backbonding in FeIII–NO compared with MnII–NO due to the higher effective charge on iron in FeIII–NO.16 Clearly, such an effect is responsible for the differences in the observed NO stretching frequencies between the Mn and Fe compounds.
We have not been able to observe the parent ions of the (por)Mn(NO)(pip) compounds in their ESI mass spectra; the spectra show ion fragments due to loss of both axial ligands. Attempts to grow crystals of the (TTP)Mn(NO)(pip) complex from dichloromethane–methanol resulted in the replacement of the piperidine ligand with methanol solvent to give (TTP)-Mn(NO)(CH3OH) (νNO 1743 cm−1), as determined from the X-ray crystal structure of the crystallization product (see later).
The (TTP)Mn(NO)(1-MeIm) and (T(p-OCH3)PP)Mn(NO)-(1-MeIm) derivatives were prepared in 64 and 77% isolated yields, respectively, from the reaction of their piperidine precursors with an excess of 1-MeIm in CH2Cl2 (eqn. (2)).
| (2) |
Prior to this work, the (por)Mn(NO)(imidazole) compounds reported in the literature were prepared in situ from exposure of NO to solutions of MnII-porphyrins containing an excess of the imidazole ligand, and the success of this synthetic procedure was very limited.16,31 We find that the preparative route described by eqn. (2) provides a convenient method by which pure samples of the nitrosyl products can be obtained in sizeable quantities.
These products of eqn. (2) are purple and have similar solubilities and stabilities as those of their piperidine precursors. The IR spectra of the (por)Mn(NO)(1-MeIm) complexes (as KBr pellets) reveal bands at 1738/1732 cm−1 (split band) and 1736 cm−1 for the TTP and T(p-OCH3)PP derivatives, respectively, assignable to νNO. The split νNO band of (TTP)Mn(NO)-(1-MeIm) is likely due to the presence of two orientations of the 1-MeIm ligand in the bulk solid. In any event, the νNO bands in (por)Mn(NO)(1-MeIm) are ~12 cm−1 lower in energy than the νNO bands of the precursor (por)Mn(NO)(pip) complexes. This feature is consistent with the replacement of piperidine ligand with the π-interacting 1-MeIm ligand, which makes more electron density available for MnII→NO backdonation, resulting in the lowering of νNO.
The low-temperature 1H NMR spectra of the (por)Mn(NO)-(1-MeIm) complexes show, in addition to the peaks due to the porphyrin rings, the peaks of the bound 1-MeIm ligands. The ESI mass spectrum of (TTP)Mn(NO)(1-MeIm) shows ion fragments assigned to loss of the NO ligand or loss of both axial ligands, whereas that of (T(p-OCH3)PP)Mn(NO)-(1-MeIm) shows ion fragments from loss of both axial ligands.
X-Ray crystallographic characterization
We were successful in obtaining suitable crystals of (TTP)-Mn(NO)(CH3OH), (TTP)Mn(NO)(1-MeIm) and (T(p-OCH3)-PP)Mn(NO)(1-MeIm) for single-crystal X-ray crystallography. The molecular structures of (TTP)Mn(NO)(CH3OH) and (T(p-OCH3)PP)Mn(NO)(1-MeIm) are shown in Figs. 1 and 2. The structure of (TTP)Mn(NO)(1-MeIm) is provided in the ESI. † Selected structural parameters for all three compounds are summarized in Fig. 3. As stated in the Experimental Section, the asymmetric units in the crystals of (TTP)Mn(NO)(CH3OH) and (TTP)Mn(NO)(1-MeIm) contain one crystallographically ordered molecule and a second disordered molecule. Only the more accurate data from the ordered components are discussed here.
Fig. 3.
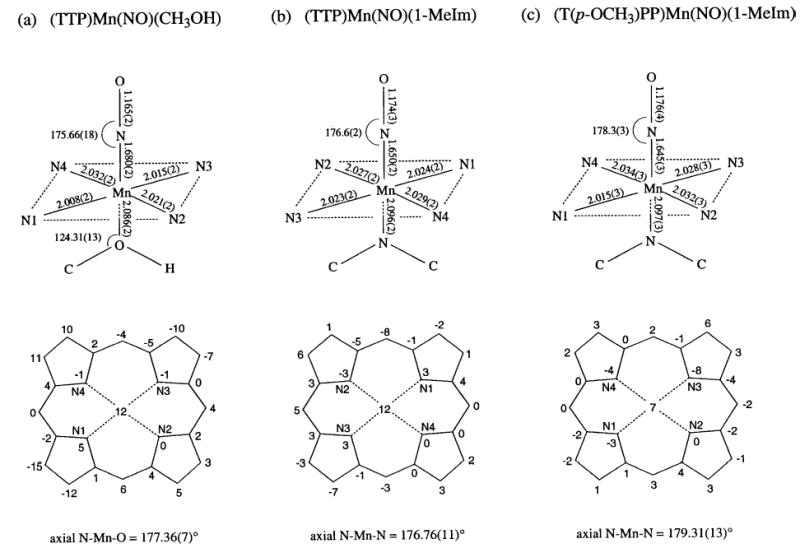
Structural data for the ordered molecule of (TTP)Mn(NO)(CH3OH), the ordered molecule of (TTP)Mn(NO)(1-MeIm) and (T(p-OCH3)PP)Mn(NO)(1-MeIm). Selected bond lengths and angles are shown in the top sketches. Perpendicular atom displacements from the 24-atom porphyrin planes (in 0.01 Å units) are shown in the bottom sketches.
The Mn–N–O moieties in all three complexes are linear, and this observed linearity is consistent with the classification of these (por)Mn(NO)(L) compounds as {MnNO} 6 species.9–11 The axial Mn–N(O) bond length in (TTP)Mn(NO)(CH3OH) is 1.680(2) Å while the N–O bond length is 1.165(2) Å. The Mn–N(por) distances fall in the 2.008(2)–2.032(2) Å range while the Mn atom is displaced by 0.12 Å from the 24-atom mean porphyrin plane toward the π-acid NO ligand. The methanol ligand in (TTP)Mn(NO)(CH3OH) is oriented essentially between two porphyrin N-atoms (Fig. 1(b)), and the Mn–O bond length is 2.086(2) Å while the Mn–O2–C49 angle is 124°. The Mn–O bond length is shorter than those observed in other structurally characterized MnII and MnIII porphyrins containing alcohol ligands: [(TPP)Mn(CH3OH)2]ClO4 (2.252(2) and 2.270(2) Å),32 [(TPP)Mn(CH3OH)2]SbCl6 (2.283(5) Å),33 (TPP)Mn(N3)(CH3OH) (2.329(7) Å),34 (P*)Mn-(CH3OH)(OH) (2.251(7) Å; P* = D4 symmetrical chiral porphyrin),35 (TCPP)Mn(CH3OH)(H2O) (2.246 Å; TCPP = tetracarboxyphenylporphyrin),36 and [(OEP)Mn(EtOH)]ClO4 (2.145(2) Å).37
The axial Mn–N(O) bond lengths in the two (por)Mn(NO)-(1-MeIm) complexes are in the 1.645(3)–1.650(2) Å range and are similar to that observed previously in (TPP)Mn(NO)-(4-Mepip) (1.644(5) Å),25,28 but are shorter than that determined for (TTP)Mn(NO)(CH3OH) (1.680(2) Å). This feature is suggestive of greater Mn→NO π-backdonation in these (por)Mn(NO)(N-base) compounds. The lower νNO of the 1-methylimidazole complexes compared with that of the methanol complex supports this view of a greater Mn→NO π-backdonation in (por)Mn(NO)(1-MeIm). Importantly, the axial trans Mn–N(imidazole) bond lengths of 2.096(2) and 2.097(3) Å for the (TTP)Mn(NO)(1-MeIm) and (T(p-OCH3)-PP)Mn(NO)(1-MeIm) compounds, respectively, are shorter than that reported for the five-coordinate high-spin (TPP)-Mn(1-MeIm) complex (2.192(2) Å),38 reflecting the influence of the strong field π-acid NO ligand on these trans Mn–N(imidazole) bond lengths. A comparison of the structural data of the (por)Mn(NO)(1-MeIm) compounds with those of the previously reported and isoelectronic [(OEP)Fe(NO)(1-MeIm)]+ shows that in both classes of compounds, the metal–NO groups are linear. Furthermore, the Mn–N(O) bond lengths (1.645(3)–1.650(2) Å) are similar to that in [(OEP)Fe(NO)(1-MeIm)]+ (1.6465(17) Å),30 although the N–O bond lengths in (por)-Mn(NO)(1-MeIm) (1.174(3)–1.176(4) Å) are slightly longer than that observed in [(OEP)Fe(NO)(1-MeIm)]+ (1.135(2) Å).
In summary, we have prepared several isolable six-coordinate manganese nitrosyl porphyrins and have characterized them by spectroscopy. Our successful crystallization of three of these derivatives, which were characterized by single-crystal X-ray crystallography, significantly increases the number of available structures of manganese nitrosyl porphyrins. In particular, the trans influence of the linear NO group observed in these structures provides a convenient entry into further studies of this structural effect. Comparisons between these manganese(II) nitrosyl derivatives and the ferric nitrosyl derivatives suggest that there are structural similarities between these formally isoelectronic MnII–NO and FeIII–NO species. However, care must be taken in the intepretation of these results, since subtle π-backbonding differences exist between them. Further work to explore these electronic differences is currently in progress.
Acknowledgments
We thank the National Institutes of Health of the USA (Grant No. GM 64476) for financial support of this work. Z. N. Z. is grateful to the Government of Egypt for a graduate fellowship.
Footnotes
Electronic supplementary information (ESI) available: Molecular structure of (TTP)Mn(NO)(1-MeIm). See http://www.rsc.org/suppdata/dt/b3/b308143p/
References
- 1.L. Cheng and G. B. Richter-Addo, Binding and Activation of Nitric Oxide by Metalloporphyrins and Heme, in The Porphyrin Handbook, ed. R. Guilard, K. Smith and K. M. Kadish, Academic Press, New York, 2000, vol. 4, ch. 33, pp. 219–291.
- 2.Deinum G, Stone JR, Babcock GT, Marletta MA. Biochemistry. 1996;35:1540–1547. doi: 10.1021/bi952440m. [DOI] [PubMed] [Google Scholar]
- 3.Ballou DP, Zhao Y, Brandish PE, Marletta MA. Proc. Natl. Acad. Sci., USA. 2002;99:12097–12101. doi: 10.1073/pnas.192209799. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dierks EA, Hu S, Vogel KM, Yu AE, Spiro TG, Burstyn JN. J. Am. Chem. Soc. 1997;119:7316–7323. doi: 10.1007/s007750050354. [DOI] [PubMed] [Google Scholar]
- 5.Kharitonov VG, Sharma VS, Magde D, Koesling D. Biochemistry. 1997;36:6814–6818. doi: 10.1021/bi970201o. [DOI] [PubMed] [Google Scholar]
- 6.Ford PC, Lorkovic IM. Chem. Rev. 2002;102:993–1017. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 7.Walker FA, Monfort WR. Adv. Inorg. Chem. 2001;51:295–358. [Google Scholar]
- 8.Wyllie GRA, Scheidt WR. Chem. Rev. 2002;102:1067–1089. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]
- 9.B. L. Westcott and J. H. Enemark, in Transition Metal Nitrosyls, in Inorganic Electronic Structure and Spectroscopy, ed. A. B. P. Lever and E. I. Solomon, Wiley and Sons, New York, 1999, vol. 2, ch. 7.
- 10.Enemark JH, Feltham RD. Coord. Chem. Rev. 1974;13:339–406. [Google Scholar]
- 11.Feltham RD, Enemark JH. Top. Stereochem. 1981;12:155–215. [Google Scholar]
- 12.Yonetani T, Yamamoto H, Erman JE, Leign JJS, Reed GH. J. Biol. Chem. 1972;247:2447–2455. [PubMed] [Google Scholar]
- 13.Dickinson LC, Chien JCW. J. Biol. Chem. 1977;252:6156–6162. [PubMed] [Google Scholar]
- 14.Hemmens B, Gorren ACF, Schmidt K, Werner ER, Mayer B. Biochem. J. 1998;332:337–342. doi: 10.1042/bj3320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bender AT, Kamada Y, Kleaveland PA, Osawa Y. J. Inorg. Biochem. 2002;91:625–634. doi: 10.1016/s0162-0134(02)00430-0. [DOI] [PubMed] [Google Scholar]
- 16.Parthasarathi N, Spiro TG. Inorg. Chem. 1987;26:2280–2282. [Google Scholar]
- 17.Hori H, Ikeda-Saito M, Lang G, Yonetani T. J. Biol. Chem. 1990;265:15028–15033. [PubMed] [Google Scholar]
- 18.Masuya F, Hori H. Biochim Biophys Acta. 1993;1203:99–103. doi: 10.1016/0167-4838(93)90041-o. [DOI] [PubMed] [Google Scholar]
- 19.Gibson QH, Hoffman BM. J. Biol. Chem. 1979;254:4691–4697. [PubMed] [Google Scholar]
- 20.Lan EH, Dave BC, Fukuto JM, Dunn B, Zink JI, Valentine JS. J. Mater. Chem. 1999;9:45–53. [Google Scholar]
- 21.Diab N, Schuhmann W. Electrochim Acta. 2001;47:265–273. [Google Scholar]
- 22.Spasojevic I, Batinic-Haberle I, Fridovich I. Nitric Oxide: Biol. Chem. 2000;4:526–533. doi: 10.1006/niox.2000.0303. [DOI] [PubMed] [Google Scholar]
- 23.Ferrer-Sueta G, Quijano C, Alvarez B, Radi R. Methods Enzymol. 2002;349:23–37. doi: 10.1016/s0076-6879(02)49318-4. [DOI] [PubMed] [Google Scholar]
- 24.Shimanovich R, Hannah S, Lynch V, Gerasimchuk N, Mody TD, Magda D, Sessler J, Groves JT. J. Am. Chem. Soc. 2001;123:3613–3614. doi: 10.1021/ja005856i. and references therein. [DOI] [PubMed] [Google Scholar]
- 25.Scheidt WR, Hatano K, Rupprecht GA, Piciulo PL. Inorg. Chem. 1979;18:292–299. [Google Scholar]
- 26.Adler AD, Longo FR, Finarelli JD, Goldmacher J, Assour J, Korsakoff L. J. Org. Chem. 1967;32:476. [Google Scholar]
- 27.Adler AD, Longo FR, Kampas F, Kim J. J. Inorg. Nucl. Chem. 1970;32:2443–2445. [Google Scholar]
- 28.Piciulo PL, Rupprecht G, Scheidt WR. J. Am. Chem. Soc. 1974;96:5293–5295. [Google Scholar]
- 29.Piciulo PL, Scheidt WR. Inorg. Nucl. Chem. Lett. 1975;11:309–311. [Google Scholar]
- 30.Ellison MK, Scheidt WR. J. Am. Chem. Soc. 1999;121:5210–5219. [Google Scholar]
- 31.Yu NT, Lin SH, Chang CK, Gersonde K. Biophys. J. 1989;55:1137–1144. doi: 10.1016/S0006-3495(89)82910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatano K, Anzai K, Iitaka Y. Bull. Chem. Soc. Jpn. 1983;56:422–427. [Google Scholar]
- 33.Scheidt WR, Pearson WB, Gosal N. Acta Crystallogr Sect C. 1988;44:927–929. doi: 10.1107/s0108270187012411. [DOI] [PubMed] [Google Scholar]
- 34.Day VW, Stults BR, Tasset EL, Day RO, Marianelli RS. J. Am. Chem. Soc. 1974;96:2650–2652. doi: 10.1021/ja00815a075. [DOI] [PubMed] [Google Scholar]
- 35.T.-S. Lai, H.-L. Kwong, C.-M. Che, S.-M. Peng, Chem. Commun., 1997, 2373–2374.
- 36.Y. Diskin-Posner, G.K. Patra I. and Goldberg, Eur. J. Inorg. Chem., 2001, 2515–2523.
- 37.Cheng B, Scheidt WR. Acta Crystallogr Sect C. 1996;52:585–588. doi: 10.1107/s0108270195010705. [DOI] [PubMed] [Google Scholar]
- 38.Kirner JF, Reed CA, Scheidt WR. J. Am. Chem. Soc. 1977;99:2557–2563. doi: 10.1021/ja00450a025. [DOI] [PubMed] [Google Scholar]