

Abstract
In evaluating potential indicators of biotin status, we quantitated the expression of biotin-related genes in leukocytes from human blood of normal subjects before and after inducing marginal biotin deficiency. Biotin deficiency was induced experimentally by feeding an egg-white diet for 28 d. Gene expression was quantitated for the following biotin-related proteins: methylcrotonyl-CoA carboxylase chains A (MCCA) and B (MCCB); propionyl-CoA carboxylase chains A (PCCA) and B (PCCB); pyruvate carboxylase (PC); acetyl-CoA carboxylase isoforms A (ACCA) and B (ACCB); holocarboxylase synthetase (HCS); biotinidase; and 2 potential biotin transporters: sodium-dependent multivitamin transporter (SMVT) and solute carrier family 19 member 3 (SLC19A3). For 7 subjects who successfully completed the study, the abundance of the specific mRNAs was determined by quantitative real-time RT-PCR at d 0 and 28. At d 28, SLC19A3 expression had decreased to 33% of d 0 (P < 0.02 by two-tailed, paired t test). Expression of MCCA, PCCA, PC, ACCA, ACCB, HCS, biotinidase, and SMVT decreased to ~80% of d 0 (P < 0.05). Expression of the MCCB and PCCB chains that do not carry the biotin-binding motif did not change significantly; we speculate that expression of the biotin-binding chains of biotin-dependent carboxylases is more responsive to biotin status changes. These data provide evidence that expression of SLC19A3 is a relatively sensitive indicator of marginal biotin deficiency.
Keywords: leukocytes, blood, humans, biotin, gene expression
Biotin deficiency is teratogenic in mice (1,2) and may be teratogenic in humans (3). Valid indicators of marginal and moderate biotin deficiency would be useful in investigating the role of biotin deficiency in birth defects and in other illnesses hypothesized to be biotin related (4 –7). Emerging evidence indicates that biotin plays a role in gene expression (8 –12). In addition to acting as a cofactor for biotin-dependent carboxylases, biotin stimulates expression of hepatic glucokinase (8) and represses expression of hepatic phosphoenolpyruvate carboxylase (9) in vivo, and expression of the biotin-related enzymes propionyl-CoA carboxylase chain A (PCCA),4 acetyl-CoA carboxylase isoform A (ACCA), and holocarboxylase synthetase (HCS) in cultured human hepatoblastoma cells and normal fibroblasts (10). However, no such studies have been performed in humans in vivo. In this study, we examined the expression of specific biotin-related genes as indicators of marginal, asymptomatic biotin deficiency and assessed gene response to marginal biotin deficiency.
In mammals, biotin is a coenzyme for 5 biotin-dependent carboxylases: methylcrotonyl-CoA carboxylase (MCC), propionyl-CoA carboxylase (PCC), pyruvate carboxylase (PC), and the 2 isoforms of ACC (ACCA and ACCB). The active forms of the enzymes (holocarboxylases) contain biotin covalently bound to lysine residues; the attachment of biotin to the corresponding apocarboxylase is catalyzed by HCS.
Biotin is transported into eukaryotic cells by biotin transporters located in cell membranes. Three biotin transporters have been proposed in human cells: 1) the sodium-dependent multivitamin transporter (SMVT) (13,14); 2) the solute carrier family 19 member 3 (SLC19A3) (15,16); and 3) the monocarboxylate transporter R1 (17). This third transporter was proposed after this study was initiated and was not examined here.
Biotinidase catalyzes the release of covalently bound biotin from biotinyl-peptides generated by the turnover of intracellular proteins and releases biotin from dietary proteins during digestion (18). Biotinidase is also likely important in catalyzing the covalent binding of biotin to histones (19).
In this study, we evaluated expression of biotin-related genes as potential indicators of marginal, asymptomatic biotin deficiency. Gene expression was quantitated in leukocytes of 7 healthy humans after 28 d of progressive biotin deficiency.
SUBJECTS AND METHODS
Subjects and diet
This study was approved by the University of Arkansas for Medical Sciences Human Research Advisory Committee. Informed consent was obtained at enrollment. Subjects were selected as described previously (20,21). Initially, 10 healthy adult volunteers (4 women) were enrolled in the study. Two subjects voluntarily withdrew from the study at d 2 due to difficulties in complying with the diet and housing protocols.
Eight healthy adult volunteers (3 women) completed the study. All subjects consumed a multivitamin supplement without biotin for the duration of this study (d −21 to d +65). The supplement contained 46 μmol pantothenic acid, 341 μmol vitamin C, 5 μmol thiamin, 162 μmol niacin, 0.91 μmol folate, 4.5 μmol riboflavin, 10 μmol vitamin B-6, 4.4 nmol vitamin B-12, 7.5 μmol vitamin A (5000 IU), 26 nmol vitamin D (400 IU), 70 μmol vitamin E (30 IU), plus 11 mmol calcium, 483 μmol iron, and 229 μmol zinc.
As with our previous studies (21), to prevent inadvertent study of a subject with unsuspected marginal biotin deficiency or biotin excess, a biotin “loading and washout” phase was instituted at d –21. To avoid biotin excess, the amount of biotin in the loading phase was 123 nmol (30 μg), the recommended daily adequate intake. On d –1, subjects were admitted to the General Clinical Research Center (GCRC) and resided there through d 28. The GCRC study regimen and biotin-deficient diet provided the subjects were as previously described (21).
The egg-white diet provided avidin sufficient to bind ~7 times the analyzed dietary biotin intake as described previously (20). Dietary intake and egg-white drink consumption were monitored daily by a dietitian. Despite this rigorous control, one subject admitted non-compliance with the dietary regimen. The data of that subject were excluded.
At 0730 h on d 0 and 28, after overnight fasting, blood was collected in PAXgene blood RNA tubes (PreAnalytiX) for the gene expression study and in heparinized vaccutainer tubes (Becton Dickinson) for determination of lymphocyte PCC activity. Each subject collected urine for 24 h from 0600 of one day to 0600 h of the next day beginning on d –1 and again on d 27; these collections are referred to as d 0 and d 28, respectively. Urine was analyzed for quantitation of biotin and 3-hydroxyisovaleric acid (3HIA). Records of body weight and urine volumes were maintained by the GCRC nurses.
RNA isolation
Total RNA was isolated from whole-blood samples (collected in PAXgene blood RNA tubes) using the PAX-geneTM Blood RNA kit (PreAnalytiX) according to the manufacture’s protocol. These tubes contained a proprietary blend of reagents that protect RNA from degradation by RNases and preserve the in vivo RNA expression profile by preventing ex vivo induction of gene expression during and after blood drawing. The term “leukocytes” is used here although total RNA was isolated from whole blood. Our tacit assumption is that the contributions of mRNA from erythrocytes and platelets is negligible. Residual DNA was removed by an additional on-column DNase digestion with RNase-free DNase (Qiagen). The purity of isolated RNA with respect to protein contamination was assessed by the ratio of optical absorbance values at 260 –280 nm; values ranged from 1.9 to 2.1. The integrity of the purified RNAs was assessed by denaturing agarose gel electrophoresis and ethidium bromide staining. All 18s and 28s ribosomal RNA appeared as sharp bands on the stained glyoxal-agarose gels; the 28s rRNA bands were present at an intensity at least twice that of the corresponding 18s RNA bands, indicating isolation of intact RNA.
Quantitation of gene expression by real-time PCR
Gene expression was assessed by quantitation of cDNA converted from the messenger RNA corresponding to each gene using a two-step RT-PCR gene expression method. In the first step, synthesis of total cDNA from total RNA was performed using the SYBR Green RT-PCR Reagent Kit (Applied Biosystems) according to the manufacture’s protocol. In this step, random hexamer primers were used to generate cDNA from total RNA using MultiScribe Reverse Transcriptase. In the second step, the abundance of the specific cDNAs was determined by real-time PCR using gene-specific primers (Table 1) and SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacture’s protocol. Primer Express software was used for primer design (Applied Biosystems). The primer pairs for most of the genes spanned intron/exon boundaries. To control for DNA contamination, negative RT controls were performed for all primer pairs; no products were detected. PCR (40 cycles) was performed in optical 96-well reaction plates using the following temperatures and times per cycle: 95°C for 15 s and 60°C for 1 min; the first PCR cycle was preceded by denaturation at 95°C for 10 min. For each gene, PCR for each sample of total cDNA was performed in the same plate. PCR products were detected by the ABI Prism 7700 Sequence Detection System (Applied Biosystems). Nonspecific amplification was assessed by dissociation curve analysis; none was detected.
TABLE 1.
Real-time PCR primers for biotin-related genes
| Gene | Accession # | Primer sequence | Primer site |
|---|---|---|---|
| PCCA | BC000140 | F 5′-CAGTGATGTTGATGCTAGTTCTGTTC-3′ | 269 |
| R 5′-GGGAGCTGGGCCAACAC-3′ | 338 | ||
| PCCB | S67325 | F 5′-GGGCCAACGCCGTATTG-3′ | 180 |
| R 5′-ATCCTCTCCCTGGCTGTTAGC-3′ | 239 | ||
| MCCA | BC004214 | F 5′-CATGGGTGTGGAGGCAAAG-3′ | 233 |
| R 5′-TGCAATGAGGACCTTGGTAATG-3′ | 306 | ||
| MCCB | NM_022132 | F 5′-TGTACGCAAGCAGGGTACCAT-3′ | 795 |
| R 5′-TCCCCAGTTGCCGCTTTA-3′ | 857 | ||
| PC | NM_000920 | F 5′-GCGACTCTGTGAAACTCGCTAA-3′ | 1079 |
| R 5′-TGCCTGTCCACCAGGAACTC-3′ | 1151 | ||
| ACCA | NM_000664 | F 5′-CAGAGATGGTGGCTGATGTCA-3′ | 1346 |
| R 5′-GAGGAATCCCCATGGCAAT-3′ | 1408 | ||
| ACCB | U89344 | F 5′-GCCGCTGGCCATATTCG-3′ | 1617 |
| R 5′-CTACATAGCCCACGGTCTTG-3′ | 1684 | ||
| HCS | D23672 | F 5′-GTTTGCGTCTGCCGAGAAC-3′ | 834 |
| R 5′-GTCTCATCAGCAACACTCTCCAA-3′ | 908 | ||
| Biotinidase | U03274 | F 5′-ACCCAACTGCCTGGATGAAC-3′ | 838 |
| R 5′-CAAAGGCAACAGCAAAAGCTT-3′ | 912 | ||
| SMVT | AF069307 | F 5′-CACATCTTCATCCCCGTTTTCT-3′ | 734 |
| R 5′-TGAATCGAAGCTCCAGGTACTCA-3′ | 803 | ||
| SLC19A3 | AF271633 | F 5′-AGAGCAGAAACCCACATCAGAAAT-3′ | 730 |
| R 5′-GCTTGGTTTCAGGCTGTTCAG-3′ | 808 |
Expression of the following biotin-related genes was quantitated: MCCA, MCCB, PCCA, PCCB, PC, ACCA, ACCB, HCS, biotinidase, SMVT, and SLC19A3. 18s rRNA expression served as an endogenous control to account for minor variability in the initial amount of total RNA and for variability in the conversion efficiency of the reverse transcription reaction.
Assessment of biotin status
To assess the effectiveness of induction of biotin deficiency, we chose to quantitate urinary excretion of biotin and 3HIA and lymphocyte activity of PCC on d 0 and 28. Urinary excretion of biotin and 3HIA were reported previously (20,21) as reliable indicators of marginal biotin deficiency in 2 studies; ~90% of study subjects exhibited abnormally decreased urinary excretion of biotin and abnormally increased urinary excretion of 3HIA after 3 (20) or 4 (21) wk of consuming the egg-white diet. Increased excretion of 3HIA reflects decreased hepatic activity of the biotin-dependent enzyme MCC as verified by studies in mouse and rat models (1,22) and in individuals with inborn errors producing deficiency of MCC (23–25). Decreased activity of PCC in lymphocytes was shown to be a sensitive measure of biotin deficiency in animals (26) and humans (27).
Determination of urinary biotin and 3HIA
Urinary biotin was quantitated by HPLC separation followed by an avidin-binding assay performed as described previously (28). Urinary 3HIA was quantitated by GC-MS as described previously (20). Urine concentrations of biotin and 3HIA were assayed at least in triplicate for each subject at each time point.
Determination of lymphocyte PCC activity
PCC activity was measured by 14CO2 incorporation as described previously (27,29). Lymphocyte PCC activity was determined in triplicate for each subject at each time point.
Statistical analysis
For comparison of these study subjects to a larger, free-living population of normal adults, normal ranges of these indicators of biotin status were defined as follows. For urinary biotin excretion, the normal range was calculated from 19 normal subjects (21). The normal range for urinary 3HIA excretion was calculated from 17 normal subjects (21). Because existing lymphocyte PCC activity data in normal subjects are limited, the normal range for PCC activity was calculated by using values from 17 subjects from a combination of a previous study (27) and from d 0 of this study. Because these indicators were not normally distributed, the 10th and 90th percentiles for each variable were chosen as the lower and upper limits of the normal ranges. For statistical analysis, means of the replicate analytical data for biotin, 3HIA, and PCC activity for each subject were log transformed. Significances of differences between d 0 and 28 for these log-transformed means were tested by two-tailed, paired t test.
For each gene, 3 replicate RT-PCR determinations were performed for each subject at each time point; all data were normalized by 18s expression. Data of gene expression for the 7 subjects were not normally distributed. For the group of 7 subjects, the measure of central tendency for expression of each gene at d 0 and 28 was chosen to be the geometrical mean. The ratios of gene expression of d 28 to d 0 were calculated from the differences between the log-transformed data, which were then reverse transformed to obtain the ratios in terms of original RT-PCR measurements. Significance of the differences between the log-transformed data at d 28 and d 0 was tested by two-tailed, paired t test for each gene. The 95% CIs were determined for the log-transformed data and were reverse transformed to obtain the 95% CIs in terms of the original RT-PCR measurements. We evaluated the diagnostic sensitivity of expression data for each gene by determining the number of subjects whose values at d 28 fell outside the range of the d 0 values.
RESULTS
Initial biotin status of subjects
After 7 d of biotin loading and 14 d of washout, initial excretion of biotin was within the normal range for 5 of 7 subjects (Fig. 1A). Initial lymphocyte PCC activity was in the normal range in all subjects (Fig. 1B). Initial excretion of 3HIA was within the normal range for 5 of 7 subjects (Fig. 1C). For at least 2 of the 3 indicators of biotin status, 7 of 7 subjects were in the normal range.
FIGURE 1.
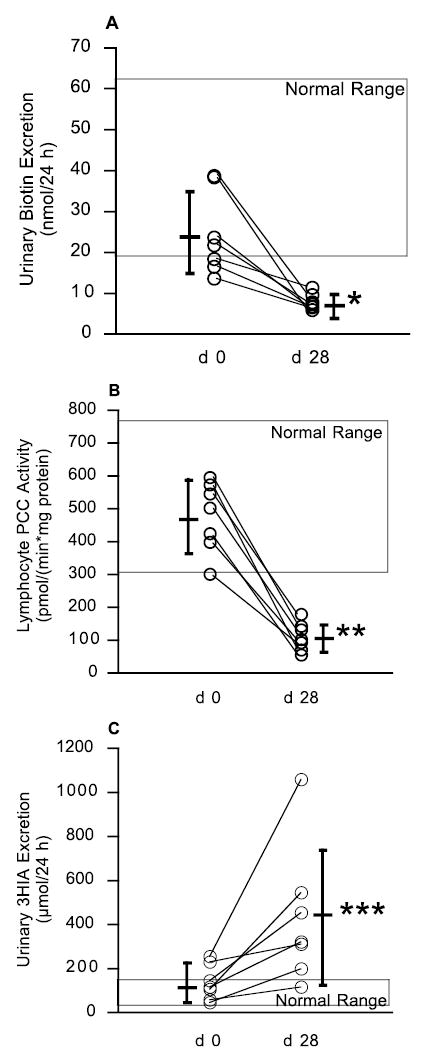
Evidence of biotin deficiency in humans as assessed by urinary biotin excretion (A), lymphocyte PCC (B), and urinary excretion of 3HIA (C). These data demonstrate that marginal biotin deficiency develops in subjects fed an egg-white diet. Individual subject data are shown as open circles; means are shown as horizontal bar with SD as vertical lines. Differences of d 28 from d 0 were tested by two-tailed, paired t test after log transformation of the corresponding original data. *P < 0.005, **P < 0.0001, ***P < 0.001.
Evidence of biotin deficiency in humans
On d 28, both mean urinary excretion of biotin and mean lymphocyte activity of PCC were strikingly decreased from d 0 (P < 0.005); in each subject, the d 28 values decreased from d 0 and were less than the lower limits of the corresponding normal ranges (Fig. 1A, B). On d 28, mean urinary excretion of 3HIA was increased from d 0 (P < 0.001); in each subject, the d 28 value was increased from d 0. The d 28 values were greater than the upper limit of the normal range for 6 of the 7 subjects (Fig. 1C). Urinary excretion of 3HIA for 1 subject remained within the normal range.
Effect of marginal biotin deficiency on expression of biotin-related genes
For each biotin-related gene, the abundance of specific mRNAs was determined in leukocytes of 7 healthy subjects before (d 0) and after (d 28) consuming the egg-white diet. Expression of SLC19A3 was decreased in 6 of 7 biotin-deficient subjects (Table 2); in 1 subject (# 4) expression of SLC19A3 was increased 1.7-fold. The mean of the d 28 expression for 7 subjects decreased to 33% of d 0 (P < 0.02) (Table 3). At d 28, values of SLC19A3 expression were less than the lower limit of the range of d 0 values for 5 of 7 subjects ( Fig. 2).
TABLE 2.
Expression of biotin-related genes in leukocytes of 7 subjects before and after consuming the egg-white diet12
| Relative gene expression
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject | Day | MCCA | MCCB | PCCA | PCCB | PC | ACCA | ACCB | HCS | Biotinidase | SMVT | SLC19A3 |
| 1 | 0 | 33 | 24 | 52 | 24 | 18 | 26 | 50 | 28 | 28 | 25 | 320 |
| 28 | 20 | 16 | 24 | 17 | 12 | 15 | 28 | 16 | 16 | 15 | 40 | |
| 2 | 0 | 22 | 20 | 25 | 20 | 14 | 19 | 40 | 17 | 22 | 20 | 152 |
| 28 | 17 | 16 | 18 | 17 | 12 | 16 | 34 | 14 | 17 | 16 | 33 | |
| 3 | 0 | 39 | 33 | 44 | 28 | 20 | 28 | 81 | 31 | 41 | 31 | 392 |
| 28 | 23 | 17 | 29 | 19 | 14 | 17 | 60 | 23 | 24 | 18 | 145 | |
| 4 | 0 | 18 | 15 | 20 | 15 | 14 | 15 | 38 | 11 | 18 | 17 | 112 |
| 28 | 17 | 14 | 17 | 15 | 12 | 13 | 41 | 11 | 16 | 12 | 188 | |
| 5 | 0 | 18 | 14 | 15 | 17 | 13 | 14 | 34 | 11 | 16 | 15 | 103 |
| 28 | 18 | 16 | 14 | 17 | 11 | 12 | 30 | 10 | 16 | 15 | 33 | |
| 6 | 0 | 29 | 26 | 25 | 23 | 16 | 21 | 60 | 20 | 26 | 26 | 435 |
| 28 | 25 | 22 | 24 | 24 | 14 | 19 | 48 | 19 | 24 | 25 | 72 | |
| 7 | 0 | 20 | 14 | 15 | 15 | 9 | 12 | 34 | 14 | 15 | 14 | 144 |
| 28 | 18 | 14 | 12 | 12 | 7 | 11 | 30 | 11 | 14 | 12 | 78 | |
Values are means, n = 3, of 18s-normalized RT-PCR data. The overall CV was 5.5 ± 5.9%; the CV for SLC19A3 was 17 ± 11%.
d 0: before consuming egg-white diet; d 28: after consuming egg-white diet.
TABLE 3.
Effect of biotin deficiency on expression of biotin-related genes in human leukocytes
| Gene | d 01 | d 281 | d 28/d 02 | P3 |
|---|---|---|---|---|
| MCCA | 25 | 20 | 0.80 (0.67,0.95) | 0.029 |
| MCCB | 20 | 16 | 0.82 (0.65,1.03) | 0.093 |
| PCCA | 25 | 19 | 0.76 (0.61,0.94) | 0.027 |
| PCCB | 20 | 17 | 0.86 (0.75,1.00) | 0.067 |
| PC | 14 | 12 | 0.82 (0.74,0.91) | 0.004 |
| ACCA | 18 | 14 | 0.78 (0.67,0.91) | 0.011 |
| ACCB | 46 | 38 | 0.82 (0.69,0.97) | 0.040 |
| HCS | 17 | 14 | 0.82 (0.69,0.97) | 0.038 |
| Biotinidase | 23 | 18 | 0.79 (0.65,0.96) | 0.037 |
| SMVT | 20 | 16 | 0.77 (0.64,0.93) | 0.019 |
| SLC19A3 | 203 | 68 | 0.33 (0.16,0.70) | 0.016 |
Values are the means of gene expression determined as geometrical averages of RT-PCR data for 7 subjects.
Values are ratios of gene expression of d 28 to d 0 and the corresponding 95% CI (shown in parentheses). To calculate the ratio, RT-PCR data were log transformed and the difference between d 28 and d 0 was determined for each of 7 subjects; the mean of 7 differences was then calculated for each gene and this mean was reverse transformed to obtain the ratio of gene expression. The corresponding 95% CI was determined in the log scale and its low and high values were then reverse transformed.
Significance of the differences between the log-transformed data at d 28 and d 0 was tested by two-tailed, paired t test; values represent P-values for each gene.
FIGURE 2.
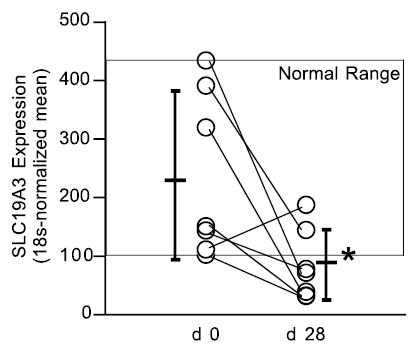
Effect of marginal biotin deficiency in humans on expression of SLC19A3. Subjects, symbols, and statistics as in Figure 1. *P < 0.02.
Marginal biotin deficiency also significantly decreased expression of MCCA, PCCA, PC, ACCA, ACCB, HCS, biotinidase, and SMVT, but the magnitudes of the changes were not large (Tables 2 and 3). At d 28, mean expression for these genes had decreased to ~80% of d 0 (P < 0.05; Table 3). Values for expression of these 8 genes at d 28 were less than the range of the d 0 values for only 2 of 7 subjects. Thus, diagnostic sensitivity was poor.
Marginal biotin deficiency did not alter the expression of MCCB or PCCB (Table 3), suggesting that expression of the biotin-binding chains of the biotin-dependent carboxylases may be more responsive to changes in biotin status than expression of the chains that do not bind biotin.
DISCUSSION
We investigated the in vivo response of gene expression in leukocytes to marginal biotin deficiency as well as assessing sensitivity for the identification of marginal biotin deficiency. Marginal biotin deficiency caused only moderate decreases in the expression of PCCA, MCCA, PC, ACCA, ACCB, HCS, biotinidase, and SMVT. No changes occurred in the expression of PCCB or MCCB. Thus, in vivo expression of some genes was responsive to even marginal biotin deficiency. However, the sensitivity of these measurements in detecting marginal biotin deficiency was considerably less than that of established indicators such as urinary biotin, urinary 3HIA, and lymphocyte PCC activity (21,27).
Unlike the moderate changes in expression of the other biotin-related genes, expression of SLC19A3 was strikingly repressed by marginal biotin deficiency in 6 of the 7 subjects. Values for SLC19A3 expression were less than the range of the beginning values for 5 of 7 subjects. Thus, the sensitivity of this measurement is similar to that of established indicators. Specificity remains to be established, but the expense and the labor argue against adoption of this measure as an indicator of marginal biotin deficiency in clinical studies. These data are consistent with the hypothesized role for SLC19A3 as a biotin transporter (16) in addition to the proposed role for SLC19A3 as a thiamin transporter (30 –32). The decrease in SLC19A3 expression observed in 6 of the 7 subjects differs from our observations of SLC19A3 expression in cell culture (33). We cultured Epstein-Barr virus-transformed lymphocytes (EBVL) for 6 d in a biotin-deficient medium; we observed a significant increase in expression of SLC19A3 in 2 of 3 EBVL cell lines (33) similar to the one subject who responded differently from the others. We speculate that differences between results of our EBVL study and the results reported here may be attributed to differences in 1) the inherent properties of the EBVL vs. peripheral blood lymphocytes or 2) culture medium biotin concentrations (25 pmol/L) compared with the plasma biotin concentrations experienced by the peripheral blood lymphocytes (~250 pmol/L), or time of induction of deficiency (6 vs. 28 d). We speculate that 28 d of egg-white diet produced a lesser degree of biotin deficiency in leukocytes, whereas culture for 6 d in biotin-deficient medium produced a more severe degree of biotin deficiency in EBVL. The unusual response of 1 subject could reflect that the lymphocytes this subject became more deficient than those of the other subjects or that his regulatory response was inherently different. Such discordant observations suggest that opposing mechanisms are at work. We hypothesize that early biotin deficiency might decrease intracellular concentrations of a critical biotin metabolite such as biotinyl-AMP, thereby repressing expression of SLC19A3 and other biotin-responsive genes. With a more severe degree of biotin deficiency, we speculate that the concentration of biotin in intracellular pools decreases, thereby stimulating expression of SLC19A3, increasing synthesis of SLC19A3 protein, and increasing biotin transport.
By what mechanism(s) could decreased biotin affect gene expression? In eukaryotic and prokaryotic cells, biotinyl-AMP, an activated form of biotin, is synthesized by HCS or BirA, a prokaryotic analog of HCS. In bacterial systems in vivo and in vitro, biotinyl-AMP binds with high affinity to DNA in the presence of BirA (34 –36) and exerts a regulatory effect on the expression of the genes that synthesize biotin. Thus, by analogy, biotin deficiency might act through an effect on biotinylate-AMP. In eukaryotic cells, HCS is present in the nucleus of several eukaryotic cell lines and is associated with chromatin and the nuclear lamina (37) in addition to the established location in cytoplasm and mitochondria of mammalian cells (38,39). Location of HCS in the nucleus allows for the possibility that biotinyl-AMP is synthesized in the nucleus and participates in gene regulation. However, unlike BirA, mammalian holocarboxylase synthetases do not carry DNA-binding motifs (40) and therefore cannot promote binding of biotinyl-AMP to DNA. We speculate that biotinyl-AMP (or the complex between biotinyl-AMP and HCS) may bind reversibly to specific DNA-binding protein(s) in the nucleus followed by interaction with regulatory elements of the genes. There are regulatory elements in the SLC19A3 gene. Nabokina and Said (32) recently reported the presence of regulatory elements for Sp1 transcriptional factor on the SLC19A3 gene. Sp1 belongs to a large family of mammalian transcription factors that can promote (41) or repress gene expression (42).
Published studies of biotin-dependent gene regulation both confirm and conflict with the observations of this study. Leon-Del-Rio and co-workers (10) observed decreased expression of HCS, PCCA, and ACCA in human hepatoblastoma cells and normal fibroblasts in response to biotin deficiency. Velazquez and co-workers (43,44) reported decreased expression of HCS in liver, kidney, muscle, and brain of rats after progressive biotin deficiency; no significant changes in expression of PC and PCCA were observed. Zempleni and co-workers (45) detected no significant change in the expression of PCCA in biotin-deficient Jurkat cells. Studies from our laboratory observed little or no effect of marginal deficiency on the expression of PCCA, PCCB, MCCA, MCCB, PC, ACCA, ACCB, HCS, biotinidase, or SMVT in EBVL (33). We speculate that differences between results of others and the results reported here may be attributed to differences in the cell lines, species, organs, and degree of deficiency as well as the techniques used for quantitation of gene expression.
Alternative mechanisms for the regulation of biotinyl-AMP may include regulation of gene expression via signaling mediators such as guanylate cyclase, cGMP, and cGMP-dependent protein kinases. The involvement of guanylate cyclase and cGMP-dependent protein kinase in the regulation of gene expression by biotin was proposed by Leon-Del-Rio and co-workers (10). Biotin may also affect gene expression by changing histone biotinylation (46), thereby causing structural alteration in chromatin and affecting nucleosomal access of transcription machinery and linker proteins in analogy with other covalent histone modifications (47). Reduced histone biotinylation in HCS-deficient patients was reported recently by Gravel and co-workers (37).
In summary, this study indicates that among all biotin-related genes examined, expression of SLC19A3 is most responsive to reduced biotin status in vivo. However, SLC19A3 expression does not appear to be more sensitive than established indicators of marginal biotin deficiency.
Acknowledgments
We thank R. A. Dennis for assistance in conducting quantitative real-time PCR, Teresa Evans for measurement of 3HIA, Matthew Mock for measurement of biotin and lymphocyte PCC, and Cecil Bogy for serving as Study Coordinator.
Footnotes
Presented at Experimental Biology 2004, April 17–21, 2004, Washington, DC [Vlasova, T. I. & Mock, D. M. (2004) Biotin deficiency reduces expression of SLC19A3, a potential biotin transporter, and decreases expression of several biotin-related genes in the leukocytes of normal subjects. FASEB J. 18: select. biosis.org/faseb/eb2004_data/FASEB009278.html].
Supported by the National Institutes of Health, DK-36823, and University of Arkansas for Medical Sciences General Clinical Research Center, M01RR14288.
Abbreviations used: ACCA, acetyl-CoA carboxylase isoform A; ACCB, acetyl-CoA carboxylase isoform B; EBVL, Epstein-Barr virus-transformed lymphocytes; GCRC, General Clinical Research Center; 3HIA, 3-hydroxyisovaleric acid; HSC, holocarboxylase synthetase; MCC, methylcrotonyl-CoA carboxylase; MCCA, MCC chain A; MCCB, MCC chain B; PC, pyruvate carboxylase; PCC, propionyl-CoA carboxylase; PCCA, PCC chain A; PCCB, PCC chain B; SLC19A3, solute carrier family 19 member 3; SMVT, sodium-dependent multivitamin transporter.
References
- 1.Mock DM, Mock NI, Stewart CW, LaBorde JB, Hansen DK. Marginal biotin deficiency is teratogenic in ICR mice. J Nutr. 2003;133:2519–2525. doi: 10.1093/jn/133.8.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watanabe T, Endo A. Teratogenic effects of maternal biotin deficiency in mouse embryos examined at midgestation. Teratology. 1990;42:295–300. doi: 10.1002/tera.1420420313. [DOI] [PubMed] [Google Scholar]
- 3.Zempleni J, Mock DM. Marginal biotin deficiency is teratogenic. Proc Soc Exp Biol Med. 2000;223:14–21. doi: 10.1046/j.1525-1373.2000.22303.x. [DOI] [PubMed] [Google Scholar]
- 4.Velazquez A, Martin-del-Campo C, Baez A, Zamudio S, Quiterio M, Aguilar JL, Perez-Ortiz B, Sanchez-Ardines M, Guzman-Hernandez &, Casanueva E. Biotin deficiency in protein-energy malnutrition. Eur J Clin Nutr. 1988;43:169–173. [PubMed] [Google Scholar]
- 5.Krause KH, Berlit P, Bonjour JP. Impaired biotin status in anticonvulsant therapy. Ann Neurol. 1982;12:485–486. doi: 10.1002/ana.410120513. [DOI] [PubMed] [Google Scholar]
- 6.Mock DM, Dyken ME. Biotin catabolism is accelerated in adults receiving long-term therapy with anticonvulsants. Neurology. 1997;49:1444–1447. doi: 10.1212/wnl.49.5.1444. [DOI] [PubMed] [Google Scholar]
- 7.Mock DM, Mock NI, Lombard KA, Nelson RP. Disturbances in biotin metabolism in children undergoing long-term anticonvulsant therapy. J Pediatr Gastroenterol Nutr. 1998;26:245–250. doi: 10.1097/00005176-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Chauhan J, Dakshinamurti K. Transcriptional regulation of the glucokinase gene by biotin in starved rats. J Biol Chem. 1991;266:10035–10038. [PubMed] [Google Scholar]
- 9.Dakshinamurti K, Li W. Transcriptional regulation of liver phosphoenolpyruvate carboxykinase by biotin in diabetic rats. Mol Cell Biochem. 1994;132:127–132. doi: 10.1007/BF00926921. [DOI] [PubMed] [Google Scholar]
- 10.Solorzano-Vargas RS, Pacheco-Alvarez D, Leon-Del-Rio A. Holocarboxylase synthetase is an obligate participant in biotin-mediated regulation of its own expression and of biotin-dependent carboxylases mRNA levels in human cells. Proc Natl Acad Sci USA. 2002;99:5325–5330. doi: 10.1073/pnas.082097699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMahon RJ. Biotin in metabolism and molecular biology. Annu Rev Nutr. 2002;22:221–239. doi: 10.1146/annurev.nutr.22.121101.112819. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Melendez R, Zempleni J. Regulation of gene expression by biotin. J Nutr Biochem. 2003;14:680–690. doi: 10.1016/j.jnutbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Grassl S. Human placental brush-border membrane Na(+)-biotin cotransport. J Biol Chem. 1992;267:17760–17765. [PubMed] [Google Scholar]
- 14.Balamurugan K, Ortiz A, Said HM. Biotin uptake by human intestinal and liver epithelial cells: role of the SMVT system. Am J Physiol. 2003;285:G73–G77. doi: 10.1152/ajpgi.00059.2003. [DOI] [PubMed] [Google Scholar]
- 15.Ozand PT, Gascon GG, Al Essa M, Joshi S, Al Jishi E, Bakheet S, Al Watban J, Al-Kawi MZ, Dabbagh O. Biotin-responsive basal ganglia disease: a novel entity. Brain. 1999;121:1267–1279. doi: 10.1093/brain/121.7.1267. [DOI] [PubMed] [Google Scholar]
- 16.Zeng, W., Al-Yamani, E., Acierno, J. S., Ozand, P. & Gusella, J. F. (2001) Mutations in SLC19A3 encoding a novel transporter cause biotin-responsive basal ganglia disease [Online] July 21, 2004. American Society of Human Genetics Meeting http://faseb.org/genetics/ashg01/f101.htm [DOI] [PMC free article] [PubMed]
- 17.Daberkow RL, White BR, Cederberg RA, Griffin JB, Zempleni J. Monocarboxylate transporter 1 mediates biotin uptake in human peripheral blood mononuclear cells. J Nutr. 2003;133:2703–2706. doi: 10.1093/jn/133.9.2703. [DOI] [PubMed] [Google Scholar]
- 18.Hymes J, Wolf B. Biotinidase and its roles in biotin metabolism. Clin Chim Acta. 1996;255:1–11. doi: 10.1016/0009-8981(96)06396-6. [DOI] [PubMed] [Google Scholar]
- 19.Hymes J, Wolf B. Human biotinidase isn’t just for recycling biotin. J Nutr. 1999;129(suppl):485S–489S. doi: 10.1093/jn/129.2.485S. [DOI] [PubMed] [Google Scholar]
- 20.Mock NI, Malik MI, Stumbo PJ, Bishop WP, Mock DM. Increased urinary excretion of 3-hydroxyisovaleric acid and decreased urinary excretion of biotin are sensitive early indicators of decreased status in experimental biotin deficiency. Am J Clin Nutr. 1997;65:951–958. doi: 10.1093/ajcn/65.4.951. [DOI] [PubMed] [Google Scholar]
- 21.Mock DM, Henrich CL, Carnell N, Mock NI. Indicators of marginal biotin deficiency and repletion in humans: validation of 3-hydroxy-isovaleric acid excretion and a leucine challenge. Am J Clin Nutr. 2002;76:1061–1068. doi: 10.1093/ajcn/76.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mock NI, Mock DM. Biotin deficiency in rats: disturbances of leucine metabolism are detectable early. J Nutr. 1992;122:1493–1499. doi: 10.1093/jn/122.7.1493. [DOI] [PubMed] [Google Scholar]
- 23.Wolf, B. (2001) Disorders of biotin metabolism. In: The Metabolic and Molecular Basis of Inherited Disease, 8th ed. (Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D., eds.). McGraw-Hill, New York, NY.
- 24.Sweetman, L. & Williams, J. C. (1995) Branched chain organic acid-urias. In: The Metabolic and Molecular Basis of Inherited Disease, 7th ed. (Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D., eds.). McGraw-Hill, New York, NY.
- 25.Bonjour JP. Biotin-dependent enzymes in inborn errors of metabolism in human. World Rev Nutr Diet. 1981;38:1–88. [Google Scholar]
- 26.Mock DM, Mock NI. Lymphocyte propionyl-CoA carboxylase activity is an early and sensitive indicator of biotin deficiency in rats, but urinary excretion of 3-hydroxyisopropionic acid is not. J Nutr. 2002;132:1945–1950. doi: 10.1093/jn/132.7.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mock DM, Henrich CL, Carnell N, Mock NI, Swift L. Lymphocyte propionyl-CoA carboxylase and accumulation of odd-chain fatty acid in plasma and erythrocytes are useful indicators of marginal biotin deficiency. J Nutr Biochem. 2002;13:462–470. doi: 10.1016/s0955-2863(02)00192-4. [DOI] [PubMed] [Google Scholar]
- 28.Mock, D. M. (1997) Determinations of biotin in biological fluids. In: Methods in Enzymology (McCormick, D. B., Suttie, J. W. & Wagner, C., eds.). Academic Press, New York, NY. [DOI] [PubMed]
- 29.Zempleni J, Trusty TA, Mock DM. Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J Nutr. 1997;127:1776–1781. doi: 10.1093/jn/127.9.1776. [DOI] [PubMed] [Google Scholar]
- 30.Rajgopal A, Edmondnson A, Goldman I, Zhao R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim Biophys Acta. 2001;1537:175–178. doi: 10.1016/s0925-4439(01)00073-4. [DOI] [PubMed] [Google Scholar]
- 31.Ganapathy V, Smith S, Prasad P. SLC19: the folate/thiamine transporter family. Pflugers Arch. 2004;447:641–646. doi: 10.1007/s00424-003-1068-1. [DOI] [PubMed] [Google Scholar]
- 32.Nabokina, S. M. & Said, H. M. (2004) Characterization of the 5′-regulatory region of the human thiamin transporter SLC19A3: in vitro and in vivo studies. Am. J. Physiol. http://ajpgi.physiology.org/papbyrecent.shtml [accessed June 24, 2004]. [DOI] [PubMed]
- 33.Vlasova, T. I., Stratton, S. L. & Mock, D. M. (2004) In vivo biotin deficiency and supplementation at a pharmacological dose causes differential expression of certain biotin-related genes in EBV-transformed lymphocytes (EBVL). FASEB J. 18: A501 (abs.).
- 34.Chapman-Smith A, Cronan JE., Jr Molecular biology of biotin attachment to proteins. J Nutr. 1999;129:477S–484S. doi: 10.1093/jn/129.2.477S. [DOI] [PubMed] [Google Scholar]
- 35.Prakash O, Eisenberg MA. Biotinyl 5′-adenylate: corepressor role in the regulation of the biotin genes of Escherichia coli K-12. Proc Natl Acad Sci USA. 1979;76:5592–5595. doi: 10.1073/pnas.76.11.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, Matthews BW. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc Natl Acad Sci USA. 1992;89:9257–9261. doi: 10.1073/pnas.89.19.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narang MA, Dumas R, Ayer LM, Gravel RA. Reduced histone biotinylation in multiple carboxylase deficiency patients: a nuclear role for holocarboxylase synthetase. Hum Mol Genet. 2004;13:15–23. doi: 10.1093/hmg/ddh006. [DOI] [PubMed] [Google Scholar]
- 38.Chang HI, Cohen ND. Regulation and intracellular localization of the biotin holocarboxylase synthetase of 3T3–L1 Cells. Arch Biochem Biophys. 1983;225:237–247. doi: 10.1016/0003-9861(83)90026-7. [DOI] [PubMed] [Google Scholar]
- 39.Chiba Y, Suzuki Y, Aoki Y, Ishida Y, Narisawa K. Purification and properties of bovine liver holocarboxylase synthetase. Arch Biochem Biophys. 1994;313:8–14. doi: 10.1006/abbi.1994.1351. [DOI] [PubMed] [Google Scholar]
- 40.Chapman-Smith A, Cronan J. The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity. Trends Biochem Sci. 1999;24:359–363. doi: 10.1016/s0968-0004(99)01438-3. [DOI] [PubMed] [Google Scholar]
- 41.Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagen G, Muller S, Beato M, Suske G. Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J. 1994;13:3843–3851. doi: 10.1002/j.1460-2075.1994.tb06695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Melendez R, Perez-Andrade ME, Diaz A, Deolarte A, Camacho-Arroyo I, Ciceron I, Ibarra I, Velazquez A. Differential effects of biotin deficiency and replenishment on rat liver pyruvate and propionyl-CoA carboxylases and on their mRNAs. Mol Genet Metab. 1999;66:16–23. doi: 10.1006/mgme.1998.2777. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Melendez R, Cano S, Mendez ST, Velazquez A. Biotin regulates the genetic expression of holocarboxylase synthetase and mitochondrial carboxylases in rats. J Nutr. 2001;131:1909–1913. doi: 10.1093/jn/131.7.1909. [DOI] [PubMed] [Google Scholar]
- 45.Manthey KC, Griffin JB, Zempleni J. Biotin supply affects expression of biotin transporters, biotinylation of carboxylases and metabolism of interleukin-2 in Jurkat cells. J Nutr. 2002;132:887–892. doi: 10.1093/jn/132.5.887. [DOI] [PubMed] [Google Scholar]
- 46.Stanley JS, Griffin JB, Zempleni J. Biotinylation of histones in human cells: effects of cell proliferation. Eur J Biochem. 2001;268:5424–5429. doi: 10.1046/j.0014-2956.2001.02481.x. [DOI] [PubMed] [Google Scholar]
- 47.Strahl BD, Allis CD. The language of covalent histone modifications. Nature (Lond) 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]