

Abstract
HMGB1, a non-histone nuclear factor, acts extracellularly as a mediator of delayed endotoxin lethality, which raises the question of how a nuclear protein can reach the extracellular space. We show that activation of monocytes results in the redistribution of HMGB1 from the nucleus to cytoplasmic organelles, which display ultrastructural features of endolysosomes. HMGB1 secretion is induced by stimuli triggering lysosome exocytosis. The early mediator of inflammation interleukin (IL)-1β is also secreted by monocytes through a non-classical pathway involving exocytosis of secretory lysosomes. However, in keeping with their respective role of early and late inflammatory factors, IL-1β and HMGB1 respond at different times to different stimuli: IL-1β secretion is induced earlier by ATP, autocrinally released by monocytes soon after activation; HMGB1 secretion is triggered by lysophosphatidylcholine, generated later in the inflammation site. Thus, in monocytes, non-classical secretion can occur through vescicle compartments that are at least partially distinct.
Introduction
Cytokines, the soluble mediators of immune and inflammatory response (Dinarello, 2000), are externalized through the classical endoplasmic reticulum (ER)–Golgi exocytotic route. However, some lack a signal peptide and are secreted through non-classical secretory mechanisms (Rubartelli and Sitia, 1997). The prototype of leaderless cytokines is interleukin (IL)-1β, a major pro-inflammatory factor produced by activated monocytes. From the cytosol, where it accumulates, IL-1β translocates in part into secretory lysosomes and is secreted upon exocytosis of these organelles induced by exogenous ATP (Andrei et al., 1999). Secretory lysosomes are Ca2+-regulated secretory organelles displaying features of both lysosomes and secretory granules. Particularly abundant in hemopoietic cells such as leukocytes and platelets, they defend tissue homeostasis, repair injuries and participate in inflammatory and immune response by mobilizing their content into the external milieu in response to triggering signals (Andrews, 2000; Blott and Griffiths, 2002). Recent studies have shown that HMGB1, widely known as a nuclear protein (Bianchi and Beltrame, 2000), can also behave as a pro-inflammatory cytokine (Wang et al., 1999; reviewed by Müller et al., 2001). HMGB1 release is induced in monocytes by lipopolysaccharide (LPS), tumor necrosis factor (TNF)-α or IL-1. Its detection in serum is delayed by several hours compared with the early pro-inflammatory cytokines IL-1 and TNF-α; furthermore, anti-HMGB1 antibodies protect against endotoxin lethality even when passive immunization is delayed until after the early cytokine response (Wang et al., 1999). Thus, HMGB1 seems to occupy a position downstream of IL-1 and TNF-α in the inflammatory mediator cascade. The observation that a constitutively expressed nuclear protein that lacks secretory signal sequences works as a cytokine raises two questions: how does it move from the nucleus to the extracellular space, and which signals trigger its secretion?
Here, we show that, in activated monocytes, HMGB1 relocalizes from the nucleus to cytoplasmic organelles whose exocytosis is induced by lysophosphatidylcholine (LPC), a bioactive lipid that promotes many inflammatory effects (Quinn et al., 1988; Kabarowski et al., 2001). IL-1β secretion, although mediated similarly by secretory lysosomes (Andrei et al., 1999), is significantly different, because it is activated early and responds to extracellular ATP: this nucleotide is rapidly released at high concentrations under inflammatory conditions by monocytic and endothelial cells (Bodin and Burnstock, 1998; Di Virgilio et al., 2001) and acts autocrinally or paracrinally on monocytes. In contrast, LPC appears later in the site of inflammation, as it is generated from phosphatidylcholine through the action of the secretory phospholipase sPLA2 (Quinn et al., 1988), which is produced by monocytes several hours after activation (Ribardo et al., 2001). Our data justify the different kinetics of HMGB1 and IL-1β release and imply that their secretion occurs via similar but specialized vesicles.
Results
HMGB1 relocalizes from nucleus to cytoplasm in monocytes activated by inflammatory stimuli
The intracellular localization of HMGB1 in monocytes freshly isolated or treated with LPS was investigated by immunofluorescence. Cells were double stained with anti-HMGB1 antibodies and with the nuclear dye ethidium homodimer-2 (EthD-2) and analyzed by confocal microscopy. Non-stimulated monocytes (Figure 1A) display a strong staining for HMGB1 mostly restricted to the nucleus, as indicated by the colocalization with EthD-2, well evident in the central section. In contrast, the cytoplasm (peripheral section) is negative. Eighteen hours after stimulation with LPS (Figure 1B), HMGB1 appears to move from the nucleus, which is still partly positive (central section), to the periphery of the cell, where it displays a punctuate staining in the cytoplasm (peripheral section). Similar results were obtained when monocytes were incubated with TNF-α or IL-1β (data not shown).
Figure 1.
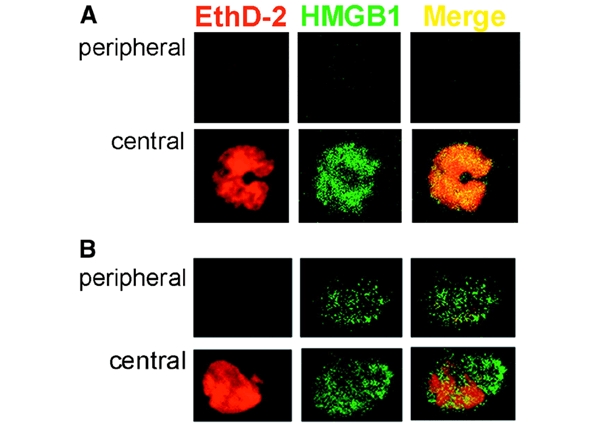
Immunofluorescence analysis of HMGB1 in resting and LPS-activated monocytes. Monocytes, freshly isolated (A) or cultured for 18 h with LPS (B), were fixed, permeabilized, stained with ethidium homodimer-2 (EthD-2, red channel) and anti-HMGB1 antibody (HMGB1, green channel) and analyzed by confocal microscopy. Two sections of the same cells (peripheral and central) are shown, which allows the nuclear (central) and cytoplasmic (peripheral, absent at time 0, and evident after 18 h of activation) staining to be appreciated. The merged images (Merge) verify the almost complete colocalization of HMGB1 and EthD-2 at time 0 (A, yellow) which decreases after LPS activation (B).
To investigate the nature of the cytoplasmic HMGB1 positive spots, resting and activated monocytes were subcellular fractionated and the distribution of HMGB1 analyzed by western blotting. As shown in Figure 2A a shift of HMGB1 from the nuclear to the postnuclear fraction is observed at 18 h from LPS stimulation. In resting monocytes (t0), the small amount of cytoplasmic HMGB1 is all contained in the soluble fraction (cyt); in contrast, in activated monocytes (t18), most of the postnuclear HMGB1 (70–90% in the different experiments) is detected in a particulated fraction (P50), which protects it from protease digestion (Figure 2B, upper, lane 3). A trypsin-resistant IL-1β band is also present in P50 from activated but not resting monocytes (middle, lane 3), in keeping with our previous observations (Andrei et al., 1999). Both HMGB1 and IL-1β are accessible to trypsin digestion after detergent solubilization of P50 (lane 4).
Figure 2.

Relocalization of HMGB1 in activated monocytes. (A) Aliquots of nuclei (lanes 1 and 2), postnuclear supernatants (PNS, lanes 3 and 4) and soluble cytosol (lanes 5 and 6) (corresponding to 5 × 106 cells) from resting (t0, lanes 1, 3 and 5) or 18 h LPS-activated monocytes (t18, lanes 2, 4 and 6) were analyzed by western blotting with anti-HMGB1 antibody. One experiment out of six performed is shown. (B) Aliquots (50 μg) from P50 fractions from resting (t0, lanes 1 and 2) or activated monocytes (t18, lanes 3 and 4), treated with trypsin (Try, lanes 1 and 3) or with trypsin and Triton X-100 (TryTx, lanes 2 and 4) were analyzed by western blotting with anti-HMGB1 (upper), anti-IL-1β (middle) or anti-CD (lower) antibodies. One experiment of six performed is shown. (C) Confocal microscopy analysis of LPS-activated monocytes, double stained with anti-HMGB1 (red channel) and anti-EEA1 (green channel, left) or anti-LAMP1 (green channel, right) antibodies.
P50 fraction is enriched in endolysosomes (Pitt et al., 1992; Andrei et al., 1999): it contains a large amount of the endolysosomal marker cathepsin D (CD), equally represented in resting and activated monocytes (Figure 2B, lower). The partial colocalization with the lysosomal marker LAMP1 revealed by double immunofluorescence experiments (Figure 2C, right) supports the idea that HMGB1-containing organelles belong to the endolysosomal compartment. The lack of costaining with EEA1 excluded the presence of HMGB1 in early endosomes (Figure 2C, left).
Immunoelectron microscopy confirmed that HMGB1 relocalizes to cytoplasmic organelles following LPS activation (Figure 3). Quantitative analysis performed by counting HMGB1 gold particles revealed that 65% of HMGB1 is confined to the nucleus of resting monocytes; in contrast, in activated monocytes, only 26% of HMGB1 is nuclear and 74% appears associated to cytoplasmic organelles.
Figure 3.

Immunoelectron microscopy analysis of freshly isolated (A) or 24 h activated monocytes (B and C), immunogold labeled for HMGB1. Arrows point to HMGB1 staining, restricted to the nucleus in (A) and localizing into vesicles in (B) and (C). In activated monocytes, HMGB1 appears associated to dense material in structures displaying the morphology of endosomes (C). The partial loss of the dense matrix is probably due to the fixation technique used to allow staining with anti-HMGB1 (see Methods), which led to partial extraction of vesicle content. Counting of gold particles allowed us to determine that, in resting monocytes (A), 65% of HMGB1 is in the nucleus and 35% in the cytoplasm; in activated monocytes (B and C), 26% of gold particles are nuclear and 74% are cytoplasmic, associated to endosomal structures.
These HMGB1 positive organelles are vesicular structures of different size that have a uniform, electron-lucent appearence and contain electron-dense materials. Double staining was performed in order to determine whether the cofractionation with CD and IL-1β shown in Figure 2 corresponds to colocalization. As shown in Figure 4A and B, HMGB1 and CD do not colocalize, whereas a partial colocalization with IL-1β is detected (Figure 4C–E).
Figure 4.
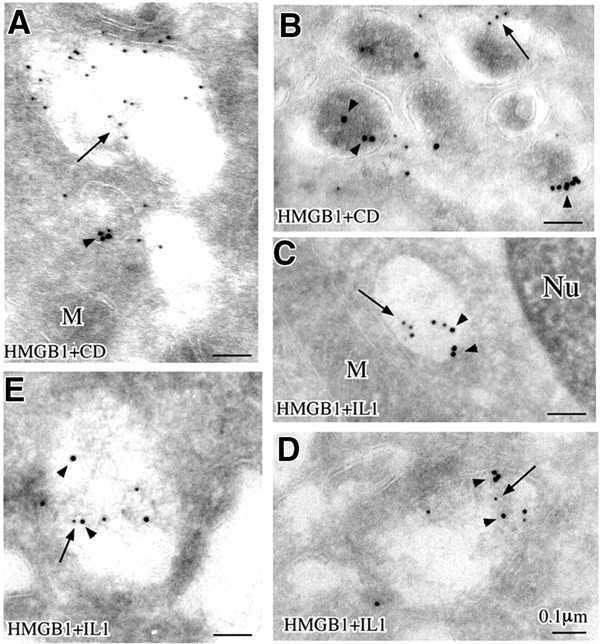
Double immunogold labeling of HMGB1 and CD (A and B) and HMGB1 and IL-1β (C–E). Arrows point to small gold particles (HMGB1), and arrowheads point to large gold particles (CD in A and B, IL-1β in C–E)
Secretion of HMGB1 by LPS-activated monocytes is a late event induced by LPC
Figure 5A and B shows the kinetics of secretion of mature IL-1β (17 kDa) and HMGB1 by LPS-activated monocytes. A secreted IL-1β band is detected after 3 h from LPS stimulation, and increases after 6 h, in keeping with previous observations (Rubartelli et al., 1990). In contrast, HMGB1 secretion starts to be detected at 6 h and is still increasing at 18 and 30 h, when secretion of IL-1β is decreasing and then stops. Secretion of IL-1β at 3 and 6 h is strongly induced by exogenous ATP (Figure 5A): in contrast, ATP triggers very little secretion of HMGB1 (Figure 5B). To investigate whether other inflammatory stimuli are stronger inducers of HMGB1 secretion, we tested the effects of bioactive phospholipids that are generated by inflammatory cells at different times in the inflammatory fluids (Dart and Chin-Dusting, 1999). Whereas neither phosphatidic acid (PA) nor lysophosphatidic acid (LPA) induce HMGB1 secretion (data not shown), a short exposure (10 min) to 20 μM LPC resulted in a strong increase of secretion, well evident at late time points (Figure 5B). Interestingly, exogenous LPC also induced IL-1β secretion, but only at earlier time points (3 and 6 h of LPS stimulation, Figure 5A). After 18 h, LPC-induced HMGB1 secretion is ∼30% of the total HMGB1 cell content and is paralleled by the release of a similar amount of CD and β-hexosoaminidase, two markers of lysosome exocytosis; in contrast, the cytoplasmic marker lactate dehydrogenase (LDH) is low in supernatants, ruling out the possibility that LPC-induced HMGB1 release is due to cell lysis (Figure 5C).
Figure 5.
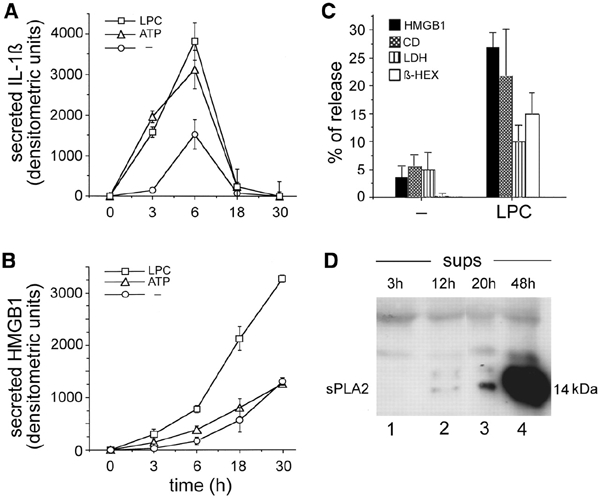
Kinetics of basal and induced secretion of mature IL-1β (17 kDa) and HMGB1. Densitometric analysis of IL-1β (A) and HMGB1 (B) secreted at different times in the absence of stimuli (circles) or after the addition of 1 mM ATP (triangles) or 20 μM LPC in the last 10 min (squares). The mean of a triplicate experiment ± SD is shown. One experiment of six perfomed is shown. (C) Release of HMGB1, CD, LDH and β-hexosoaminidase by 18 h LPS-activated monocytes, non-stimulated (−) or stimulated with 20 μM LPC for the last 10 min. Data are expressed as a percentage of secretion according to the following formula: secreted protein/intracellular + secreted protein × 100 ± SD. One experiment of four performed is shown. (D) Supernatants (sups) from monocytes at different times after LPS stimulation as indicated were analyzed for the presence of sPLA2 by western blotting.
As sPLA2 is responsible for the generation of extracellular LPC, we investigated the kinetics of release of this enzyme by activated monocytes. As shown in Figure 5D, sPLA2 is barely detectable in monocyte supernatants after 12 h of stimulation, becomes clearly visible after 20 h and is abundant after 48 h. This indicates that sPLA2 secretion by activated monocytes is a late event, consistent with a late generation of extracellular LPC and a consequent delayed secretion of HMGB1.
Discussion
We have shown here that HMGB1 relocalizes in LPS-activated monocytes from the nucleus to secretory organelles; its secretion is rather inefficient in the presence of LPS alone and is strongly and rapidly induced by LPC, a bioactive lipid generated at the inflammation site. These conclusions are based on the following evidence: (i) during LPS activation, HMGB1 gradually disappears from the nucleus and moves to cytoplasmic organelles possibly belonging to the endolysosomal compartment, as assessed by immunofluorescence, subcellular fractionation and ultrastructural analyses; (ii) secretion of HMGB1 occurs only after HMGB1 relocalization from nucleus to cytoplasm; (iii) the addition of LPC to activated monocytes strongly and rapidly stimulates secretion, which is paralleled by the release of the lysosomal enzymes β-hexosoaminidase and CD, but not of the cytosolic marker LDH; and (iv) consistent with the role of late mediator of inflammation proposed for HMGB1 (Wang et al., 1999), secretion of sPLA2, responsible for LPC generation, is detected 24 h after monocyte activation (a similar activation time is required for the relocalization of HMGB1 from the nucleus to peripheral organelles).
Thus, we postulate that the secretion of HMGB1 requires at least three steps: (i) exit from the nucleus into the cytoplasm, (ii) translocation from the cytosol into cytoplasmic organelles and (iii) exocytosis.
HMGB1 is a molecule of 25 kDa, small enough to diffuse through the nuclear pores. It contains nuclear localization signals and binds to chromatin in a more or less stable way, depending on the acetylation state of the chromatin (Scaffidi et al., 2002). Thus, the relocalization to the cytoplasm in activated monocytes might be due to a differentiation-induced change in the acetylation of some chromatin components. Activated monocytes might either restrict nuclear import or increase nuclear export. In any case, HMGB1 is low in the cytosol of activated monocytes and is concentrated in its cytoplasmic target organelles: this implies that these organelles express on their cytosolic surface a dedicated and efficient translocation machinery, the molecular nature of which deserves further investigation.
The ultrastructural features of HMGB1-containing organelles, their sedimentation coefficient and the partial colocalization with IL-1β suggest that they belong to the compartment of secretory lysosomes. However, as no colocalization is detected with CD, and only partially with LAMP1, it is conceivable that HMGB1 is contained in a specialized subset of secretory lysosomes, different from mature lysosomes. The lack of a fixed structure is a peculiarity of secretory lysosomes, which, in the different cell types, differ from conventional lysosomes both in morphology and content (Blott and Griffiths, 2002). Similarly, IL-1β is contained in endolysosomal-related vesicles, which only in part are positive for lysosomal markers and display less mature morphological features (Andrei et al., 1999). Thus, not only lysosomes but also less mature vesicles may undergo exocytosis, in agreement with previous reports showing that, in some cells, including activated monocytes, also prelysosomal compartments fuse with the plasma membrane upon stimulation (Raposo et al., 1997). Interestingly, secretory lysosomes are present almost exclusively in hemopoietic cells, in keeping with their major role in immune and inflammatory reactions (Blott and Griffiths, 2002). In cells lacking these structures, the exit of HMGB1 from the nucleus leads to cytosolic accumulation of the protein. These cells release HMGB1 when damaged, promoting uncontrolled inflammation (Scaffidi et al., 2002). In contrast, in professional inflammatory cells, such as monocytes, the compartmentalization of HMGB1 in cytoplasmic secretory structures allows the regulated secretion of the protein. Furthermore, the differences in terms of time and stimuli observed in HMGB1 and IL-1β secretion suggest the existence of specific subcompartments within secretory lysosomes of monocytic cells, whose exocytosis is tightly regulated in order to ensure a strict control of the inflammatory reaction.
The microenvironment is important in the modulation of the inflammatory response, which is finely tuned by soluble factors such as ATP and cytokines, osmolarity, bioactive lipids and cell–cell interactions (Andrei et al., 1999; Dart and Chin-Dusting, 1999; Andrews, 2000; Dinarello, 2000). Our observation that LPC induces the release of HMGB1 and lysosomal enzymes is in keeping with previous reports demonstrating a role for this lipid in promoting exocytosis both in yeast and in mammals (Grote et al., 2000; Gilon and Henquin, 2001) and highlights a new mechanism underlying the pro-inflammatory effects of LPC. The enzyme responsible for the generation of LPC from phosphatidylcholine, sPLA2, has been reported to promote exocytosis (Enomoto et al., 2000) and induce release of lysosomal enzymes (Triggiani et al., 2000): whether these effects are direct or mediated by LPC generation is unclear. We show here that the secretion of sPLA2 is delayed by several hours after LPS induction, in agreement with the observation that PLA2 synthesis does not take off until 4–24 h after LPS induction (Ribardo et al., 2001): thus, it is likely that the late secretion of PLA2 leads to a late extracellular accumulation of LPC. This observation, together with the slower kinetics of appearance in endolysosomes of HMGB1 in comparison with IL-1β (Andrei et al., 1999), may provide the explanation for the different kinetics of secretion of the two cytokines: a rapid secretion for IL-1β, induced by ATP, promptly released after activation (Bodin and Burnstock, 1998; Di Virgilio et al., 2001); and a delayed release for HMGB1, triggered by LPC, generated later in the site of inflammation.
Methods
Cells.
Human monocytes were enriched from buffy coats from healthy donors by adherence on plastic plates or glass coverslips and cultured for different periods of time in the presence of 1 μg/ml LPS (Sigma, St. Louis, MO), 50 ng/ml recombinant TNF-α (Genzyme, Cinisello Balsamo, Italy) or IL-1β (NCI Biological Resources Branch, Frederick, MD) in RPMI medium (Biochrom, Berlin, Germany) supplemented with 20% FCS (Gibco, S. Giuliano Milanese, Italy), as described previously (Andrei et al., 1999). To analyze secretion, culture media were replaced with RPMI supplemented with 1% Nutridoma-HU (Sigma) 6 h before harvesting. When indicated, 1 mM ATP, 20 μM LPC, 100 μM LPA or 100 μg/ml PA (all from Sigma) were added to the cultures for the last 10 min. At the end of the incubation time, supernatants were removed and precipitated in 10% TCA; cells were lysed in 1% Triton X-100-containing lysis buffer (Andrei et al., 1999)
Immunofluorescence.
Freshly isolated or activated monocytes, cultured on glass coverslips, were fixed in 4% formaldehyde for 1 h at room temperature prior to detergent extraction with 0.5% Triton X-100, 3 min at 4°C (Degryse et al., 2001). Coverslips were saturated with phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA) for 30 min at room temperature and processed for immunofluorescence with rabbit anti-HMGB1 antibody (Pharmingen, Torrey Pines, CA) followed by FITC-conjugated goat anti-rabbit Ig (Southern Biotechnology Associates, Birmingham, UK). EthD-2 (200 nM; Molecular Probes Europe BV, Leiden, The Netherlands) was added with the secondary antibody to visualize nuclei. Between all incubation steps, cells were washed three times for 5 min with PBS containing 0.2% BSA. In other experiments, double labeling was performed, using rabbit anti-HMGB1 antibody (Pharmingen) together with anti-LAMP1 monoclonal antibodies (IgG1, DSHB, Iowa City, IA) or anti-EEA1 monoclonal antibodies (IgG1, BD Bioscience, Milan, Italy), followed by TRITC-conjugated donkey anti-rabbit Ig (Jackson ImmunoResearch Laboratories, Baltimore Pike, PA) and FITC-conjugated goat anti-mouse Ig (Southern Biotechnology Associates). Coverslips were mounted on slides using Moviol (Sigma). Fluorescence signals were analyzed by confocal microscopy using a Zeiss Invert Laser Scan Microscope (Zeiss, Oberkochen, Germany). Optical slices (0.5 μm) were examined with a 63× oil immersion objective.
Subcellular fractionation.
Resting or 18 h LPS-activated monocytes were homogenized in 250 mM sucrose, 5 mM EGTA, 20 mM HEPES-KOH (pH 7.2) in a Dounce homogenizer and centrifuged at 800 g. Pellets were loaded onto a cushion of 700 mM sucrose in PBS and spun for 15 min at 800 g to purify nuclei. Nuclei were treated with 50 μg/ml DNase (Sigma) for 10 min at 37°C, sonicated and boiled in Laemmli sample buffer. Postnuclear supernatants were subjected to differential ultracentrifugation (50 000 g for 5 min) as described previously (Pitt et al., 1992) to obtain a pellet enriched in endolysosomes (P50). This pellet was treated with 100 μg/ml trypsin (Sigma) for 30 min on ice, in the absence or presence of 1% Triton X-100, and boiled in Laemmli sample buffer. Cytosol was purified from P50 supernatants by 100 000 g ultracentrifugation. The protein content of the different fractions was evaluated by a colorimetric Lowry method (Bio-Rad, Milan, Italy).
Western blot analysis.
Aliquots from the subcellular fractions or TCA-concentrated supernatants or cell lysates were resolved on 12% SDS–PAGE under reducing conditions and electrotransferred onto PVDF filters (Hybond-P, Amersham Pharmacia Biotech, Milan, Italy), which were stained with Ponceau S (Sigma) and de-stained prior to blocking overnight with 5% non-fat dry milk in PBS containing 0.05% Tween (Sigma). Filters were stained with rabbit anti-HMGB1 antibody, anti-CD (IgG2a, Calbiochem, Milan, Italy), anti-IL-1β monoclonal antibody (3ZD, IgG1, NCI Biological Resources Branch) or rabbit anti-sPLA2 antibody (Vinci Biochem, Vinci, Italy) followed by a horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG (DAKO A/S, Denmark) and developed with ECL-Plus (Amersham Pharmacia Biotech) according to the manufacturer's instructions. Densitometric analysis of the blots was performed by analyzing at least three different exposures of the same blot.
Ultrathin cryosections.
Cells and fractions were fixed with 2% paraformaldehyde in 0.1 M phosphate buffer for 2 h at 25°C, washed and embedded in 10% gelatin (Sigma) in 0.1 M phosphate buffer that was solidified on ice. Gelatin blocks were infused with 2.3 M sucrose overnight at 4°C, frozen in liquid nitrogen and cryosectioned. Ultrathin cryosections were collected with sucrose and methyl cellulose and incubated with rabbit anti-HMGB1 antibody followed by 10 nm diameter protein A–colloidal gold conjugates (British BioCell International, Cardiff, UK). In double-labeling experiments, after several washes in PBS-0.1% BSA, the sections were post-fixed with 1.0% glutaraldehyde for 5 min (Gardella et al., 2001) and then incubated with the anti-IL-1β or anti-CD monoclonal antibodies followed by rabbit anti-mouse IgG (DAKO) and 18 nm diameter colloidal gold particles (prepared by the citrate method) conjugated with protein A (Amersham Pharmacia Biotech). Control experiments were performed using as primary antibodies a rabbit pre-immune serum and an unrelated isotype matched (IgG1) monoclonal antibody (both kindly provided by Dr A. Santoni, Rome, Italy). All ultrathin cryosections were finally stained with a solution of 2% methyl cellulose and 0.4% uranyl acetate. Quantitative evaluation of immunolabeling was performed by comparing the number of gold particles in nuclei and cytoplasms of resting or activated monocytes. Results are expressed as percentages of the total gold particles present in nuclei and in cytoplasmic organelles. The areas counted were 47.674 μm2 for resting nuclei; 63.72 μm2 for resting cytoplasm; 32.74 μm2 for activated nuclei; and 58.33 μm2 for activated cytoplasm. Ten images of resting or activated monocyes, randomly photographed from three different immunolabeling experiments, were analyzed.
β-Hexosaminidase and lactate dehydrogenase determination.
LDH release was quantified using a commercially available colorimetric kit (Sigma), according to the manufacturer's instructions; β-hexosaminidase release was quantified according to Storrie and Madden (1990). The release of both enzymes was expressed as a percentage of the total cellular enzyme, according to the following formula: percentage of release=released enzyme/intracellular + released enzyme × 100.
Acknowledgments
We thank Dr A. Santoni and the NCI Biological Resources Branch for the generous gift of antibodies; the Blood Center of Galliera Hospital for providing buffy coats; and Drs F. Belleudi and M. Passalacqua and Prof. E. Melloni for the help with confocal microscopy analysis. This work was supported in part by grants from AIRC, CNR (target project Biotechnology), Ministero Salute, and FIRB. C.A. is supported by a fellowship from FIRC.
References
- Andrei C., Dazzi C., Lotti L., Torrisi M.R., Chimini G. and Rubartelli A. (1999) The secretory route of the leaderless protein IL-1β involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell, 10, 1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews N.W. (2000) Regulated secretion of conventional lysosomes. Trends Cell Biol., 10, 316–321. [DOI] [PubMed] [Google Scholar]
- Bianchi M.E. and Beltrame M. (2000) Upwardly mobile proteins. Workshop: the role of HMG proteins in chromatin structure, gene expression and neoplasia. EMBO rep., 1, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blott E.J. and Griffiths G.M. (2002) Secretory lysosomes. Nat. Rev. Mol. Cell Biol., 3, 122–31. [DOI] [PubMed] [Google Scholar]
- Bodin P. and Burnstock G. (1998) Increased release of ATP from endothelial cells during acute inflammation. Inflamm. Res., 47, 351–354. [DOI] [PubMed] [Google Scholar]
- Dart A.M. and Chin-Dusting J.P. (1999) Lipids and the endothelium. Cardiovasc. Res., 43, 308–22. [DOI] [PubMed] [Google Scholar]
- Degryse B., Bonaldi T., Scaffidi P., Müller S., Resnati M., Sanvito F., Arrigoni G. and Bianchi M.E. (2001) The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell Biol., 152, 1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C.A. (2000) Proinflammatory cytokines. Chest, 118, 503–508. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F., Chiozzi P., Ferrari D., Falzoni S., Sanz J.M., Morelli A., Torboli M., Bolognesi G. and Baricordi O.R. (2001) Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood, 97, 587–600. [DOI] [PubMed] [Google Scholar]
- Enomoto A., Murakami M., Valentin E., Lambeau G., Gelb M.H. and Kudo I. (2000) Redundant and segregated functions of granule-associated heparin-binding group II subfamily of secretory phospholipases A2 in the regulation of degranulation and prostaglandin D2 synthesis in mast cells. J. Immunol., 165, 4007–4014. [DOI] [PubMed] [Google Scholar]
- Gardella S., Andrei C., Lotti L.V., Poggi A., Torrisi M.R., Zocchi M.R. and Rubartelli A. (2001) CD8+ T lymphocytes induce polarized exocytosis of secretory lysosomes by dendritic cells with release of interleukin-1β and cathepsin D. Blood, 98, 2152–2159. [DOI] [PubMed] [Google Scholar]
- Gilon P. and Henquin J.C. (2001) Mechanisms and physiological significance of the cholinergic control of pancreatic β-cell function. Endocr. Rev., 22, 565–604. [DOI] [PubMed] [Google Scholar]
- Grote E., Baba M., Ohsumi Y. and Novick P.J. (2000) Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J. Cell Biol., 151, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabarowski J.H.S., Zhu K., Le L.Q., Witte O. and Xu Y. (2001) Lysophosphatidylcholine as a ligand for the immunoregulatory receptor G2A. Science, 293, 702–705. [DOI] [PubMed] [Google Scholar]
- Müller S., Scaffidi P., Degryse B., Bonaldi T., Ronfani L., Agresti A., Beltrame, M. and Bianchi, M.E. (2001) The double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J., 16, 4337–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt A., Mayorga L.S., Schwartz A.L. and Stahl P.D. (1992). Assay for phagosome–endosome fusion and phagosome protein recycling. Methods Enzymol. 219, 21–31. [DOI] [PubMed] [Google Scholar]
- Quinn M.T., Parthasarathy S. and Steinberg D. (1988) Lyso-phosphatidylcholine: a chemotactic factor for human monocytes and its potential role in atherogenesis. Proc. Natl Acad. Sci. USA, 85, 2805–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G., Tenza D., Mecheri S., Peronet R., Bonnerot C. and Desaymard C. (1997) Accumulation of major histocompatibility complex class II molecules in mast cell secretory granules and their release upon degranulation. Mol. Biol. Cell, 8, 2631–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribardo D.A., Crowe S.E., Kuhl K.R., Peterson J.W. and Chopra A.K. (2001) Prostaglandin levels in stimulated macrophages are controlled by phospholipase A2-activating protein and by activation of phospholipase C and D. J. Biol. Chem., 276, 5467–5475. [DOI] [PubMed] [Google Scholar]
- Rubartelli A. and Sitia R. (1997) Secretion of mammalian proteins that lack a signal sequence. In Kuchler, K., Rubartelli, A. and Holland, B.I. (eds), Unusual Secretory Pathways: From Bacteria to Man. RG Landes, Austin, TX, USA, pp. 87–104. [Google Scholar]
- Rubartelli A., Cozzolino F., Talio M. and Sitia R. (1990) A novel secretory pathway for interleukin-1β, a protein lacking a signal sequence. EMBO J., 9, 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P., Misteli T. and Bianchi M.E. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature, 418, 191–195. [DOI] [PubMed] [Google Scholar]
- Storrie B. and Madden E.A. (1990) Isolation of subcellular organelles. Methods Enzymol., 182, 203–225. [DOI] [PubMed] [Google Scholar]
- Triggiani M., Granata F., Oriente A., De Marino V., Gentile M., Calabrese C., Palumbo C. and Marone G. (2000) Secretory phospholipases A2 induce β-glucuronidase release and IL-6 production from human lung macrophages. J. Immunol., 164, 4908–4915. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science, 285, 248–251. [DOI] [PubMed] [Google Scholar]