

Abstract
The cellular response to hypoxia is, at least in part, mediated by the transcriptional regulation of hypoxia-responsive genes involved in balancing the intracellular ATP production and consumption. Recent evidence suggests that the transcription factor, HIF-1α, functions as a master regulator of oxygen homeostasis by controlling a broad range of cellular events in hypoxia. In normoxia, HIF-1α is targeted for destruction via prolyl hydroxylation, an oxygen-dependent modification that signals for recognition by the E3 ubiquitin ligase complex containing the von Hippel-Lindau tumor suppressor (VHL). Three HIF prolyl hydroxylases (EGLN1, EGLN2, and EGLN3) have been identified in mammals, among which, EGLN1 and EGLN3, are hypoxia-inducible at their mRNA levels in a HIF-1α-dependent manner. In this study, we demonstrate that apart from promoting HIF-1α proteolysis in normoxia, EGLN1 specifically represses HIF-1α transcriptional activity in hypoxia. Ectopic expression of EGLN1 inhibited HIF-1α transcriptional activity without altering its protein levels in a VHL-deficient cell line, indicating a discrete activity of EGLN1 in transcriptional repression. Conversely, silencing of EGLN1 expression augmented HIF-1α transcriptional activity and its target gene expression in hypoxia. Hence, we propose that the accumulated EGLN1 in hypoxia acts as a negative-feedback mechanism to modulate HIF-1α target gene expression. Our finding also provides new insight into the pharmacological manipulation of the HIF prolyl hydroxylase for ischemic diseases.
Hypoxia-inducible factor 1α (HIF-1α1), a basic helix-loop-helix transcription factor of the PAS superfamily (1), plays a central role in cellular adaptation to reduced oxygen availability (2–9). Under hypoxic stress, activated HIF-1α strives for oxygen homeostasis by not only maintaining intracellular energy production via the induction of angiogenesis and glycolysis, but also limiting energy consumption by virtue of the inhibition of cell proliferation and DNA repair (10–12). In general, HIF-1α activates its target genes such as EPO, VEGF, and PGK1 through dimerization with ARNT (also known as HIF-1β) (13), recruitment of the transcription co-activator p300/CBP (14–16), and binding to the hypoxia-responsive element in the promoter (17). Alternatively, HIF-1α functionally antagonizes the oncogene Myc via protein-protein interactions, resulting in CDKN1A/p21cip1 up-regulation (10) and MSH2 and MSH6 down-regulation (12). Hence, HIF-1α functions as a master regulator of oxygen homeostasis for cell survival.
HIF-1α activity depends primarily on hypoxia-induced HIF-1α stabilization (18–21) and p300/CBP recruitment (14–16). HIF-1α is regulated at the posttranslational level via oxygen-dependent hydroxylation (22–24). Human HIF-1α is constitutively hydroxylated at Pro-402 and Pro-564 in normoxia, resulting in recognition by the von Hippel-Lindau (VHL) ubiquitin ligase that leads to polyubiquitination and proteasomal degradation (25–29). Recently, three human prolyl-4-hydroxylases, EGLN1, EGLN2, and EGLN3 (30) (also known as PHD2/HPH2, PHD1/HPH3, and PHD3/HPH1, respectively) have been identified, each of which catalyzes oxygen-dependent hydroxylation of HIF-1α (31,32). All three EGLNs are 2-oxoglutarate- and iron-dependent dioxygenases that utilize molecular oxygen as a co-substrate for prolyl hydroxylation. Another dioxygenase, HIF1AN/FIH1, initially identified as an inhibitor of HIF-1α transcriptional activation (33), also promotes hydroxylation of Asn-803 in the HIF-1α C-terminal activation domain (CAD) (34–36), thereby precluding the recruitment of p300/CBP (37). Thus, both EGLNs and HIF1AN serve as cellular oxygen sensors to regulate HIF-1α activity.
Interestingly, both EGLN1 and EGLN3 genes are up-regulated by hypoxia at their mRNA levels (31,38) in a HIF-1α-dependent manner (39,40). These observations have led to the hypothesis that the buildup of the prolyl hydroxylases during hypoxia primes for rapid degradation of HIF-1α upon re-oxygenation. In this study, however, we sought the role of EGLN1 in hypoxia. Our results indicate that EGLN1 binds HIF-1α in hypoxia as well as in normoxia, and functionally inhibits HIF-1α N-terminal transcriptional activity.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293 and U-2 OS cells were cultured in DMEM (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT). Hep3B cells were grown in MEM medium with 10 % fetal bovine serum. A VHL-deficient renal clear cell carcinoma cell line, UMRC2 or C2, was obtained from L. Neckers (NCI, Bethesda, MD). C2 cells were cultured in DMEM, supplemented with 10% fetal bovine serum. For hypoxic treatment, cells were incubated in a hypoxic chamber (NAPCO, Winchester, VA), maintained with 1% O2 and 5% CO2, and balanced with N2.
Prolyl hydroxylase binding
Gal4-HIF-1α(C-ODD) (amino acids 498–603) or the corresponding L574S mutant (1.5 μg) was transfected with or without the V5-tagged EGLN1 expression plasmid (0.2 μg). Cells were exposed to hypoxic conditions or treated with 12.5 μM Cbz-LLL (Sigma-Aldrich, St. Louis, MO) for 4 h, and then were lyzed in a buffer containing 25 mM Tris (pH 7.5), 300 mM NaCl, and 1 % Triton-X. The lysate was immunoprecipitated with anti-Gal4 monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), followed by immunoblotting with anti-EGLN1 antibody (Novus Biologicals, Littleton, CO) and polyclonal anti-Gal4 antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
RNA interference
The design of the 21-nucleotide siRNA duplexes against EGLN1, EGLN2, and EGLN3 is as described previously (40). All of them were chemically synthesized by Qiagen (Valencia, CA). SMARTpool siRNA against VHL was purchased from Dharmacon (Lafayette, CO). A siRNA targeting the firefly luciferase coding sequence of the pGL2 vector (41) was employed as a negative control. Cells were seeded at 30% confluence in antibiotic-free medium 24 h prior to transfection. Oligofectamine reagent (Invitrogen, Carlsbad, CA) was used for transfection with 20 nM siRNA duplex twice at a 24-h interval according to the manufacturer’s instructions. The efficacy of the siRNA transfection in each experiment was determined by Western blot. Polyclonal antibodies against EGLN isoforms were purchased from Novus Biologicals (Littleton, CO), and anti-VHL monoclonal antibody was from BD Biosciences Pharmingen (San Jose, CA).
HIF-1α transcriptional activity
Transient transfection of pGal4-luc and pEpoE-luc reporter plasmids and luciferase assays were essentially as described previously (18,42). In brief, 0.25 μg reporter plasmid and 0.1 μg of each effecter plasmid were used for each well in a 12-well plate with FuGENE 6 (Roche, Indianapolis, IN). 0.1 μg of pEYFP-Nuc expression vector (Clontech, Palo Alto, CA) was co-transfected for normalization of transfection efficiency. For reporter experiments involving siRNAs, cells were first transfected with the plasmid DNA at a confluency of ~30–40%. The next day, cells were fed with Opti-MEM (Invitrogen, Carlsbad, CA) and transfected with siRNAs as described above. Luciferase and EYFP were measured as described previously (43). The data are presented as means plus standard deviations from three independent experiments in duplicate.
Western blot analysis
Western blot was performed essentially as described previously (18,43). The effect of EGLN1 on HIF-1α C-ODD protein level was examined after transfection of Hep3B and C2 cells respectively with 1 μg Gal4-HIF-1α (498–603) and 0.2 μg EGLN1. pEYFP-Nuc (0.05 μg) was also included as an internal control. Cells were treated in hypoxia for 4 h before harvest.
Quantitative Real-time RT-PCR
RNA was extracted with Trizol (Invitrogen, Carlsbad, CA). Total RNA (500 ng) was reverse-transcribed with the Taqman Reverse Transcription Reagents (Applied Biosystems, Branchburg, NJ) and 1 μg of cDNA was then amplified using the Taqman Universal PCR Master Mix (Applied Biosystems). The PGK1 probe and primers were designed using ABI Primer Express version 2.0 software: probe, FAM5′-ATTTATCTAATTGTCCCATCTCTCCACTGCTGCT-MGBNFQ-3′; forward primer, 5′-TCTTGAGGAACGGATCAGATGTC-3′; and reverse primer, 5′-AGTAGGCCCTTGATAAAGAATGGA-3′. Human ACTB (Taqman primers and probe, ABI 7700) was used as an endogenous control (VIC). Gene-specific PCR products were measured continuously by an ABI PRISM 7700 Sequence Detection System (Applied Biosystems) during 40 cycles. The difference in threshold number of cycles between PGK1 and ACTB for each sample was then expressed relative to the normoxic mock-transfected samples and converted into fold difference. All experiments were repeated three times in triplicates, and representative results were presented as means plus/minus standard errors.
RESULTS
EGLN1 binds HIF-1α irrespective of oxygen tension
HIF-1α possesses an oxygen-dependent degradation domain (ODD, amino acids 401–603) (21) that can be divided into N-terminal and C-terminal fragments (43). Previously, we demonstrated that in HEK293 cells EGLN1 co-precipitated with the C-terminal ODD (C-ODD, amino acids 498–603) after treatment with the proteasome inhibitor, Cbz-LLL (44) (Fig. 1A). In contrast, no co-precipitation could be detected when Leu-574, a molecular determinant of Pro-564 hydroxylation (44), was mutated (Fig. 1B). Moreover, no such interaction was shown when EGLN2 or EGLN3 was tested (44). Interestingly, in this study, we observed that EGLN1 also bound C-ODD during a 4-h hypoxic treatment (Fig. 1C). A similar result was obtained when cells were treated with desferrioxamine, a hypoxia mimetic (Fig. 1D). To rule out the possibility that the detected EGLN1 binding was a remnant of hydroxylated HIF-1α from normoxia, we increased the hypoxic treatment to 8 h, given the turnover rate of HIF-1α in hypoxia being less than 2 h (21). Again, the binding of EGLN1 to C-ODD was readily detectable from cells with the prolonged hypoxic treatment (data not shown). Together, these findings indicate that EGLN1 interacts with HIF-1α irrespective of oxygen tension. EGLN1 is presumably inactive in hypoxia because of the strict requirement of oxygen for its enzymatic activity. This observation prompted us to search for additional functions of EGLN1.
Fig. 1.
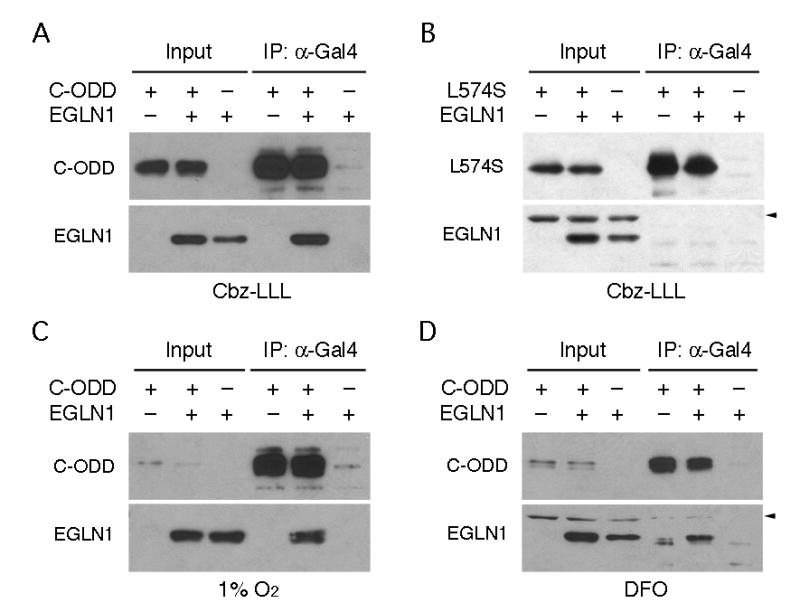
EGLN1 interacts with HIF-1α C-ODD under hypoxic conditions. Gal4-C-ODD (C-ODD) and its L574S mutant were expressed, as indicated, in HEK293 cells in the absence or presence of ectopic EGLN1. After 4-h treatment with Cbz-LLL, hypoxia (1% O2), or desferrioxamine (DFO), C-ODD was immunoprecipitated with anti-Gal4 antibody (IP: α;-Gal4), followed by Western blotting with anti-Gal4 and anti-EGLN1 antibodies. One tenth of cell extract was subjected to Western blot directly (Input) to ensure the level of ectopic expression. Arrowhead denotes nonspecific detection.
EGLN1 inhibits HIF-1α N-terminal transcriptional activity
In addition to the CAD, C-ODD also possesses transcriptional activity (19,43,45), referred to here as N-terminal transcriptional activity. To examine whether EGLN1 modulates HIF-1α transcriptional activities, we tested effects of transient expression of EGLN1 in Hep3B cells in a Gal4 reporter system (42,43). Results in Fig. 2A show that EGLN1 markedly inhibited the transcriptional activity of C-ODD, but not of CAD. Even under hypoxic conditions, there was >50% decrease of the N-terminal transcriptional activity. To further test the specificity of such inhibition, we took advantage of mutations (P564A and L574S) that disrupt EGLN1 binding. As expected, each mutation gave rise to an increased reporter activity and a loss of hypoxic induction. Of note, the elevated transcriptional activity of each mutant was apparently correlated well with its increased protein levels (data not shown). However, EGLN1 had no significant effects on these mutants in hypoxia (Fig. 2A). Similar results were obtained in U-2 OS cells (Fig. 2B). In addition, overexpression of EGLN2 and EGLN3 also inhibited the N-terminal transcriptional activity (data not shown).
Fig. 2.
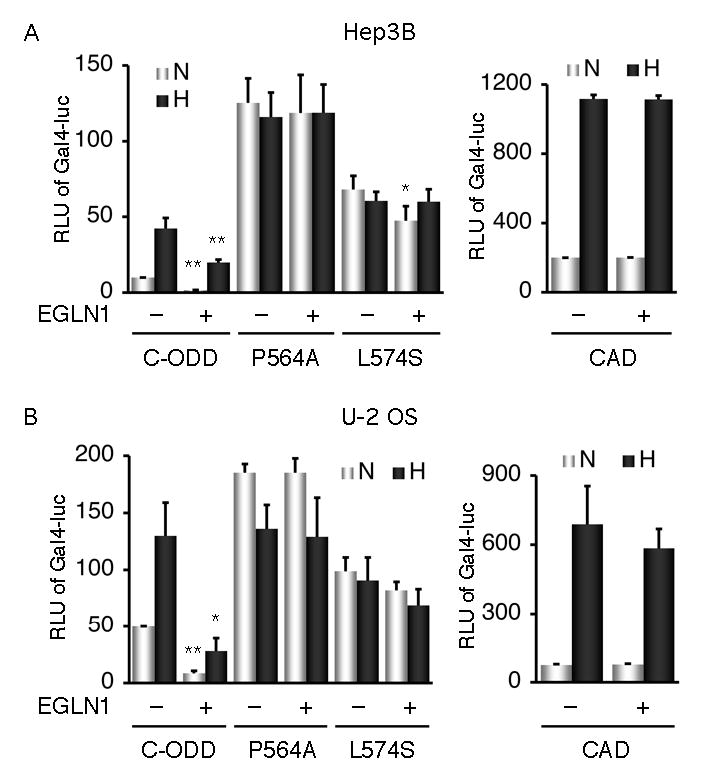
EGLN1 specifically inhibits HIF-1α N-terminal transcriptional activity. A, and B, The effects of EGLN1 on HIF-1α transcriptional activities of C-ODD and its mutants as indicated, and of CAD were tested in Hep3B and U-2 OS cells in a Gal4 reporter system. The cells grown in normoxic (N) or hypoxic (H) conditions were lysed and assayed for their luciferase activities. Relative luciferase units (RLU) of Gal4-luc in y-axis were plotted as means ± SD from three independent experiments in duplicate. *p < 0.01, **p < 0.001 significant difference in reference to the transfection controls.
EGLN1 inhibition of HIF-1α transcriptional activity is independent of the VHL degradation pathway
As mentioned above, HIF-1α hydroxylation leads to VHL mediated polyubiquitination and proteasomal degradation. It is possible therefore that the EGLN1 repression of HIF-1α transcriptional activity in hypoxia is a result of increased HIF-1α proteolysis despite the scarce hydroxylase activity. To test this possibility, we used the RCC cell line C2 (46) that lacks a functional VHL protein for HIF-1α proteolysis. However, the N-terminal transcriptional activity was still inhibited significantly by EGLN1 irrespective of oxygen tension (Fig. 3A). Of note, EGLN1 bound C-ODD in both normoxic and hypoxic C2 cells (Fig. 3B) as in HEK293 cells. Again, the transcriptional activities of the P564A and the L574S mutant was unaffected by EGLN1 overexpression. These results suggest a novel function of EGLN1 for inhibiting HIF-1α transcriptional activity.
Fig. 3.
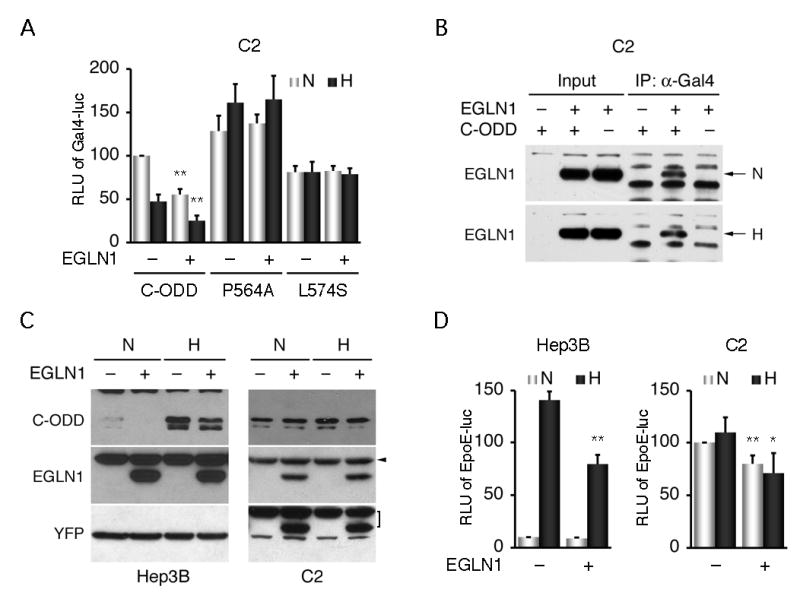
EGLN1 inhibition of HIF-1α transcriptional activity is not a result of increased HIF-1α proteolysis. A, EGLN1 inhibition of HIF-1α transcriptional activity was tested as above with C-ODD and its mutants as indicated, in the VHL-deficient C2 (C2) cell line. B, EGLN1 binding to C-ODD was examined in normoxic and hypoxic C2 cells expressed ectopically with EGLN1 and Gal4-C-ODD, as described in Fig. 1. Arrows denote detected protein complexes. C, The effect of ectopic EGLN1 on HIF-1α C-ODD expression levels was analyzed in VHL-proficient (Hep3B) and deficient (C2) cell lines respectively. After 4-h hypoxic treatment, Western blot was performed with anti-Gal4 and anti-EGLN1 antibodies. Co-transfected yellow fluorescence protein (YFP) served as an internal control. Arrowhead denotes nonspecific detection, and bracket specifies remaining signals from the blot above. D, Hep3B and C2 cell lines were transfected with the pEpoE-luc reporter in the absence or presence of ectopic EGLN1. EGLN1 overexpression inhibited endogenous HIF-1α transcriptional activity under hypoxic conditions. Relative luciferase units (RLU) of EpoE-luc in y-axis were plotted as means ± SD from three independent experiments in duplicate. *p < 0.01, **p < 0.001.
To provide further evidence, we determined C-ODD protein levels in cells co-expressed with EGLN1 by Western analysis. In Hep3B, ectopic expression of EGLN1 led to a >20% reduction of the protein levels in hypoxic conditions (Fig. 3C; also see Fig. 1). However, EGLN1 overexpression failed to do so in the VHL-deficient C2 cells. Therefore, these results support that EGLN1, in addition to promoting HIF-1α proteolysis, also independently represses the N-terminal transcriptional activity.
We also examined the effect of EGLN1 on endogenous HIF-1α mediated transcription with a reporter plasmid driven by the human EPO hypoxia-responsive element (18). EGLN1 over-expression significantly inhibited hypoxia-induced EPO reporter activity in Hep3B and C2 cells (Fig. 3D). Thus, these results indicate the biological relevance of EGLN1 as a repressor for HIF-1α mediated transcription.
EGLN1 silencing enhances HIF-1α transcriptional activity and its target gene expression in hypoxia
To corroborate the role of EGLN1 in inhibiting HIF-1α transcriptional activity, we took a complementary approach where EGLN1 expression was silenced by small interfering RNA (siRNA). Knockdown of EGLN1 in Hep3B cells resulted in a significant increase in HIF-1α N-terminal transcriptional activity under normoxic and hypoxic conditions (Fig. 4A). However, siRNA targeting EGLN2 or EGLN3 showed no effect, which differs from the EGLN2 and EGLN3 overexpression results above (see Discussion).
Fig. 4.
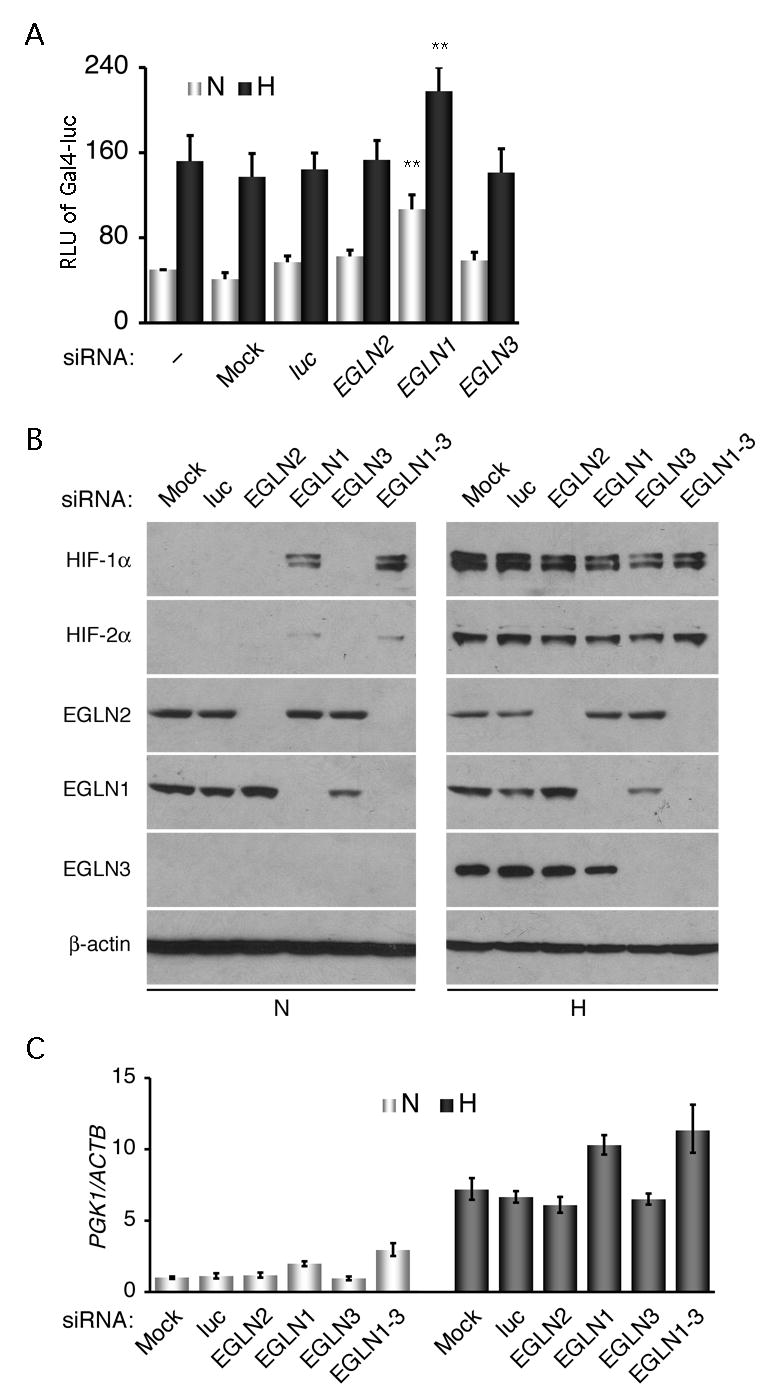
Knockdown of EGLN1 expression enhances HIF-1α N-terminal transcriptional activity in hypoxia. A, Hep3B cells were transfected with C-ODD for analyzing the effect of EGLN1 siRNA on the N-terminal transcriptional activity. Mock siRNA transfection, together with siRNAs targeting the respective firefly luciferase gene (luc), EGLN2, and EGLN3, served as controls. Relative luciferase units (RLU) of Gal4-luc in y-axis were plotted as means ± SD from three independent experiments in duplicate. **p < 0.001. B, EGLN isoforms in Hep3B cells were knocked down with their corresponding siRNA as indicated. EGLN1-3 denotes a mix of siRNAs targeting all three EGLN isoforms. Various antibodies as specified were used sequentially for Western blot analysis. Endogenous β-actin was used as a loading control. Note that HIF-1α protein expression was increased in normoxic conditions (N) by EGLN1 silencing. However, none of the siRNAs further elevated HIF-1α protein levels in hypoxic conditions (H). C, Effects of EGLN1 siRNA were examined on the HIF-1α target gene PGK1 expression. Hep3B cells were transfected with the corresponding siRNAs as above, and subjected to analysis by real-time RT-PCR after a hypoxic treatment. PGK1 mRNA levels in reference to those of endogenous ACTB are presented in means ± SE.
To confirm the effectiveness of the siRNA, we determined protein levels of targeted genes by Western blot analysis. The extent and specificity of siRNA knockdown are illustrated in Fig. 4B. The protein level of each EGLN was strikingly and specifically reduced by its corresponding siRNA; neither control siRNA nor those against its isoforms had any significant effect. Importantly, knockdown of EGLN1 expression rendered HIF-1α stable in normoxia, whereas knockdown of the other two showed no obvious effect, consistent with that EGLN1, generally most abundant (47), is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia (40). Likewise, simultaneous knockdown of EGLN1, EGLN2, and EGLN3 gave rise to similar results as the knockdown of EGLN1 alone.
To demonstrate that EGLN1 inhibits HIF-1α target gene expression in hypoxia, we asked whether EGLN1 siRNA alters hypoxic induction of PGK1, a hypoxia-responsive gene. Results in Fig. 4C from real-time RT-PCR show that only EGLN1 siRNA further elevated PGK1 mRNA levels in hypoxia. This result supports that EGLN1 modulates the HIF-1α target gene expression under hypoxic conditions, even though its enzymatic activity is inactivated by limited oxygen. It is noted that although the relative abundance of the EGLN isoforms and the efficiency of corresponding siRNAs may vary in the cells, neither EGLN2 nor EGLN3 siRNA altered PGK1 expression under these conditions. Taken together, these results argue that only EGLN1 inhibits HIF-1α transcriptional activity in hypoxia by virtue of binding to HIF-1α.
Stimulation of HIF-1α transcriptional activity and enhancement of its target gene expression by EGLN1 siRNA is VHL-independent
To demonstrate further that the increased expression of PGK1 in hypoxia by EGLN1 siRNA is not simply a result of increased HIF-1α stability, we asked whether knockdown of VHL expression in Hep3B cells would produce a similar result. Fig. 5A shows that the VHL siRNA effectively down-regulated VHL protein levels, in concomitance with an increased level of HIF-1α. However, in contrast to the augmentation by EGLN1 siRNA, VHL silencing failed to further enhance PGK1 mRNA levels in hypoxia, despite a marked increase in normoxia (Fig. 5B). Of note, EGLN1 expression was essentially unaffected by the VHL siRNA (Fig. 5A).
Fig. 5.
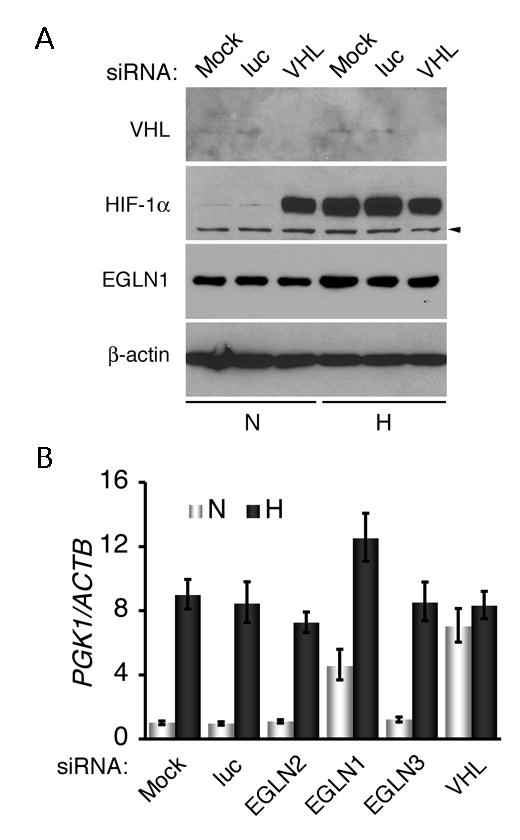
Knockdown of VHL expression fails to enhance HIF-1α transcriptional activity in hypoxia. A, VHL siRNA was used in Hep3B cells to silence VHL expression. The loss of VHL expression was shown by Western analysis, in concomitance with the elevation of HIF-1α levels in normoxia. EGLN1 levels were also determined. Endogenous β-actin was used as a loading control. Arrowhead denotes nonspecific detection. B, PGK1 expression in Hep3B cells was analyzed by real-time RT-PCR after transfection with siRNAs as indicated. PGK1 mRNA levels in reference to those of endogenous ACTB are presented in means ± SE.
Likewise, in VHL-deficient C2 cells only EGLN1 siRNA was able to stimulate HIF-1α N-terminal transcriptional activity (Fig. 6A), and to further elevated PGK1 expression under both normoxic and hypoxic conditions (Fig. 6B) without an additional increase in HIF-1α protein levels (Fig. 6C). Again, siRNA against EGLN2 or EGLN3 had no effects on the transcriptional activity and PGK1 expression. These results confirm that EGLN1 specifically represses HIF-1α transcriptional activity in hypoxia.
Fig. 6.
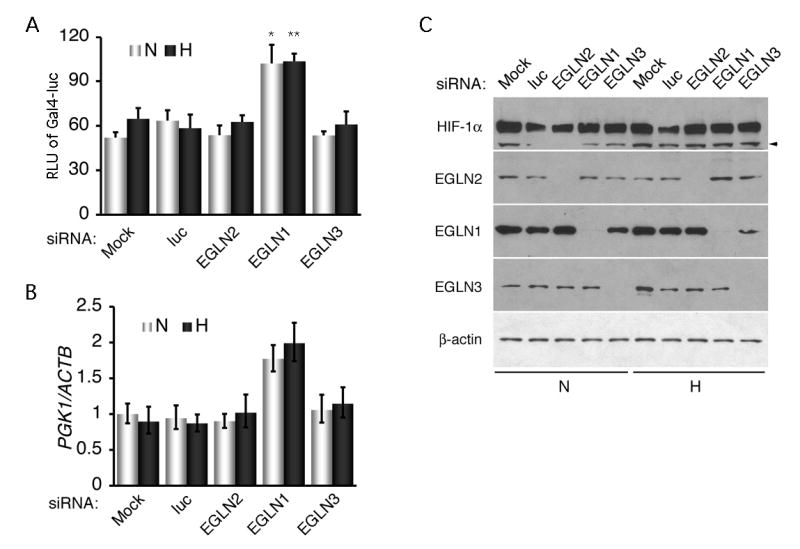
Stimulation of HIF-1α transcriptional activity and augmentation of PGK1 expression by EGLN1 siRNA in VHL-deficient cells. A, C2 cells were transfected with C-ODD to analyze the effect of EGLN1 siRNA on HIF-1α N-terminal transcriptional activity. Mock siRNA transfection, together with siRNAs targeting the respective firefly luciferase gene (luc), EGLN2, and EGLN3, served as controls. Relative luciferase units (RLU) of Gal4-luc in y-axis were plotted as means ± SD from three independent experiments in duplicate. *p < 0.01, **p < 0.001. B, PGK1 mRNA levels in C2 cells were determined by real-time RT-PCR after transfection with the indicated siRNAs. PGK1 mRNA levels in reference to those of endogenous ACTB are presented in means ± SE. C, Western blot analyses were performed to examine the specific effect of each siRNA as indicated. Endogenous β-actin was used as a loading control. None of the siRNAs increased HIF-1α protein levels under either normoxic or hypoxic conditions. Arrowhead denotes nonspecific detection.
DISCUSSION
The known function of the HIF prolyl-4-hydroxylases is to sense and transduce oxygen signals via prolyl hydroxylation of HIF-1α, thereby triggering VHL binding, polyubiquitination, and proteasomal degradation (31,32). The observation that EGLN1 and EGLN3 themselves are hypoxia-inducible (31) has prompted investigations of the biological relevance of the increased hydroxylase levels. It has been shown that the rate of HIF-1α proteolysis, upon re-oxygenation, depends on the duration of hypoxic stress; after prolonged hypoxia, accelerated HIF-1α degradation supervenes (48). Therefore, the hypoxic up-regulation of EGLN1 and EGLN3 apparently explains the buildup of the prolyl hydroxylases during hypoxia for rapid degradation of HIF-1α upon re-oxygenation (40). However, the biological role for the accumulated hydroxylases during prolonged hypoxia remains unclear. It is conceivable that a negative-feedback mechanism would take place by modulating HIF-1α transcriptional activities. In fact, it has been demonstrated that HIF-1α CAD activity is inhibited during hypoxia by CITED2/p35srj, a hypoxia-inducible gene product that competes for p300 binding (15). In contrast, the regulation of the HIF-1α N-terminal transcriptional activity remains obscure. Here we demonstrate that EGLN1 binds HIF-1α in hypoxia as well. Apart from its role in regulating HIF-1α protein levels, EGLN1 also modulates HIF-1α N-terminal transcriptional activity as a feedback mechanism to avoid excessive hypoxic response.
During this study, the possibility of altered HIF-1α stability in hypoxia resulting from the manipulation of EGLN1 expression was always a concern. Although the increased protein degradation overshadowed the direct transcriptional repression by EGLN1, the effect of transcriptional repression was uncovered, nevertheless, with the use of VHL-deficient cells defective in HIF-1α degradation. Moreover, EGLN1 inhibited endogenous HIF-1α transcriptional activity. EGLN1 silencing specifically enhanced HIF-1α transcriptional activity and PGK1 expression, especially under hypoxic conditions without further increase in HIF-1α levels. Furthermore, EGLN1 silencing in VHL-deficient cells confirmed the inhibitory effect of EGLN1 on HIF-1α transcriptional activity. In addition, the lack of enhanced PGK1 expression, when VHL expression was knocked down, further support that EGLN1 mediated HIF-1α degradation in normoxia and its inhibition of HIF-1α transcriptional activity in hypoxia are two distinct functions. Accordingly, we propose that both EGLN1 and CITED2 fine-tune HIF-1α transcriptional activities under hypoxic conditions.
It is noteworthy that forced expression of the HIF prolyl hydroxylases in cells often down-regulates HIF-1α, as shown in this study as well as in a previous report (32). Obviously, results from such approach often ‘violate’ in vivo rules that govern the specificity of intracellular biochemical reactions. Therefore, the data should be interpreted with extreme caution. Although overexpression of the three hydroxylases all inhibited HIF-1α N-terminal transcriptional activity in our initial reporter assays, only EGLN1 silencing augmented HIF-1α transcriptional activity and PKG1 expression. These findings are consistent with our co-immunoprecipitation result that only EGLN1, but not EGLN2 and EGLN3, interacts with HIF-1α C-ODD (44). Thus, alternative approaches to gene overexpression are necessary to avoid potential pitfalls.
While this manuscript was in preparation, Baek et al. (49) reported that the protein OS-9 interacts with both HIF-1α and EGLN1 in normoxic and hypoxic conditions. OS-9 promotes HIF-1α hydroxylation, VHL binding, and proteasomal degradation. In addition, it inhibits HIF-1 mediated transcription. Apparently, OS-9 is not involved in EGLN1 inhibition of HIF-1α transcriptional activity, because HIF-1α stability was not altered in this study. Nevertheless, further studies are warranted to identify the molecular mechanism(s) by which EGLN1 inhibits HIF-1 mediated transcription. Our current data seemingly suggest that the EGLN1 hydroxylase activity per se is not necessarily required for inhibiting HIF-1α transcriptional activity, and therefore it would be interesting to search for additional factors that interact with EGLN1 for mechanistic dissections. Very recently, Ozer et al. reported that EGLN1, in addition to regulating HIF stability, modulates HIF function through the recruitment of the candidate tumor suppressor ING4 (50).
Targeting the HIF prolyl hydroxylases is regarded as a potential therapeutic principle for manipulating HIF activity (5,51). The differential effects on the HIF-1α target gene expression by the respective silencing of EGLN1 and VHL also suggest that even though inhibiting EGLN1 activity alone may give rise to a marked increase in HIF-1α levels, the resulting HIF-1α transcriptional activity may depend on the intracellular oxygen tension, i.e. hypoxia specifically augments HIF-1α target gene expression. By contrast, inhibiting VHL activity would render HIF-1α stable and constitutively active regardless of oxygen tension. Therefore, understanding the regulation of HIF-1α transcriptional activity by the prolyl hydroxylase may provide new insight into the pharmacological manipulation of the HIF prolyl hydroxylase activities for the treatment of ischemic/hypoxic disease.
Footnotes
We wish to thank Richard Bruick for the expression vectors encoding the HIF prolyl hydroxylases, Len Neckers for the VHL-deficient cells, and Dorothea Dudek for the editorial assistance. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. L.E.H. is an NCI Scholar of the National Institutes of Health.
The abbreviations used are: HIF-1α, hypoxia-induced factor 1α; ODD, oxygen-dependent degradation domain; C-ODD, C-terminal ODD; VHL, von Hippel-Lindau; CAD, C-terminal activation domain.
References
- 1.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunn HF, Poyton RO. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 4.Wenger RH. Faseb J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- 5.Giaccia A, Siim BG, Johnson RS. Nat Rev Drug Discov. 2003;2:803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- 6.Huang LE, Bunn HF. J Biol Chem. 2003;278:19575–19578. doi: 10.1074/jbc.R200030200. [DOI] [PubMed] [Google Scholar]
- 7.Pugh CW, Ratcliffe PJ. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 8.Poellinger L, Johnson RS. Curr Opin Genet Dev. 2004;14:81–85. doi: 10.1016/j.gde.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 10.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. Embo J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koshiji M, Huang LE. Cell Cycle. 2004;3:853–854. doi: 10.4161/cc.3.7.990. [DOI] [PubMed] [Google Scholar]
- 12.Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE. Mol Cell. 2005;17:793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Wang GL, Semenza GL. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 14.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. Proc Natl Acad Sci U S A. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Embo J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang GL, Semenza GL. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- 18.Huang LE, Arany Z, Livingston DM, Bunn HF. J Biol Chem. 1996;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 19.Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. J Biol Chem. 1997;272:11205–11214. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- 20.Salceda S, Caro J. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 21.Huang LE, Gu J, Schau M, Bunn HF. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 23.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 24.Yu F, White SB, Zhao Q, Lee FS. Proc Natl Acad Sci U S A. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 26.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 27.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 29.Tanimoto K, Makino Y, Pereira T, Poellinger L. Embo J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor MS. Gene. 2001;275:125–132. doi: 10.1016/s0378-1119(01)00633-3. [DOI] [PubMed] [Google Scholar]
- 31.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 32.Bruick RK, McKnight SL. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 33.Mahon PC, Hirota K, Semenza GL. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 35.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 37.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 38.Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Burgel T, Jelkmann W, Acker H, Fandrey J. J Cell Sci. 2003;116:1319–1326. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- 39.del Peso L, Castellanos MC, Temes E, Martin-Puig S, Cuevas Y, Olmos G, Landazuri MO. J Biol Chem. 2003;278:48690–48695. doi: 10.1074/jbc.M308862200. [DOI] [PubMed] [Google Scholar]
- 40.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. Embo J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elbashir SM, Harborth J, Weber K, Tuschl T. Methods. 2002;26:199–213. doi: 10.1016/S1046-2023(02)00023-3. [DOI] [PubMed] [Google Scholar]
- 42.Gu J, Milligan J, Huang LE. J Biol Chem. 2001;276:3550–3554. doi: 10.1074/jbc.M009522200. [DOI] [PubMed] [Google Scholar]
- 43.Huang LE, Pete EA, Schau M, Milligan J, Gu J. J Biol Chem. 2002;277:41750–41755. doi: 10.1074/jbc.M207280200. [DOI] [PubMed] [Google Scholar]
- 44.Kageyama Y, Koshiji M, To KK, Tian YM, Ratcliffe PJ, Huang LE. Faseb J. 2004;18:1028–1030. doi: 10.1096/fj.03-1233fje. [DOI] [PubMed] [Google Scholar]
- 45.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 46.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. J Biol Chem. 2002;277:29936–29944. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 47.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 48.Berra E, Richard DE, Gothie E, Pouyssegur J. FEBS Lett. 2001;491:85–90. doi: 10.1016/s0014-5793(01)02159-7. [DOI] [PubMed] [Google Scholar]
- 49.Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M, Chan DA, Giaccia AJ, Semenza GL. Mol Cell. 2005;17:503–512. doi: 10.1016/j.molcel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 50.Ozer A, Wu LC, Bruick RK. Proc Natl Acad Sci U S A. 2005;102:7481–7486. doi: 10.1073/pnas.0502716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hewitson KS, Schofield CJ. Drug Discov Today. 2004;9:704–711. doi: 10.1016/S1359-6446(04)03202-7. [DOI] [PubMed] [Google Scholar]