

Abstract
Cholera toxin travels from the cell surface of affected mammalian cells to the endoplasmic reticulum (ER), where the A1 chain is released and retro-translocated across the ER membrane into the cytosol. We have tested whether, as in other cases, retro-translocation requires poly-ubiquitination. We show that an A1 chain mutant that lacks lysines and has a blocked N-terminus, and therefore cannot be ubiquitinated, remains active in vivo. The A1 chain is not degraded in the cytosol, as demonstrated by the fact that proteasome inhibitors do not stimulate its activity. When additional lysines are introduced into the A1 chain, moderate degradation by the proteasome is observed. The unfolded A1 chain rapidly refolds in vitro. These results show that poly-ubiquitination is not required for retro-translocation of all proteins across the ER membrane and indicate that the reason why the toxin escapes degradation in the cytosol may be both its paucity of lysines and its rapid refolding.
Introduction
Cholera toxin (CT) is made and secreted by the Gram-negative bacterium Vibrio cholerae. It consists of a ring of five B subunits and a single A subunit that is cleaved into A1 and A2 chains. The holotoxin is taken up by human intestinal cells and travels backwards along the secretory pathway to the endoplasmic reticulum (ER) (Lencer, 2001). Here, the holotoxin is disassembled by the action of protein disulfide isomerase (PDI) (Tsai et al., 2001). In its reduced form, PDI binds to the A1 chain, unfolds it and releases it from the rest of the toxin. Upon oxidation of a disulfide bridge of PDI, the A1 chain dissociates. The next step is not entirely clear, but eventually the A1 chain is transferred across the ER membrane into the cytosol, a process called retro-translocation. There is evidence that retro-translocation of the A1 chain occurs through the Sec61p channel (Schmitz et al., 2000). Once in the cytosol, the A1 chain refolds into an active enzyme that ADP-ribosylates a trimeric G protein, which in turn activates adenylyl cyclase, raises cAMP and opens chloride channels at the plasma membrane (Lencer, 2001). The resulting secretion of chloride and water leads to massive diarrhea.
The pathway of retro-translocation of the A1 chain probably co-opts the cellular route used by misfolded proteins. When proteins are transported from the cytosol into the ER and cannot reach their native conformation, they are often retro-translocated into the cytosol and degraded by the proteasome. Toxins are not the only molecules that hijack this cellular pathway. For example, two proteins from the human cytomegalovirus (HCMV), called US2 and US11, trigger retro-translocation and degradation of the heavy chain of the MHC class I molecule (Wiertz et al., 1996).
Subsequent degradation of these proteins in the cytosol requires poly-ubiquitination, which serves as a signal for recognition by the proteasome. In addition, poly-ubiquitination may be required for retro-translocation per se. For example, when poly-ubiquitination is prevented in a permeabilized cell system or in certain yeast mutants, substrates that are normally exported remain inside the ER (Biederer et al., 1996; Hiller et al., 1996; Shamu et al., 2001).
In contrast to other substrates of the retro-translocation pathway, CT and related toxins are not degraded in the cytosol, at least not completely. How do they escape cytosolic degradation? It has been observed that all ER-directed toxins contain relatively few lysine residues that serve as attachment sites for poly-ubiquitin chains (Hazes and Read, 1997). However, it is unclear whether the paucity of lysines is the full explanation, because some toxins, including CT, do contain lysines and because the N-terminus might also serve as a poly-ubiquitination site. In other cases, even a single poly-ubiquitin chain has been shown to be sufficient for proteasomal degradation of a protein (Breitschopf et al., 1998; Thrower et al., 2000). This is particularly relevant for retro-translocation substrates, which are likely transported in an extended conformation through the Sec61p channel and probably arrive in the cytosol in a non-native state.
Here, we address the role of poly-ubiquitination in the retro-translocation pathway of CT. We show that the modification is not required for the movement of the A1 chain into the cytosol. In addition, our results suggest that the reason why the toxin escapes degradation in the cytosol is not only the paucity of lysines and thus of poly-ubiquitination sites, but also because it can rapidly and spontaneously refold.
Results
Retro-translocation of the CTA1 chain to the cytosol does not depend on poly-ubiquitination or proteasome function
Initial studies showed that proteasome inhibitors had no detectable effect on the ability of wild-type (wt) CT to induce a Cl− secretory response (Isc) in human intestinal T84 cells (Figure 1A). Three different proteasome inhibitors (Z-L3VS, lactacystin and MG132) were tested and had identical results. These data indicate that the proteasome does not affect CT action and imply that the A1 chain may not be degraded, perhaps because it is not poly-ubiquitinated.
Figure 1.
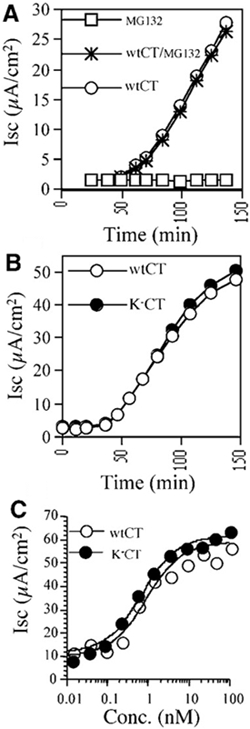
CT activity in vivo does not depend on poly-ubiquitination or proteasome function. (A) Time-course of CT-induced Cl− secretion (Isc) in T84 cells treated with MG132/DMSO, DMSO alone or MG132 but not CT. (B) Time-course of Isc induced by 0.5 nM CT and the lysine-less K−CT variant. (C) Dose response for peak Isc induced by CT or K−CT.
To test whether poly-ubiquitination is involved in retro-translocation of the toxin, we prepared a lysine-less CT variant by substituting arginine for lysine, at positions 4 and 17 in the CTA1 chain (designated K−CT). When tested in vivo, K−CT induced a Cl− secretory response with a time-course and dose-response identical to that of CT (Figure 1B and C). Pretreatment of T84 cells with MG132 had no effect on the activity of the K−CT variant (data not shown). These results indicate that lysines 4 and 17 in CTA1 cannot be required for retro-translocation and that neither sites are poly-ubiquitinated in vivo.
The only other primary amine in the CTA1 chain that may be ubiquitinated is at its N-terminus. To rule out this possibility, we blocked the N-terminus of the lysine-less variant by carbamylation. N-terminal blockade was confirmed by three methods. First, automated Edman degradation sequencing identified the predicted N-terminal residues of the K−CTA1 chain but failed to identify the N-terminal residues of the carbamylated A1 chain (data not shown). Secondly, the carbamylated K−CT variant did not serve as substrate for reaction with N-hydroxysulfosuccinimide (NHS) esters, indicating that all primary amino groups, including the N-terminus, were blocked (Figure 2A, compare lanes 1 and 2 with 3 and 4). And thirdly, stoichiometric carbamylation of the N-terminal asparagine residue was confirmed by mass spectrometry (Figure 2B and C).
Figure 2.
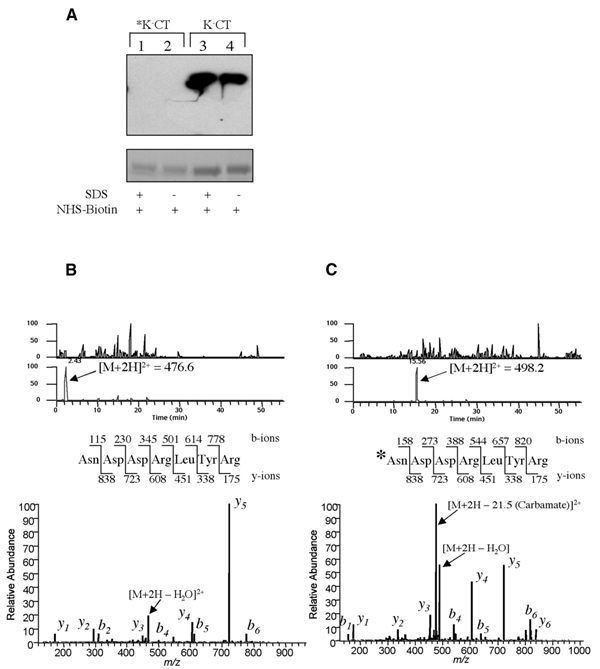
Complete blockade of the N-terminus of the CTA1 chain. (A) Upper: non-reducing SDS–PAGE and avidin-HRP blot of biotin-conjugated K−CT blocked by carbamylation (asterisk, lanes 1 and 2) or unblocked (lanes 3 and 4). Biotinylation was performed in 0.4% SDS (lanes 1 and 3) or in aqueous buffer (lanes 2 and 4). Lower: Coomassie staining of an equivalent gel shows comparable sample loading. (B) The N-terminal peptide of the unblocked K−CT was identified by mass spectroscopy analysis as a free amine. The doubly charged peptide ion (m/z = 476.6) was eluted early in the gradient, and the resulting tandem mass can be matched to the peptide sequence shown without modification. (C) The N-terminus in *K−CT is blocked by carbamylation. The m/z = 476.6 peak in (B) is missing after carbamylation, and a new peak corresponding to the modified doubly charged peptide ion (m/z = 498.2) is detected with an increased retention time. The resulting tandem mass spectrum of this ion shows an N-terminus modified by the mass of a carbamylation (43 Da).
When tested in vivo, N-terminally blocked K−CT, lacking all potential sites for ubiquitination, induced a robust chloride secretory response from T84 cells (Figure 3). Thus, retro-translocation and toxin function does not depend on poly-ubiquitination of the CTA1 chain.
Figure 3.
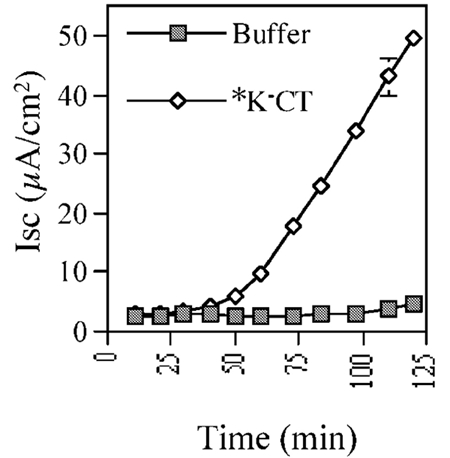
Poly-ubiquitination of CTA1 chain is not required for its retro-translocation. Time-course of Isc in T84 cells induced by 100 nM N-terminally blocked K−CT (*K−CT) or buffer alone.
The CTA1 chain avoids proteasome-dependent degradation by two independent mechanisms
To explain why the A1 chain exhibits resistance to proteasome-dependent degradation after retro-translocation to the cytosol, we introduced lysine substitutions at arginine 172 (K+172CT) or glutamine 138 (K+138CT). The amino acids in these positions are not conserved between the toxins of the cholera family; they are surface exposed and distant from the enzymic cleft. When tested in T84 cells, maximal Iscs induced by both K+172CT and K+138CT variants were attenuated by 30 and 10%, respectively, and introduction of lysines at both positions 138 and 172 [K+(138,172)CT] attenuated toxin function by 40% [Figure 4A, filled symbols; see Cl− secretion induced by K+138CT and K+(138,172)CT]. In all cases, the activities of these lysine-rich toxin variants were fully rescued by pretreatment of T84 cells with the proteasome inhibitor MG132 (Figure 4A, open symbols).
Figure 4.
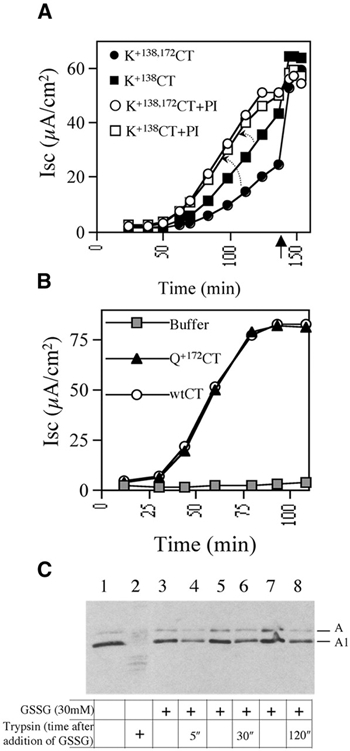
The CTA1 chain evades proteasome-dependent degradation by two mechanisms. (A) Isc induced by the lysine-rich CTA1 variants K+138CT or K+138,172CT in T84 monolayers treated with the proteasome inhibitor (PI) MG132/DMSO or DMSO alone. Forskolin (added at arrow) induces maximum Isc in all monolayers. (B) Time-course of Isc induced by 100 nM CT mutant with a glutamine substitution at position 172 in the CTA1 chain (Q+172CT), 100 nM CT or buffer alone. (C) Purified PDI was incubated in 1 mM GSH with isolated CTA subunit. Where indicated, 30 mM GSSG was added to release the A1 chain from PDI. At the indicated times, trypsin was added (lanes 2, 4, 6 and 8) or not added (lanes 1, 3, 5 and 7) to assay for the kinetics of A1 chain refolding. Lanes 1 and 2 show negative and positive control, respectively, with trypsin added 5 min after GSSG. All samples were analyzed by non-reducing SDS–PAGE and immunoblotting. The A subunit (A1 and A2 chains) and the reduced A1 chain are shown.
Two lines of evidence showed that the loss of toxin function by the introduction of lysine residues into CTA1 cannot be explained by an effect on protein folding or stability, a feature that by itself may promote proteasome-dependent degradation and thus inhibit toxin function (Verma and Deshaies, 2000). First, all toxin variants containing the lysine substitutions exhibited resistance to trypsin degradation in vitro identical to that of wtCT (data not shown). Secondly, in contrast to toxin variants carrying the lysine substitution at position 172 (K+172CT), glutamine substitution at this position (Q+172CT) had no effect on toxin action (Figure 4B). Thus, the specific substitution of lysine in certain positions in CTA1 rendered toxin action sensitive to proteasome-dependent degradation, presumably by introducing sites for poly-ubiquitination. These results explain the paucity of lysine residues in the CTA1 chain as a mechanism for evading degradation in the cytosol.
Nonetheless, the lysine-rich CT variants still induced a robust Cl− secretory response in vivo and therefore must continue to resist degradation. Since unstably folded proteins can be recognized by the proteasome in the absence of poly-ubiquitination, we hypothesized that the A1 chain may refold into a stable conformation immediately upon emerging from the translocation pore and that this might be another mechanism by which the A1 chain escapes targeting to the proteasome.
To test this idea, we measured protein refolding in vitro. We showed previously that CTA1 is unfolded by PDI (Tsai et al., 2001). In its reduced state, PDI binds and unfolds the A1 chain, rendering it sensitive to degradation by trypsin in vitro. The addition of high concentrations of oxidized glutathione induces the release of the CTA1 from PDI, which becomes resistant to trypsin degradation after refolding spontaneously. Here, we show that, when released from PDI, CTA1 refolds into a trypsin-resistant conformation in as little as 5 s (Figure 4C). Thus, the CTA1 chain may also refold very quickly in vivo.
Discussion
The results of these studies show that retro-translocation of the CTA1 chain occurs by ubiquitin- and proteasome-independent mechanisms. We also found that the A1 chain may escape proteasome-dependent degradation by virtue of a paucity of lysine residues in its primary sequence and by rapidly refolding to a stable conformation after entering the cytosol.
Although the retro-translocation of most substrates requires poly-ubiquitination, the transport of CT can occur in the absence of this modification. Thus, retro-translocation of the toxin cannot be driven by a ratcheting mechanism in which the bulky poly-ubiquitin chain would prevent backsliding of the toxin into the ER lumen. These results also exclude the possibility that the poly-ubiquitin chain serves as a downstream signal for other components, e.g. for the proteasome, which had been proposed to pull polypeptides out of the membrane (Mayer et al., 1998). Similar results were obtained by the deletion of all lysine residues in ricin toxin, but here the N-terminus was not blocked and ubiquitination at this site could not be excluded (Deeks et al., 2002). Although the poly-ubiquitin-dependent mechanisms may operate with other substrates, our results indicate that at least CT takes another route.
One possibility by which the driving force for retro-translocation of the toxin could be provided is based on its rapid folding. If the N- or C-terminal portion of the A1 chain refolded immediately upon emerging into the cytosol, as suggested by our in vitro studies, such domains could prevent the A1 chain from moving backwards into the ER lumen or could even generate an active pulling force. Another possibility by which the driving force could be provided is by a downstream component that acts independently of a poly-ubiquitin signal. A candidate is the AAA ATPase p97, which can bind to both modified and unmodified substrates and, together with its cofactors, might pull on the polypeptide chain (Ye et al., 2001; Jarosch et al., 2002). This could be a general mechanism for both poly-ubiquitinated and non-modified substrates.
The CTA1 chain escapes cytosolic degradation by the proteasome more efficiently than the plant toxin ricin that also enters the cytosol by retro-translocation across the ER membrane (Deeks et al., 2002). We identify two mechanisms by which this may occur. Generally speaking, there are two conditions that make a protein a substrate for the proteasome. First, it must be unfolded: folded proteins are not attacked by the proteasome even if they have numerous lysine residues on their surface (Thrower et al., 2000). Secondly, most proteins need lysine residues that serve as attachment sites for poly-ubiquitin chains (Hershko and Ciechanover, 1998). Although some E3 enzymes recognize specific signals in folded polypeptide chains and catalyze their poly-ubiquitination, it seems likely that the E3 enzymes involved in retro-translocation would recognize hydrophobic stretches in unfolded proteins (Laney and Hochstrasser, 1999).
One of the mechanisms by which CT avoids proteasomal degradation, the paucity of lysine residues, had been suggested before (Hazes and Read, 1997). Our data, and recent studies on ricin toxin (Deeks et al., 2002), provide evidence for this hypothesis. Unlike ricin, however, CT displays no detectable sensitivity to degradation by the proteosome. The major reason may be based on the rapid refolding of CT in the cytosol, as the additional introduction of lysine residues into CT only moderately increases its cytosolic degradation. In fact, one major difference between the toxins and all other retro-translocation substrates is that the toxins must refold in the cytosol to exert enzymic activity; in contrast, misfolded ER proteins or the heavy chain of the MHC class I molecule (in CMV-induced retro-translocation) have no role in the cytosol and thus likely remain unfolded. Although our data show that the A1 chain of CT can refold within 5 s, the effect of introducing additional lysines indicates that in vivo a significant population of toxin remains unfolded or somehow provides substrate for E3-dependent ubiquitination. This may be the reason why these bacterial toxins have evolved to contain only few lysine residues, preventing the degradation of this population and providing a doublesafety mechanism.
Methods
Materials.
The monoclonal antibody CT-17 that recognizes the CTA subunit was a gift from Dr J. Holmgren (University of Gothenburg, Sweden). Lactacystin and carboxybenzyl-Leu-Leu-Leu-vinil sulfone (Z-L3VS) were gifts from Dr H. Ploegh (Harvard Medical School), and N-carbobenzoxyl-Leu-Leu-Leucinal (MG-132) was purchased from Calbiochem. Mammalian PDI was purchased from Takara Biomedicals.
Plasmids.
The plasmid pATA14 encoding for the wtCT operon (Rodighiero et al., 1999) was used as a template for oligonucleotide-directed mutagenesis of the ctxA gene resulting in the following plasmids: pRC55 containing the double mutation K4R, K17R; pRC62 and pRC65 containing the single mutations R172K or Q138K, respectively; pRC64 containing the double mutations R172K, Q138K; and pRC66 and pRC67 containing the single mutation R172Q or the double mutation R172Q, Q138K, respectively.
Purification and characterization of recombinant toxins.
All toxins used in this study were expressed and purified from Escherichia coli TE1 periplasmic extracts as defined previously (Rodighiero et al., 1999). Expression and subunit assembly for each variant was equal to that observed with CT, and all toxins exhibited full binding affinity to GM1 as measured by modified GM1-ELISA (Rodighiero et al., 1999). The folding and stability of A subunits was assessed indirectly by trypsin sensitivity using an adaptation of the GM1-based ELISA. In brief, 96-well microtiter plates coated with GM1 ganglioside were incubated with 100 μl of toxin (0.2 nM) in PBS containing 0.1% (w/v) BSA. After washing, trypsin was added at 0.01–600 nM in PBS and incubated at room temperature for 30 min before stopping the reaction by washing with PBS containing 0.05% (v/v) Tween 20. Sensitivity to trypsin digestion was assessed by immunoreactivity to CT17.
Electrophysiology.
Measurements of short circuit current (Isc) were performed using polarized T84 cells as described previously (Rodighiero et al., 1999). All proteasome inhibitors were used at 2 μM final concentration in DMSO at 1:1000 (v/v).
Protein carbamylation and confirmation of N-terminus blockade.
Carbamylation of 1 mg of K−CT was carried out as described previously (Breitschopf et al., 1998) but in the absence of urea. Edman degradation of the carbamylated toxin gave no detectable signal in five successive cycles, whereas the un-reacted K−CT variant yielded the expected NDDRL sequence. To detect primary amines, 8 μl of 1 mg/ml solution in dH2O of NHS ester conjugated to biotin (Sulfo-NHS-LC-Biotin, Pierce) was reacted with 50 μl of unblocked and carbamylated toxin in PBS (pH 7.4) for 1 h in the presence or absence of 0.4% SDS. Then samples were resolved by SDS–PAGE, transferred onto nitrocellulose and subjected to western blotting with streptavidin–HRP conjugate (Pierce). To directly assess substitution with the carbamyl group (residue mass 43 Da) of the N-terminus, the A subunits of the unblocked and blocked toxin were excised from an SDS polyacrylamide gel, subjected to a Glu-C digestion and analyzed as described previously (Shevchenko et al., 1996).
Refolding assay.
Purified PDI (3.4 μM) and isolated CTA subunit (70 nM) were incubated in the presence of reduced glutathine (GSH) (1 mM) for 30 min at 30°C, followed by the addition of oxidized glutathione (GSSG) (30 mM) for the indicated time. Trypsin (0.1 mg/ml) was subsequently added for 30 min at 4°C and the reaction stopped by the addition of the trypsin inhibitor TLCK for 10 min at 4°C.
Acknowledgments
We thank S. Gygi, R. Tomaino and G. Bannenberg for assistance with mass spectroscopy and analysis, and E. Kern and E. Barclay for expert technical assistance. This work was supported by research grants DK48106 and DK57827 from the NIH and DK34854 from the Harvard Digestive Diseases Center (to W.I.L.). T.A.R. is a Howard Hughes Medical Institute investigator, and B.T. is a fellow of the Damon Runyon Cancer Research Foundation (DRG1579).
References
- Biederer T., Volkwein C. and Sommer T. (1996) Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin–proteasome pathway. EMBO J., 15, 2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Breitschopf K., Bengal E., Ziv T., Admon A. and Ciechanover A. (1998) A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J., 17, 5964–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks E.D., Cook J.P., Day P.J., Smith D.C., Roberts L.M. and Lord J.M. (2002) The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry, 41, 3405–3413. [DOI] [PubMed] [Google Scholar]
- Hazes B. and Read R.J. (1997) Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry, 36, 11051–11054. [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hiller M.M., Finger A., Schweiger M. and Wolf D.H. (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin–proteasome pathway. Science, 273, 1725–1728. [DOI] [PubMed] [Google Scholar]
- Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D.H. and Sommer T. (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol., 4, 134–139. [DOI] [PubMed] [Google Scholar]
- Laney J.D. and Hochstrasser M. (1999) Substrate targeting in the ubiquitin system. Cell, 97, 427–430. [DOI] [PubMed] [Google Scholar]
- Lencer W.I. (2001) Cholera: invasion of the intestinal epithelial barrier by a stably folded protein toxin. Am. J. Physiol. Gastrointest. Liver Physiol., 280, G781–G786. [DOI] [PubMed] [Google Scholar]
- Mayer T.U., Braun T. and Jentsch S. (1998) Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J., 17, 3251–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodighiero C., Aman A.T., Kenny M.J., Moss J., Lencer W.I. and Hirst T.R. (1999) Structural basis for the differential toxicity of cholera toxin and Escherichia coli heat-labile enterotoxin. J. Biol. Chem., 274, 3962–3969. [DOI] [PubMed] [Google Scholar]
- Schmitz A., Herrgen H., Winkeler A. and Herzog V. (2000) Cholera toxin is exported from microsomes by the sec61p complex. J. Cell Biol., 148, 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu C.E., Flierman D., Ploegh H.L., Rapoport T.A. and Chau V. (2001) Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol. Biol. Cell, 12, 2546–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Wilm M., Vorm O. and Mann M. (1996) Mass spectrometric sequencing of proteins silverstained polyacrylamide gels. Anal. Chem., 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Thrower J.S., Hoffman L., Rechsteiner M. and Pickart C.M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J., 19, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B., Rodighiero C., Lencer W.I. and Rapoport T. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell, 104, 937–948. [DOI] [PubMed] [Google Scholar]
- Verma R. and Deshaies R.J. (2000) A proteasome howdunit: the case of the missing signal. Cell, 101, 341–344. [DOI] [PubMed] [Google Scholar]
- Wiertz E.J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T.R., Rapoport T.A. and Ploegh H.L. (1996) Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature, 384, 432–438. [DOI] [PubMed] [Google Scholar]
- Ye Y., Meyer H.H. and Rapoport T.A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature, 414, 652–656. [DOI] [PubMed] [Google Scholar]