

Abstract
SR proteins are essential splicing factors required for constitutive splicing and function as key regulators of alternative RNA splicing. We have shown that SR proteins purified from late adenovirus-infected cells (SR-Ad) are functionally inactivated as splicing enhancer or splicing repressor proteins by a virus-induced partial de-phosphorylation. Here, we show that SR proteins purified from late vaccinia-virus-infected cells (SR-VV) are also hypo-phosphorylated and functionally inactivated as splicing regulatory proteins. We further show that incubating SR-Ad proteins under conditions that restore the phospho-epitopes to the SR proteins results in the restoration of their activity as splicing enhancer and splicing repressor proteins. Interestingly, re-phosphorylation of SR-VV proteins only partially restored the splicing enhancer or splicing repressor phenotype to the SR proteins. Collectively, our results suggest that viral control of SR protein activity may be a common strategy used by DNA viruses to take control of the host cell RNA splicing machinery.
Introduction
Gene expression in eukaryotes requires multi-component molecular machines that make up an extensively coupled network to coordinate activities such as transcription, RNA processing and RNA export (reviewed in Maniatis and Reed, 2002). It is common in nature that DNA viruses induce an inhibition of host cell gene expression to gain full access to the biosynthetic machineries in the infected cell. For example, vaccinia virus induces an inhibition of host cell RNA synthesis. This inhibition does not interfere with vaccinia virus RNA synthesis, since the virus replicates in the cytoplasm and encodes for its own transcription system (reviewed in Flint et al., 2000). Adenoviruses use an alternative strategy to achieve a selective viral gene expression. Thus, they block nuclear to cytoplasmic export of host cell mRNA while facilitating export of viral mRNA late during the infectious cycle (reviewed in Imperiale et al., 1995). Since RNA splicing is intimately coupled to mRNA transport (reviewed in Maniatis and Reed, 2002), a viral suppression of host cell RNA splicing could be a regulatory mechanism by which many viruses inhibit host cell gene expression. Most DNA viruses, like adenoviruses, replicate in the cell nucleus and encode for genes containing introns. Such viruses are dependent on a functional splicing machinery for viral gene expression. Consequently, adenoviruses appear to redirect the specificity of the splicing machinery such that splicing of pre-mRNAs with consensus-type splicing signals is reduced and splicing of late viral mRNAs containing non-consensus splicing signals is enhanced (reviewed in Akusjärvi, 1999). Vaccinia virus, in contrast, encodes for genes lacking introns. Therefore, vaccinia virus could theoretically shut off host cell RNA splicing completely without a major impact on virusspecific gene expression.
We have shown previously that adenoviruses interfere with the activity of the cellular SR family of splicing factors (Kanopka et al., 1998; Estmer Nilsson et al., 2001). SR proteins are essential splicing factors that play a crucial role in constitutive splicing and function as key regulators of alternative RNA splicing (reviewed in Fu, 1995; Graveley, 2000). They are highly phosphorylated in vivo (Roth et al., 1990), mainly at serines in the RS domain, a modification that is required for their function in spliceosome assembly (reviewed in Graveley, 2000). They have dual functions and serve as splicing enhancer or splicing repressor proteins, depending on where in the pre-mRNA they bind (reviewed in Akusjärvi, 1999). We have shown that SR proteins prepared from uninfected HeLa cells (SR-HeLa) inhibit adenovirus IIIa pre-mRNA splicing by binding to an intronic repressor element, the 3RE, located immediately upstream of the IIIa branch site (Kanopka et al., 1996). In contrast, SR proteins prepared from late adenovirus-infected HeLa cells (SR-Ad) are functionally inactivated as IIIa splicing repressor proteins by a virus-induced partial de-phosphorylation (Kanopka et al., 1998).
To test the hypothesis that other mammalian viruses also inactivate the SR family of splicing factors, we determined the status of SR proteins in late vaccinia virus-infected cells (SR-VV). We chose vaccinia virus as a good candidate virus causing SR protein inactivation, since this DNA virus replicates in the cytoplasm of the infected cell and, furthermore, encodes for genes lacking introns. The results show that SR-VV proteins are hypophosphorylated and functionally inactivated as splicing repressor or splicing enhancer proteins.
Results and Discussion
First, we determined whether SR-VV proteins were active as splicing repressor proteins. For this experiment, we compared the capacity of SR-HeLa, SR-Ad and SR-VV proteins to repress IIIa splicing. Importantly, the total amounts of all three were identical (data not shown), suggesting that the SR family of splicing factors was not targeted for degradation during infection. As shown in Figure 1A, the addition of an excess of SR-HeLa proteins to nuclear extracts prepared from uninfected HeLa cells (HeLa-NE) resulted in an efficient repression of IIIa pre-mRNA splicing (lanes 2–6) (Kanopka et al., 1996, 1998). The addition of the same amount of SR-Ad proteins resulted in a less pronounced inhibition of IIIa pre-mRNA splicing (lanes 7–11). This result was expected, since SR-Ad proteins are functionally impaired as IIIa splicing repressor proteins by a virus-induced partial de-phosphorylation (Kanopka et al., 1998). Interestingly, the addition of the same amount of SR-VV proteins did not result in an efficient inhibition of IIIa pre-mRNA splicing, even at the highest concentrations used (lanes 12–16).
Figure 1.
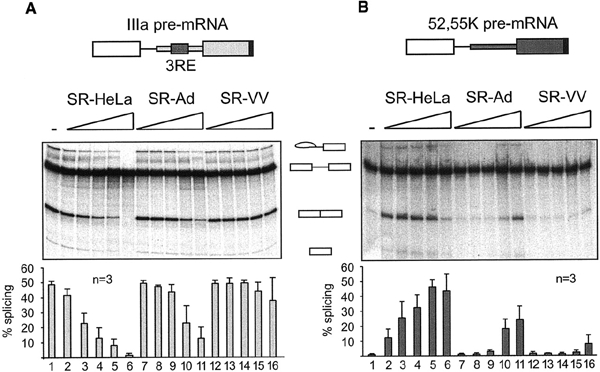
Functional inactivation of SR proteins during a vaccinia virus infection. Increasing amounts (0.2–1.0 μg) of SR-HeLa (lanes 2–6), SR-Ad (lanes 7–11) and SR-VV (lanes 12–16) proteins were pre-incubated with the IIIa and the 52,55K pre-mRNAs and then added to (A) HeLa-NE and (B) HeLa-S100 splicing reactions, respectively. The transcripts are depicted schematically at the top. Quantitative data derived from three independent experiments are shown at the bottom.
To assay for the splicing enhancer activity of SR-VV proteins, the capacity of SR-HeLa, SR-Ad and SR-VV proteins to activate 52,55K pre-mRNA splicing in splicing-deficient HeLa cytoplasmic S100 extracts (Krainer et al., 1990) was compared. As shown in Figure 1B, the addition of increasing amounts of SR-HeLa proteins resulted in an efficient activation of 52,55K pre-mRNA splicing (lanes 2–6), whereas SR-Ad proteins were much reduced as splicing enhancer factors (Kanopka et al., 1998), only showing an activity at the highest concentrations (lanes 7–11). Interestingly, SR-VV proteins were severely impaired as 52,55K splicing enhancer proteins, showing only a minute enhancer activity at the highest concentrations used (lanes 12–16). Collectively, these results show that the SR family of splicing factors are functionally inactivated during both adenovirus and vaccinia virus infections. Notably, SR-VV proteins were subjected to a more severe inactivation than SR-Ad proteins (Figure 1).
To determine whether SR-VV proteins, like SR-Ad proteins (Kanopka et al., 1998), were under-phosphorylated, equal amounts of SR-HeLa, SR-Ad and SR-VV proteins were separated by gel electrophoresis, transferred to a filter and probed with monoclonal antibody mAb104, which detects phospho-epitopes in the RS domain of SR proteins (Zahler et al., 1992). As shown in Figure 2A, SR-HeLa proteins were efficiently recognized by mAb104 (lane 1), whereas SR-Ad and SR-VV proteins were not (lanes 2 and 3). The reduced phosphate content of SR-VV and SR-Ad proteins was also observed in SR proteins prepared from 32P-labelled cell cultures (data not shown). Interestingly, incubation of SR-Ad or SR-VV proteins with a small amount of HeLa-NE restored the mAb104 phospho-epitopes on both protein preparations (lanes 5 and 6). Also, incubation of SR-Ad or SR-VV proteins in HeLa-NE resulted in a reduced mobility, such that SR-Ad and SR-VV migration was identical to that of SR-HeLa. The small amount of HeLa-NE used in this phosphorylation assay did not contribute detectable amounts of SR proteins (lane 7). Re-probing the filter with mAb96 (Hanamura et al., 1998), which recognizes the RNA binding domain in ASF/SF2, showed that re-phosphorylation produced a shift in ASF/SF2 mobility to the slower migrating hyper-phosphorylated species seen in SR-HeLa (Figure 2B). Notably, mAb96 recognition is slightly affected by phosphorylation, as seen by a stronger signal in lanes 5 and 6 than in lanes 2 and 3. Collectively, these results show that SR-VV and SR-Ad proteins are hypo-phosphorylated and, furthermore, that they can be converted to SR proteins with the same mAb104-recognizable phospho-epitopes as SR-HeLa proteins by in vitro phosphorylation.
Figure 2.
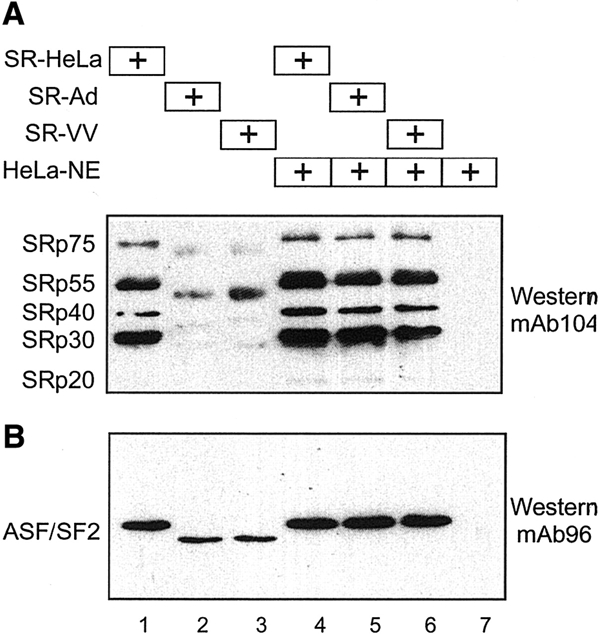
SR-VV proteins are hypo-phosphorylated. (A) SR-HeLa (lane 1), SR-Ad (lane 2) and SR-VV (lane 3) proteins were pre-incubated in HeLa-NE (lanes 4–6), separated by 10% SDS–PAGE, transferred to a nitrocellulose filter and probed with mAb104 in a western blot assay. (B) The same filter re-probed with the ASF/SF2-specific mAb96.
We have previously presented evidence suggesting that SR-Ad proteins are inactivated as splicing regulatory proteins because of their reduced level of phosphorylation (Kanopka et al., 1998). However, we have not provided more direct experimental evidence that this post-translational modification is responsible for their reduced activity as regulatory splicing factors. We therefore tested whether re-phosphorylation of SR-Ad and SR-VV proteins, in a small amount of HeLa-NE, restored their activity as both IIIa splicing repressor and 52,55K splicing enhancer proteins. As shown in Figure 3A, pre-incubation of SR-Ad proteins in a small amount of HeLa-NE resulted in the restoration of their IIIa splicing repressor activity (compare lanes 3 and 4). Similarly, re-phosphorylation of SR-Ad proteins restored their splicing enhancer activity (Figure 3B, lanes 5 and 6). This result is important and extends our previous studies by suggesting that SR-Ad proteins can be fully converted to SR proteins with the splicing regulatory properties of SR-HeLa proteins by re-phosphorylation. In contrast, re-phosphorylation of SR-VV proteins restored most, but not all, of its IIIa splicing repressor activity (Figure 3A, lanes 5 and 6) but only some of the 55,52K splicing enhancer phenotype (Figure 3B, lanes 7 and 8). Clearly, SR-Ad and SR-VV proteins differ in their capacity to be reactivated by a simple in vitro phosphorylation assay. Potentially, SR-VV proteins are subjected to an additional modification or, alternatively, require phosphorylation of a protein kinase that is not active in the HeLa-NE re-phosphorylation assay. Pre-incubation of the already hyper-phosphorylated SR-HeLa proteins in HeLa-NE had no significant effects on their splicing repressor activity (data not shown) or splicing enhancer activity (Figure 3B, lanes 3 and 4).
Figure 3.
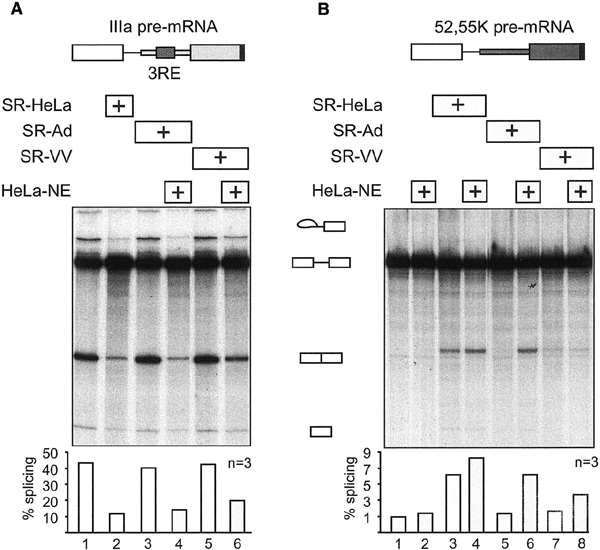
Effect of re-phosphorylation of SR-Ad and SR-VV proteins on their splicing repressor (A) and splicing enhancer (B) phenotype. SR-HeLa, SR-Ad and SR-VV proteins (0.5 μg) were pre-incubated with the IIIa or the 52,55K pre-mRNAs prior to the addition to HeLa-NE or HeLa-S100 splicing reactions. The same amount of SR-Ad and SR-VV proteins was also re-phosphorylated with a small amount of HeLa-NE (Figure 2) and then added to the HeLa-NE (A) and HeLas100 (B) splicing reactions. Quantitative data of splicing efficiencies derived from three independent experiments are shown at the bottom.
In our previous work, we speculated that SR proteins binding to the 3RE block IIIa splicing by sterically interfering with U2 snRNP recruitment to the IIIa branch site (Kanopka et al., 1996). However, our more recent work has argued against this simplistic steric interference model by showing that ASF/SF2 repression of IIIa splicing critically depends on the ASF/SF2 second RNA binding domain (Dauksaite and Akusjärvi, 2002). Thus, tethering an ASF/SF2 mutant protein lacking the second RNA binding domain to the IIIa pre-mRNA does not result in splicing repression. Here, the capacity of SR-VV proteins to bind to a short transcript containing the 3RE was tested in a UV cross-linking assay. As shown in Figure 4, the RNA binding capacity of SR-Ad proteins was slightly reduced compared with that of SR-HeLa (Figure 4, lanes 1 and 2). In contrast, SR-VV proteins, which were severely impaired as splicing repressor proteins (Figure 1A), were not affected in their RNA binding capacity (Figure 4, lanes 1 and 3). Note that SR-Ad and SR-VV proteins migrated slightly faster compared with the corresponding SR-HeLa proteins (Figure 4), a result consistent with the conclusion that they are hypo-phosphorylated (Figure 2). We conclude that inactivation of the splicing repressor phenotype of SR proteins does not require an inactivation of the RNA binding capacity. More likely, the activity of SR proteins, as splicing repressor proteins, is regulated by post-translational modification, such as phosphorylation/de-phosphorylation (Figure 3). The variations in the RNA binding capacity of SR-Ad and SR-VV proteins probably result from differences in the specific amino acid residues that are de-phosphorylated during the respective virus infection.
Figure 4.
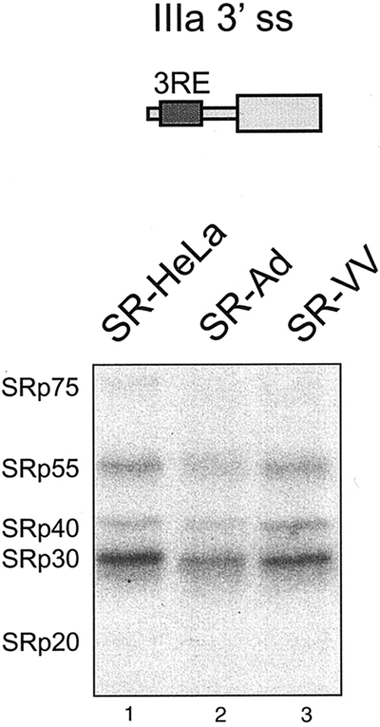
RNA binding capacity of SR-VV and SR-Ad proteins. SR-HeLa, SR-Ad and SR-VV proteins (1 μg) were UV cross-linked to the wild-type IIIa 3′ splice site fragment (shown schematically at the top) as described in Methods.
Collectively, our results show that during a vaccinia virus infection the SR family of splicing factors are hypo-phosphorylated and functionally inactivated as splicing regulatory proteins. We have shown previously that the adenovirus E4-ORF4 protein, which binds the cellular protein phosphatase 2A (reviewed in Branton and Roopchand, 2001), induces SR protein de-phosphorylation (Kanopka et al., 1998) and activates IIIa pre-mRNA splicing (Kanopka et al., 1998; Estmer Nilsson et al., 2001). However, we do not have evidence that vaccinia virus achieves the same by inducing SR protein de-phosphorylation. Thus, theoretically, vaccinia virus may inactivate SR protein functions by inhibiting a protein kinase(s) responsible for SR protein phosphorylation. However, it should be noted that vaccinia virus does encode for its own dualspecificity protein phosphatase, VH1 (Guan et al., 1991), which may be a potential candidate protein inducing SR protein de-phosphorylation during a vaccinia virus infection. The VH1 phosphatase is required for vaccinia virus gene expression and is vital for virus growth, although the mechanistic details are still, to a large extent, unknown (Liu et al., 1995; Derrien et al., 1999).
To test whether VH1 could induce SR protein inactivation, we cloned and expressed the VH1 protein in bacteria. Incubating SR-HeLa proteins with purified VH1 resulted in an efficient SR protein de-phosphorylation (data not shown). We have shown previously that pre-incubation of a bacterially produced adenovirus E4-ORF4 protein with HeLa-NE induces SR protein de-phosphorylation and activation of IIIa pre-mRNA splicing (Kanopka et al., 1998). Attempts to reproduce this experiment using the recombinant VH1 failed and resulted in a repression of IIIa pre-mRNA splicing at all concentrations tested (data not shown). The failure of VH1 to activate IIIa splicing may result from the fact that VH1 causes a more severe de-phosphorylation of SR proteins than E4-ORF4. Such a conclusion is in agreement with the observations that SR-VV proteins were more severely impaired as splicing regulatory proteins than SR-Ad proteins (Figure 1) and that re-phosphorylation of SR-VV proteins only partially restored their splicing regulatory activity (Figure 3). It should be noted that vaccinia virus does not encode for genes containing introns and, therefore, in theory, could induce a complete inactivation of the cellular splicing machinery without a negative impact on virus growth. Adenoviruses, in contrast, make extensive use of RNA splicing to produce a multitude of alternatively spliced mRNAs during virus replication and therefore need the host cell RNA splicing machinery for viral pre-mRNA splicing (reviewed in Akusjärvi, 1999). Preliminary experiments indicate that SR-VV proteins accumulate in enlarged nuclear speckles (data not shown), as has been observed in late adenovirus-infected cells (Bridge et al., 1995). It is also possible that VH1 does not function as a IIIa splicing enhancer regulator because it de-phosphorylates additional splicing factors required for spliceosome assembly and/or splicing in HeLa-NE. Also, as mentioned above, we cannot exclude the possibility that vaccinia virus encodes for a protein that blocks SR protein phosphorylation rather than induces SR protein de-phosphorylation.
Speculation
A lesson that we have learnt during the last two decades of research is that mammalian viruses typically target the same key cellular factors controlling cell growth and survival. The best examples are the multiple viruses that encode for proteins that interact, and interfere, with the activity of the tumour suppressor proteins pRb and p53 and the interferon-induced protein kinase PKR. The finding that two unrelated DNA virus families inactivate the SR family of splicing factors forecasts that SR proteins and RNA splicing may be a common target of viral interference. It is possible that future studies will show that viral interference with SR protein activity is an important mechanism in the viral remodelling of the host cell gene expression machinery.
Methods
In vitro splicing.
Preparation of extracts (HeLa-NE and HeLa-S100) and conditions for in vitro splicing were as described previously (Mühlemann and Akusjärvi, 1998). The IIIa and 52,55K pre-mRNAs were synthesized with a U1 snRNA-enhancer sequence at the 3′ end as described previously (Mühlemann and Akusjärvi, 1998). SR proteins (0.2–1.0 μg) were incubated with 10–20 fmol of RNA for 15 min at room temperature prior to the addition of HeLa-NE or HeLas100 extracts. Pre-mRNA and products were visualized and quantified by PhosphorImager scanning as described previously (Petersen-Mahrt et al., 1999).
Virus infection and SR protein purification.
Four litres of HeLa spinner cell cultures (2–4 × 105 cells/ml) were infected with adenovirus (wt900) or vaccinia virus (vTF7-3, kindly provided by Dr B. Moss) at 10 PFU/ml. Cells were harvested 20 h after infection and SR proteins purified as described previously (Zahler et al., 1992; Kanopka et al., 1998), except that 100 nM staurosporine (Sigma) was included in buffer A.
Western blot analysis.
SR proteins (0.5 μg) were separated by 10% SDS–PAGE and transferred to a nitrocellulose filter. The filter was probed with mAb104 (Zahler et al., 1992), which recognizes phospho-epitopes in the RS domain of SR proteins, in a standard western blot assay (Petersen-Mahrt et al., 1999). The filter was stripped by incubation in 0.78% mercaptoethanol, 62.5 mM Tris–HCl and 2% SDS for 20 min at 50°C and re-probed with mAb96, which recognizes RRM1 of ASF/SF2 (Hanamura et al., 1998).
In vitro phosphorylation assay.
SR-HeLa, SR-Ad or SR-VV proteins (0.5–1.0 μg) were incubated with 5–10 μg of HeLa-NE for 10–15 min at room temperature. Note that HeLa-NE contains sufficient amounts of ATP to restore phospho-epitopes to SR proteins (Estmer Nilsson et al., 2001). The re-phosphorylated SR proteins were detected by western blotting or processed for in vitro splicing in HeLa-NE or HeLas100 extracts as described above.
UV cross-linking.
Purified SR-HeLa, SR-Ad or SR-VV proteins (1 μg) were mixed with 20 fmol of a 32P-labelled IIIa 3′ splice site transcript (Kanopka et al., 1996) in buffer D supplemented with 1 μg of BSA and 1 μg of poly(U) (Pharmacia) and incubated at 30°C for 10 min. The reaction mixtures were irradiated with a R52G UV lamp at a distance of 4 cm for 5 min on ice. RNA was digested with 30 μg of RNase A and 300 U of RNase T1 in the presence of 10 mM MgCl2 at 37°C for 45 min. The samples were resolved by 10% SDS–PAGE and visualized by autoradiography.
Acknowledgments
This work was made possible by generous grants from the Swedish Cancer Society and the Göran Gustafsson foundation for Natural and Medical Research.
References
- Akusjärvi G. (1999) Remodelling of the host cell RNA splicing machinery during an adenovirus infection. Recent Res. Dev. Virol., 1, 621–630. [DOI] [PubMed] [Google Scholar]
- Branton P.E. and Roopchand D.E. (2001) The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene, 20, 7855–7865. [DOI] [PubMed] [Google Scholar]
- Bridge E., Xia D.X., Carmo-Fonseca M., Cardinali B., Lamond A.I. and Pettersson U. (1995) Dynamic organization of splicing factors in adenovirus-infected cells. J. Virol., 69, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauksaite V. and Akusjärvi G. (2002) Human splicing factor ASF/SF2 encodes for a repressor domain required for its inhibitory activity on pre-mRNA splicing. J. Biol. Chem., 277, 12579–12586. [DOI] [PubMed] [Google Scholar]
- Derrien M., Punjabi A., Khanna M., Grubisha O. and Traktman P. (1999) Tyrosine phosphorylation of A17 during vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J. Virol., 73, 7287–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estmer Nilsson C., Petersen-Mahrt S., Durot C., Shtrichman R., Krainer A.R., Kleinberger T. and Akusjärvi G. (2001) The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J., 20, 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint S.J., Enquist L.W., Krug R.M., Racaniello V.R. and Skalka A.M. (2000) The Principles of Virology: Molecular Biology, Pathogenesis and Control. ASM Press, Washington, DC. [Google Scholar]
- Fu X.D. (1995) The superfamily of arginine/serine-rich splicing factors. RNA, 1, 663–680. [PMC free article] [PubMed] [Google Scholar]
- Graveley B.R. (2000) Sorting out the complexity of SR protein functions. RNA, 6, 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K.L., Broyles S.S. and Dixon J.E. (1991) A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature, 350, 359–362. [DOI] [PubMed] [Google Scholar]
- Hanamura A., Caceres J.F., Mayeda A., Franza B.R. Jr and Krainer A.R. (1998) Regulated tissuespecific expression of antagonistic pre-mRNA splicing factors. RNA, 4, 430–444. [PMC free article] [PubMed] [Google Scholar]
- Imperiale M.J., Akusjärvi G. and Leppard K.N. (1995) Post-transcriptional control of adenovirus gene expression. Curr. Topics Microbiol. Immunol., 199, 139–171. [DOI] [PubMed] [Google Scholar]
- Kanopka A., Mühlemann O. and Akusjärvi G. (1996) Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature, 381, 535–538. [DOI] [PubMed] [Google Scholar]
- Kanopka A., Mühlemann O., Petersen-Mahrt S., Estmer C., Öhrmalm C. and Akusjärvi G. (1998) Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature, 393, 185–187. [DOI] [PubMed] [Google Scholar]
- Krainer A.R., Conway G.C. and Kozak D. (1990) The essential pre-mRNA splicing factor SF2 influences 5′ splice site selection by activating proximal sites. Cell, 62, 35–42. [DOI] [PubMed] [Google Scholar]
- Liu K., Lemon B. and Traktman P. (1995) The dualspecificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J. Virol., 69, 7823–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T. and Reed R. (2002) An extensive network of coupling among gene expression machines. Nature, 416, 499–506. [DOI] [PubMed] [Google Scholar]
- Mühlemann O. and Akusjärvi G. (1998). Preparation of splicing-competent nuclear extracts from adenovirus-infected cells. In Wold, W.S.M. (ed.), Adenovirus Methods and Protocols. Humana Press, Totowa, NJ, pp. 203–216. [Google Scholar]
- Petersen-Mahrt S.K., Estmer C., Öhrmalm C., Matthews D.A., Russell W.C. and Akusjärvi G. (1999) The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J., 18, 1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M.B., Murphy C. and Gall J.G. (1990) A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosome loops and small granules in the amphibian germinal vesicle. J. Cell Biol., 111, 2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler A.M., Lane W.S., Stolk J.A. and Roth M.B. (1992) SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev., 6, 837–847. [DOI] [PubMed] [Google Scholar]