

Abstract
The RUNX family represents a small group of heterodimeric transcription factors that master-regulate osteogenesis and hematopoiesis in mammals. Their genetic defects cause human diseases such as cleidocranial dysplasia (CCD) and acute myelogenous leukemia. However, the mechanism(s) regulating their functions are still poorly understood. Here, we report a novel observation that suggests that the osteogenesis-associated homologue RUNX2 is negatively regulated by the phosphorylation of two conserved serines (S104 and S451) in two distinct functional aspects. The phosphorylation of S104 could abolish the heterodimerization of RUNX2 with the partner subunit, PEBP2β, which enhances the metabolic stability of RUNX2. On the other hand, the phosphorylation of S451 resides within the C-terminal transcription inhibition domain of RUNX2 and hence is implicated in its functional mobilization. One CCD mutation, S104R of RUNX2, appears to mimic the phosphorylation-dependent inhibition of heterodimerization, thereby rendering RUNX2 metabolically unstable.
Introduction
The Runt domain transcription factors are composed of a larger DNA-binding subunit, α, and a smaller non-DNA-binding subunit, β (Ito, 1999). There are three mammalian genes encoding the α subunit, termed Runt-related genes 1, 2 and 3 (RUNX1, RUNX2 and RUNX3). Runx1 is essential for the generation of hematopoietic stem cells (Okuda et al., 1996; Wang et al., 1996). Mutations and translocations involving RUNX1 are frequently associated with acute leukemia (Look, 1997). Runx2 is essential for the generation of osteoblasts, and its homozygous disruption eliminates bone formation in mice (Komori et al., 1997; Otto et al., 1997). Furthermore, heterozygous mutations in RUNX2 cause the human autosomal dominant bone disease, cleidocranial dysplasia (CCD) (Mundlos et al., 1997). On the other hand, there is only one mammalian gene encoding the β subunit, termed PEBPB2/CBFB (Ito, 1999). PEBPB2 is frequently associated with acute leukemia through its translocation-mediated lesion (Look, 1997).
RUNX proteins consist of several functional modules: the Runt domain, transcription activation domains and inhibition domains (IDs) (Kanno et al., 1998; Zhang et al., 2000). The Runt domain, named after the Drosophila runt gene, is an evolutionarily conserved 128-amino-acid region that is responsible for both DNA binding and heterodimerization with PEBP2β (Ito, 1999). Although PEBP2β does not itself contact with DNA, it allosterically enhances DNA binding by RUNX proteins upon heterodimerization. In addition, we have recently found that PEBP2β has another important role in stabilizing RUNX proteins against proteolytic degradation by the ubiquitin–proteasome system (Huang et al., 2001). The heterodimerization, therefore, is a centrally important regulatory mechanism for the in vivo function of RUNX proteins.
Although the biochemical and biological functions of each subunit have been extensively characterized, the mechanisms controlling these functions are still poorly understood. We are interested in the potential importance of phosphorylation-mediated protein modifications. Previously, RUNX1 has been shown to undergo extracellular signal-regulated kinase (ERK)-dependent phosphorylation with a concomitant stimulation of its transactivation ability (Tanaka et al., 1996). With RUNX2 as the focus of analysis, we describe the identification of several conserved serine residues that are constitutively phosphorylated in cultured cells under ordinary growth conditions. Of these sites, two in particular were implicated in the negative regulation of the RUNX2 functions.
Results
To investigate the phosphorylation of RUNX2, we expressed its full-length and deletion constructs (Figure 1A) in HEK293 cells and isolated them by immunoprecipitation after labelling with 32P (Figure 1B). In the present study, the RUNX2 isoform with the N-terminal sequence MRIPV was used throughout (SWISS-PROT, NP-004339). Prominent labelling was observed with the full-length RUNX2 and two segments containing amino acids 1–376 and 425–507 (Figure 1B, lanes 5, 6, 4, respectively). The remaining two segments were labelled only weakly (214–340, lane 2) or almost not at all (341–424, lane 3). These labelling patterns suggested the presence of putative phosphorylation sites in the two regions 1–215 and 425–507. Phosphoamino acid analysis of full-length RUNX2 revealed that the phosphorylated sites consisted solely of serine (Figure 1D).
Figure 1.
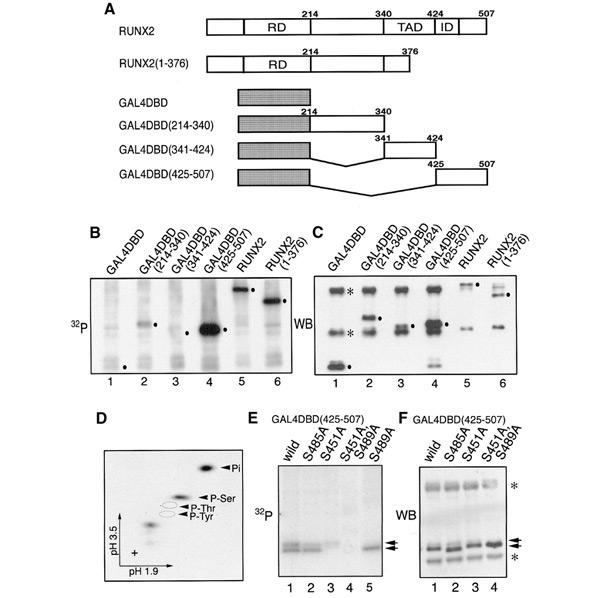
Phosphorylation of RUNX2. (A) Diagrammatic representation of RUNX2 and its derivatives. RD, Runt domain; TAD, transcription activation domain; ID, inhibition domain. (B) HEK293 cells transfected with expression plasmids of RUNX2 or its derivatives were labelled with 32P. Proteins were immunoprecipitated with either anti-GAL4DBD or anti-PEBP2αA(8G5). (C) Western blot, as described in Huang et al. (2001), of the membrane shown in (B) with the mixture of anti-GAL4DBD and anti-PEBP2αA(8G5). Dots indicate the expected positions of expressed proteins. Immunoglobulin heavy and light chains are marked with asterisks. (D) Phosphoamino acid analysis of the full-length RUNX2 protein shown in (B). (E) HEK293 cells transfected with expression plasmids as indicated were labelled with 32P and the fusion proteins immunoprecipitated with anti-GAL4DBD. (F) Western blot of the membrane shown in (E) using anti-GAL4DBD. The expressed proteins of the indicated plasmids are marked with arrows. Immunoglobulin heavy and light chains are marked with asterisks.
Within the C-terminal region (425–507), there are as many as 16 serine residues. By serial alanine-scanning mutation analyses on these serine residues (data not shown), we narrowed down the potential phosphorylation sites to three residues, S451, S485 and S489. To confirm the involvement of these serines in phosphorylation, we substituted them to alanine either separately or together in the context of the 425–507 region fused to GAL4 DNA-binding domain (DBD). With no mutation, the radiolabelled product(s) of this construct generated doublet bands upon mild exposure (Figure 1E, lane 1). Of these bands, the upper moderate one was partially weakened by either S485A (lane 2) or S451A (lane 3) but completely disappeared in S489A (lane 5), whereas the lower strong one was totally abolished by S451A (lanes 3 and 4). Furthermore, both these bands were simultaneously abolished in a double mutant, S451A/S489A (lane 4). Taken together, these results suggest that the upper band may contain multiple phosphorylated proteins (S489; S489,S451; S489,S485; and S489,S451,S485) and that their retarded mobilities resulted from the phosphorylation of S489. Conversely, the lower band contains only S451 phosphorylated protein. Considering the relationship of these phosphorylations, those between S489 and S451 may be independent events, but the phosphorylation of S485 requires prior phosphorylation of S489, and the hierarchical protein kinase GSK3 may be involved in this.
Interestingly, the serines at 451 and 489 reside in the evolutionarily conserved Ser-Pro motifs among RUNX proteins and hence may serve as substrates for proline-directed protein kinases such as MAPK, cdc2 and GSK3 (Figure 2A). Similar such Ser-Pro motifs are also present at two positions in the 1–215 region, S14 and S104. To examine directly whether these four serine residues are actually involved in phosphorylation, we overexpressed full-length RUNX2 proteins mutated at the respective sites with COS-7 cells as host and subjected them to two-dimensional tryptic phosphopeptide analysis after isolation immunoprecipitation followed by SDS–PAGE. In the case of the wild-type RUNX2, eight distinguishable spots were detected (Figure 2C). Notably, the same sets of spots with similar relative intensities were also observed with RUNX2 endogenously produced in an osteosarcoma cell line, SAOS-2 (Figure 2B). This excluded the possibility that the observed phosphorylation pattern was an artefact due to the forced expression of RUNX2. When RUNX2 was mutated, one or two spots were abolished in a manner specific to each serine residue changed: S451A, a and b; S14G, c; S104G, d (Figure 2D–F). The apparent intensity of phosphorylation is highest at S451 and lowest at S104 in both SAOS-2 cells and COS-7 cells. In this assay, however, it is likely that the intrinsic rate of phosphorylation at S104 is underestimated, because RUNX2 would undergo a rapid degradation upon this phosphorylation event, as described below. Although we also examined the mutant RUNX2 carrying S489A, no significant effect was observed on the phosphopeptide pattern or the transactivation ability to be addressed below (data not shown for either aspect). This suggests that S489 might be masked from phosphorylation in the context of full-length RUNX2.
Figure 2.
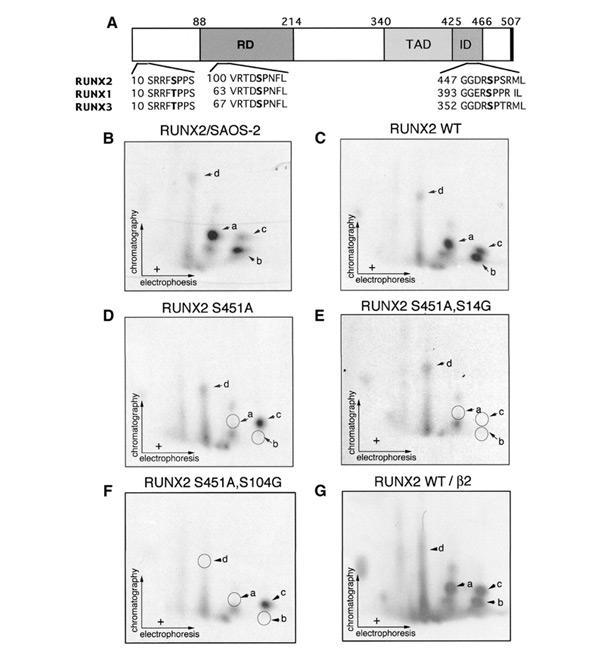
RUNX2 is phosphorylated at the conserved proline-directed serines. (A) Diagram showing the positions of the conserved serine followed by proline in RUNX2, together with surrounding amino acid sequences of RUNX1, RUNX2 and RUNX3. The numbering of the RUNX protein is based on the MRIPV forms, the accession numbers of which in SWISS-PROT are Q01196 (RUNX1), NP-004339 (RUNX2) and Q13761 (RUNX3). RD, Runt domain; TAD, transcription activation domain; ID, inhibition domain. The delineation of functional domains (TAD and ID) were from Kanno et al. (1998) and Zhang et al. (2001). (B) SAOS-2 human osteosarcoma cells were labelled with 32P. The immunopurified endogenous RUNX2 was subjected to phosphopeptide mapping analysis. (C–G) COS-7 cells were transfected with RUNX2 or its mutants and labelled with 32P. RUNX2 proteins were immunoprecipitated with anti-PEBP2αA(8G5) and subjected to autoradiography. (C) Two-dimensional tryptic phosphopeptide analysis of the wild-type RUNX2. Since the peptide map shown here and that shown in (B) is virtually identical and COS-7 cells do not express the endogenous RUNX2 protein, phophopeptide analyses for RUNX2 mutants were performed with materials obtained from COS-7 cells. (D–G) Two-dimensional tryptic phophopeptide anaylses of RUNX2(S451A), RUNX2(S451A,S14G), RUNX2(S451A,S104G) and wild-type RUNX2 coexpressed with PEBP2β2.
In order to assess the biological significance of the phosphorylation of the three serine residues, we first tested the effects of their mutations on the ability of RUNX2 to transactivate the osteocalcin promoter (Ducy and Karsenty, 1995). We tested two types of mutants, one substituted with glycine or alanine and the other with glutamic acid. As shown in Figure 3A, transactivation by the wild-type RUNX2 was strongly stimulated when PEBP2β was coexpressed (lanes 2 and 10). RUNX2(S14G) and RUNX2(S14E) showed nearly the same activity as the wild-type RUNX2 (lanes 3, 4, 11 and 12), suggesting that phosphorylation at this residue has no significant functional impact, at least in the present assay. S104G and S104E, on the other hand, showed drastically reduced activities (lanes 5 and 6). Interestingly, the coexpression of PEBP2β restored the activity of RUNX2(S104G) (lane 13) but not that of RUNX2(S104E) (lane 14). Since S104 is located within one of the important contact sites with PEBP2β (Bravo et al., 2001), its substitution to a bulkier, acidic glutamic acid residue may prevent the association of RUNX2 with PEBP2β in a manner mimicking the phosphorylation of S104. On the other hand, the substitution of S104 to the side-chain-less amino acid, glycine, may be compatible with heterodimerization, though less favourable than normal. If these interpretations are correct, phosphorylation of S104 should be inhibited in a heterodimer form. Unfortunately, the available anti-β antibody does not react with PEBP2β when it is complexed with RUNX2. Therefore, we tested whether the phosphorylation of S104 would be suppressed upon overexpression of PEBP2β. Indeed, this prediction was directly verified by phosphopeptide mapping, as presented in Figure 2G.
Figure 3.
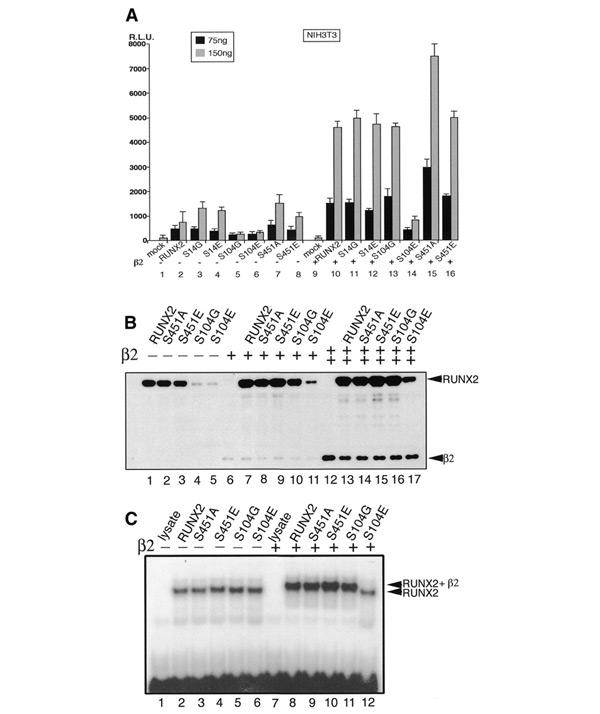
Transcription activation and heterodimerization activities of RUNX2 mutated on phophorylation sites. (A) NIH 3T3 cells were transfected with osteocalcin reporter plasmid (200 ng) together with 75 or 150 ng per well of the expression plasmids for RUNX2 or its mutated forms in the absence or presence of PEBP2β2 (100 ng). Luciferease activities are plotted. S14G indicates RUNX2(S14G). (B) Western blot of the extracts of COS-7 cells transfected with the expression plasmids for RUNX2 or its mutated forms in the absence (lanes 1–5) or presence of lower (lanes 6–11) or higher (lanes 12–17) amounts of PEBP2β. Monoclonal antibodies against PEBP2β and RUNX2 were used to detect respective proteins. (C) Electrophoretic mobility shift assay for in vitro translated RUNX2 or its mutated forms in the absence (lanes 1–6) or presence (lanes 7–12) of the purified PEBP2β.
In the case of RUNX2(S451A), the transactivation activity was significantly higher (1.5- to 2-fold) than that of the wild type. Since S451 is located within the transcription inhibitory region (ID; Kanno et al., 1998; Zhang et al., 2000), its phosphorylation could be linked to the ID activity. Accordingly, the mutation of S451 to alanine would abolish the ID activity, leaving the protein to be constitutively active. On the other hand, its substitution to glutamic acid would mimic the phosphorylated state of S451 and thus keep the activity at about the wild-type level.
We then examined the level of expression of the wild-type and mutant RUNX2 proteins by western blotting. The wild-type RUNX2, RUNX2(S451A) and RUNX2(S451E) were detected at about the same level (Figure 3B, lanes 1–3). In contrast, the amounts of RUNX2(S104G) and RUNX2(S104E) were markedly reduced (lanes 4 and 5). When PEBP2β was coexpressed, however, the amounts of the wild-type RUNX2, RUNX2(S451A) and RUNX2(S451E) increased significantly (lanes 7–9). Interestingly, the amounts of RUNX2(S104G) was also dramatically increased by the coexpression of PEBP2β to about the wild-type level (lanes 10 and 16), suggesting again that RUNX2(S104G) retains a reduced but still significant ability to interact with PEBP2β. However, RUNX2(S104E) continued to be less stable than normal, even at the highest dosage of PEBP2β, indicating that this mutant is severely defective in heterodimerization and hence becomes very unstable (lanes 11 and 17).
To confirm that the decrease in the stability of RUNX2(S104E) is due to the defect of the heterodimerization activity with PEBP2β, we conducted an electrophoretic mobility shift assay (EMSA) (Figure 3C). For the normalization of protein amounts, we used in vitro translated proteins in the EMSA instead of the proteins of RUNX2 variants from transfected cells. The wild type and all the mutant proteins bound to DNA to similar extents (lanes 2–6) in the absence of PEBP2β. When PEBP2β (type 2 isoform) was added in a saturating amount, all except RUNX2(S104E) showed supershifts, indicating that they can interact with PEBP2β (lanes 8–11). In contrast, RUNX2(S104E) did not show any supershift (lane 12). It is interesting to note that S104 is mutated to arginine in one CCD mutation (Quack et al., 1999). We compared RUNX2(S104G), RUNX2(S104E) and RUNX2(S104R) for their transactivation abilities using the same osteocalcin promoter as before with a human osteosarcoma cell line, HOS, as a more legitimate host, since HOS represents an early osteoblastic precursor showing a low endogenous expression of RUNX2 (Figure 4A). The transcriptional activation by RUNX2(S104G) was much lower than normal at no dose or a low dose of PEBP2β, but it reaches a plateau equivalent to the wild-type control at a high dose. On the other hand, RUNX2(S104E) and RUNX2(S104R) scarcely responded to PEBP2β, indicating that the substitution of S104 by arginine is as deleterious as that by glutamic acid. The EMSA also clearly demonstrated that both S104R and S104E almost completely abolish the heterodimerization activity of RUNX2 (Figure 4B). Consistent with these observations, the half-lives of RUNX2(S104R) and RUNX2(S104E) in the cells were markedly shorter (<90 min) than that of the wild-type RUNX2 (>180 min), even when PEBP2β was coexpressed (Figure 4C).
Figure 4.
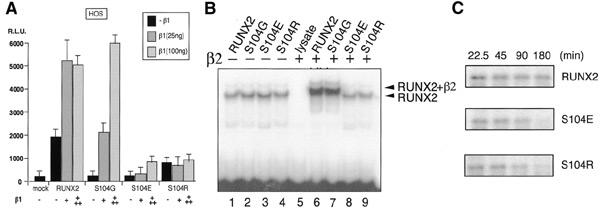
Transactivation and heterodimerization activities of one CCD mutant, RUNX2(S104R). (A) HOS cells were transfected with osteocalcin luciferase reporter (200 ng) together with the expression plasmids for RUNX2 (100 ng) and its mutated forms in the presence of 25 or 100 ng of PEBP2β1 plasmids. Relative luciferase activities are plotted. (B) Electrophoretic mobility shift assay for in vitro translated RUNX2 or its mutated forms in the absence (lanes 1–4) or presence (lanes 5–9) of the purified PEBP2β. (C) HOS cells were transfected with RUNX2, RUNX2(S104E) or RUNX2(S104R) expression plasmids together with the expression plasmids PEBP2β, labelled with [35S]methionine for 1 h and chased with unlabelled methionine for indicated lengths, as described in Huang et al. (2001).
In an attempt to find the signals involved in these phosphorylation events, we examined whether there is any change in the phosphorylation status of S104 or S451 on RUNX2 during osteoblastic differentiation. For this experiment, we used the mouse C2C12 multipotent mesenchymal precursor cell line, which is known to undergo osteoblastic differentiation by induction with BMP-2 (Katagiri et al., 1994). At an appropriate dose, BMP-2 induces various osteoblastspecific differentiation markers in a sequential manner. An early marker, alkaline phosphatase, begins to appear on day 2 and is expressed in almost all the cells on day 6. On the other hand, a late marker, osteocalcin, is strongly induced from day 3 and its expression is sustained thereafter (Katagiri et al., 1994). In the present experiment, the cells were treated with BMP-2 at 300 ng/ml for 8 days and then labelled with [32P]orthophosphate at 1 mCi/ml for 5 h. After immunoprecipitation and blotting, the bands containing phosphorylated RUNX2 proteins were cut out and subjected to phosphopeptide mapping analysis. The result presented in Figure 5 shows that, during the differentiation process, there was no significant change in the phosphorylation status of S457 on RUNX2 (spots a and b in Figure 5B and C), which corresponds to S451 of human RUNX2 proteins. On the other hand, the phosphorylation of S110 corresponding to S104 of human RUNX2 was moderately increased during the differentiation (spot d in Figure 5B and C). These results suggest that two independent pathways might be involved in regulating the phosphorylation of S451 and S104, respectively.
Figure 5.
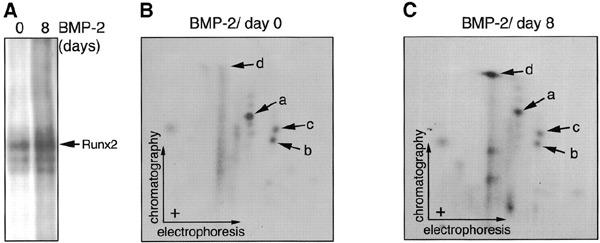
Changes in the phosphorylation pattern of RUNX2 in C2C12 cells during BMP-2-induced osteoblatic differentiation. (A) C2C12 mouse multipotent mesenchymal cells were treated with BMP-2 at 0 or 300 ng/ml for 8 days. These cells were labelled with [32P]orthophosphate at 1 mCi/ml for 5 h in the absence or presence of BMP-2. Labelled endogenous RUNX2 proteins were immunoprified. (B and C) Two-dimensional tryptic phosphopeptide analysis of samples in (A) was performed as described in Figure 2.
Discussion
In this study, we demonstrated that RUNX2 is phosphorylated at evolutionarily conserved serines preceding proline (S14, S104 and S451). Of these, S104 and S451 were implicated in the negative regulation of the RUNX2 functions and probably also the RUNX1 and RUNX3 functions in two distinct functional contexts, respectively, although we have no direct experimental evidence for this supposition yet.
First, it is suggested that the phosphorylation of S104 abolishes the heterodimerization of RUNX2 with the obligatory partner subunit, PEBP2β, which is required not only for enhancing DNA binding by RUNX proteins, but also for protecting them against the ubiquitin–proteasome-mediated degradation. In conjunction with the proteolytic machinery, this phosphorylation event would act as a mechanism to accentuate the dependency of the RUNX function on the intracellular availability of PEBP2β. Indeed, we observed a good consistent correlation between the intracellular level of RUNX2 and its apparent transactivation ability when the wild-type RUNX2 and different S104 mutants were coexpressed with PEBP2β at varying dosages. This corroborates and further extends our previous proposal that the PEBP2β is a crucial determinant of the RUNX activity in vivo (Huang et al., 2001). Moreover, a mutation hitting this very site, S104R, has been identified in one CCD patient. S104R, as well as an artificially generated mutant, S104E, caused a nearly total loss of the heterodimerization ability, plausibly mimicking a phosphorylated serine. The destruction of heterodimerization appears to be one effective way to impair the function of RUNX2 to cause the disease.
Secondly, the phosphorylation of S451, which resides within the ID region, could act to downregulate the RUNX2-dependent transactivation. This is somewhat similar to the case of HSF1, in which the phosphorylation of two specific proline-directed serines in the regulatory domain results in repression at control temperature (Knauf et al., 1996). The repressive effect of the corresponding ID region in RUNX1 is higher in cultured cells of lymphoid and myelogenous lineages than in an undifferentiated embryonal cell line, P19 (Kanno et al., 1998). These observations suggest that the putative S451-linked regulation might work as a mechanism to keep the RUNX2 activity low until an appropriate time either during the process of cell differentiation or in the cell cycle.
To further understand the mechanisms and biological significance of the observed phosphorylation events, we have been investigating the potential signal(s) or kinase(s) involved. Because the MAPK family is very well known for its regulation on various transcription factors through the phosphorylation of Ser-Pro motifs, we have examined whether the transactivation ability of RUNX2 might be affected by the cotransfection of well-known members of the proline-directed kinases, ERK1, p38 and JNK1, or their regulatory proteins, raf, MKK4 and MKK7. However, no differential responses were observed between the wild-type RUNX2 and the mutants substituted at S104 or S451, indicating that those extracellular signal-dependent kinases would not be responsible.
As another possibility, we examined whether differentiation would influence the phosphorylation status of S104 or S451 on RUNX2. Interestingly, the osteoblastic differentiation induced by BMP-2 could increase the phosphorylation of S110 but not S457 on RUNX2, which corresponds to S104 or S451 of human RUNX2 proteins, respectively. These data suggest the existence of at least two independent pathways in regulating the phosphorylation of S451 and S104, respectively. Because it is conceivable that the transcriptional activity of RUNX2 is enhanced to transcribe large amounts of differentiation marker mRNA during the differentiation, it seems paradoxical that the phosphorylation of S104 of RUNX2 was increased during the differentiation, in contrast to the expectation that the osteoblastic differentiation would inhibit this phosphorylation. However, it has been reported that transcription factors are often very unstable when they are in the transcription-competent state. Accordingly, the phosphorylation of S104 might occur on the DNA region where the transcription is actively progressed: the phosphorylation of S104 may be transcription-coupled. Therefore, we suppose that a kinase like CDK7, which is a component of TFIIH, might be involved in the phosphorylation of S104.
There are various other osteogenic signals that could affect these two phosphorylation events as an alternative or additional regulatory mechanism. Considering the tight conservation of the corresponding serine residues in all three RUNX paralogues, we suspect that a common regulatory process such as the cell cycle might be related to these phosphorylation events. Interestingly, S451 resides in a typical phosphorylation site of the cdc2/CDK1 family, S/TPXR/K. Moreover, in the cases of RUNX1 and RUNX3, there is an additional consensus sequence for the CDK family, which might make each RUNX member differentially sensitive to various CDK members. Accumulating studies about the RUNX family reveal the new emerging aspect that this family might be linked to oncogenesis, in addition to their well-known roles, the specifications of cell lineages. In the case of RUNX1, it is well known that haploinsufficiency of RUNX1 and, more frequently, the chromosomal translocations of RUNX1 are implicated in the development of acute myeloid leukemia. Furthermore, our recent report showed that the blocking of RUNX3 function is linked to the progression of gastric cancer (Li et al., 2002). It will be worth investigating whether the enhanced CDK activity in cancer negatively regulates the transcriptional activity of RUNX proteins through the conserved Ser-Pro motifs described in this paper.
RUNX2 contains further phosphorylation sites, the locations and functional effects of which remain to be defined. We await explorations into the whole repertoire of phosphorylation-mediated regulation acting on RUNX proteins. In addition, searches for pharmacological modulators of these phosphorylation reactions may provide novel therapeutics to treat CCD or other RUNX-associated diseases.
Methods
Reagents and plasmids.
Mouse monoclonal anti-PEBP2αA (8G5) and anti-β (clone122) antiserum were generated by using bacterially produced recombinant full-length RUNX2 (Zhang et al., 2000) and PEBP2β (amino acids 1–141) proteins, respectively. Monoclonal anti-GAL4DBD (RK5C1) was purchased from Santa Cruz Biotechnology, monoclonal anti-tubulin from Roche Molecular Biochemicals, polyclonal anti-luciferase from Biogenesis, protein A and G immobilized on beads from Pharmacia, and recombinant human BMP-2 from Genetic Institute. The pEF-RUNX2, pEF-RUNX2 (1–376), pEF-β1, pEF-β2 and pEF-GAL4DBD series (RUNX2 214–340, 341–424 and 425–507) have been described previously (Kanno et al., 1998; Zhang et al., 2000). The reporter plasmid p147-luc which contains the sequence extending from −147 to +13 of the mouse osteocalcin gene 2 (mOG2) promotor region has been described previously (Ducy and Karsenty, 1995). The various serine mutants were generated by PCR mutagenesis using Pfu polymerase (Stratagene).
Cell culture and transfection.
NIH 3T3, HOS, HEK293 and COS-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). SAOS-2 HOS cells and C2C12 mouse multipotent mesenchymal cells were cultured in DMEM supplemented with 15% FBS. Transfection of cells with plasmids was carried out by using FuGENE 6 (Roche Molecular Biochemicals) according to the manufacturer's recommendations.
Reporter assay.
Cells were lysed 36 h after transfection of NIH 3T3 or HOS cells, and luciferase assays were performed using dual luciferase reagents (Promega).
In vivo phosphorylation and immunoprecipitation.
SAOS-2 or transiently transfected HEK293 or COS-7 cells were labelled for 6 h with 0.75 mCi/ml [32P]orthophosphate in phosphate-free DMEM supplemented with 10% dialysed FBS. Cell lysates were prepared, and RUNX2 was immunoprecipitated and transferred to an Immobilon-P membrane (Millipore). Membranes were subjected to immunoblotting or autoradiography. The membrane region harbouring radioactive RUNX2 was excised and subjected to phosphoamino acid analysis or phosphopeptide mapping as described below.
Phosphopeptide mapping.
The excised membrane was incubated with TPCK-trypsin (Sigma). Digested products were oxidized with performic acid for 1 h at 4°C, followed by lyophilization, and applied to a cellulose-coated TLC plate. Electrophoresis in the first dimension was performed in pH 1.9 buffer for 40 min at 1 kV, and ascending liquid chromatography for the second dimension was performed in the regular chromatography buffer for 7 h.
Electrophoretic mobility shift assay (EMSA).
RUNX2 and its point mutants were in vitro translated using the TNT reticulocyte-lysate system (Promega). That all proteins were produced in similar amounts was confirmed by western blotting. PEBP2β2 protein was produced and purified from Escherichia coli. The annealed double-stranded oligonucleotide derived from polyomavirus enhancer (5′-CATGGTAACTGACCGCAGAGGGC-3 ′ and the complementary strand) was end-labelled with 32P by T4 kinase.
Acknowledgments
We thank N. Nozaki, K. Sasaguri and Y. Yamagi for producing monoclonal anti-β antibody (clone122), and G. Karsenty for p147-luc plasmid. This work was supported in part by grant-in-aid 12213058 for Priority Areas in Cancer Research and grant-in-aid 12309005 for Scientific Research (A) (to Y.I.) from the Ministry of Education, Culture, Science, Sports and Technology of Japan.
References
- Bravo J., Li Z., Speck N.A. and Warren A.J. (2001) The leukemia-associated AML1 (Runx1)–CBFβ complex functions as a DNA-induced molecular clamp. Nat. Struct. Biol., 8, 371–378. [DOI] [PubMed] [Google Scholar]
- Ducy P. and Karsenty G. (1995) Two distinct osteoblastspecific cis-acting elements control expression of a mouse osteocalcin gene. Mol. Cell. Biol., 15, 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Shigesada K., Ito K., Wee H.J., Yokomizo T. and Ito Y. (2001) Dimerization with PEBP2β protects RUNX1/AML1 from ubiquitin–proteasome-mediated degradation. EMBO J., 20, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y., (1999) Molecular basis of tissuespecific gene expression mediated by the Runt domain transcription factor PEBP2/CBF. Genes Cells, 4, 685–696. [DOI] [PubMed] [Google Scholar]
- Kanno T., Kanno Y., Chen L.F., Ogawa E., Kim W.Y. and Ito Y. (1998) Intrinsic transcriptional activation–inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor β subunit revealed in the presence of the α subunit. Mol. Cell. Biol., 18, 2444–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T. et al. (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol., 127, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U., Newton E.M., Kyriakis J. and Kingston R.E. (1996) Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev., 10, 2782–2793. [DOI] [PubMed] [Google Scholar]
- Komori T. et al. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell, 89, 755–764. [DOI] [PubMed] [Google Scholar]
- Li Q.L. et al. (2002) Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell, 109, 113–124. [DOI] [PubMed] [Google Scholar]
- Look A.T. (1997) Oncogenic transcription factors in the human acute leukemias. Science, 278, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Mundlos S. et al. (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell, 89, 773–779. [DOI] [PubMed] [Google Scholar]
- Okuda T., van Deursen J., Hiebert S.W., Grosveld G. and Downing J.R. (1996) AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell, 84, 321–330. [DOI] [PubMed] [Google Scholar]
- Otto F. et al. (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell, 89, 765–771. [DOI] [PubMed] [Google Scholar]
- Quack I. et al. (1999) Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am. J. Hum. Genet., 65, 1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. et al. (1996) The extracellular signal-regulated kinase pathway phosphorylates AML1, an acute myeloid leukemia gene product and potentially regulates its transactivation ability. Mol. Cell. Biol., 16, 3967–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Stacy T., Binder M., Marin-Padilla M., Sharpe A.H. and Speck N.A. (1996) Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl Acad. Sci. USA, 93, 3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.W. et al. (2000) A RUNX2/PEBP2A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc. Natl Acad. Sci. USA, 97, 10549–10554. [DOI] [PMC free article] [PubMed] [Google Scholar]