

Abstract
RIP1 and its homologs, RIP2 and RIP3, form part of a family of Ser/Thr kinases that regulate signal transduction processes leading to NF-κB activation. Here, we identify RIP4 (DIK/PKK) as a novel member of the RIP kinase family. RIP4 contains an N-terminal RIP-like kinase domain and a C-terminal region characterized by the presence of 11 ankyrin repeats. Overexpression of RIP4 leads to activation of NF-κB and JNK. Kinase inactive RIP4 or a truncated version containing the ankyrin repeats have a dominant negative (DN) effect on NF-κB induction by multiple stimuli. RIP4 binds to several members of the TRAF protein family, and DN versions of TRAF1, TRAF3 and TRAF6 inhibit RIP4-induced NF-κB activation. Moreover, RIP4 is cleaved after Asp340 and Asp378 during Fas-induced apoptosis. These data suggest that RIP4 is involved in NF-κB and JNK signaling and that caspase-dependent processing of RIP4 may negatively regulate NF-κB-dependent pro-survival or pro-inflammatory signals.
Introduction
Activation of the transcription factor NF-κB is induced by the triggering of pro-inflammatory cytokine receptors or antigen receptors but also in response to microbial and viral infections (Karin and Lin, 2002). NF-κB is a collective term describing a family of dimeric transcription factors that are kept in an inactive form in the cytoplasm by binding to specific inhibitors, the IκBs (Karin and Delhase, 2000). Upon cell stimulation, IκB proteins undergo rapid phosphorylation by the IκB kinase (IKK) complex and are subsequently ubiquitylated and degraded by the proteasome.
The IKK complex is composed of two IκB kinases, IKKα and IKKβ, and a regulatory subunit called NEMO (IKKγ) (Karin and Delhase, 2000). Many of the upstream regulators of the IKK complex are characterized by the presence of homotypic interaction motifs with a so-called death domain (DD) fold, composed of six α-helical bundles with Greek key topology (Hofmann, 1999).
The DD-containing Ser/Thr kinase RIP1 is part of a multicomponent signaling complex assembled upon binding of TNF to TNF receptor 1 (TNFR1) (Hsu et al., 1996). RIP1-deficient cells from knockout mice fail to activate NF-κB in response to TNF, indicating that RIP1 plays a critical role in TNFR1-induced NF-κB activation (Kelliher et al., 1998).
RIP1 belongs to a family of related kinases that includes RIP2 (also known as CARDIAK/RICK) (Inohara et al., 1998; McCarthy et al., 1998; Thome et al., 1998) and RIP3 (Sun et al., 1999; Yu et al., 1999). These kinases share significant similarity in their N-terminal kinase domains, but possess distinct C-termini. RIP1 has a DD at its C-terminus, whereas RIP2 has a related caspase recruitment domain (CARD) motif (see Figure 1A). Ectopic expression of RIP2 causes activation of NF-κB and Jun N-terminal kinase (JNK) (Inohara et al., 1998; McCarthy et al., 1998; Thome et al., 1998). Cytokine production induced by IL-1, IL-18 and Toll receptors is reduced in RIP2-deficient cells (Chin et al., 2002; Kobayashi et al., 2002). Furthermore, T-cell-receptor-triggered proliferation is severely compromised in the RIP2-deficient animals.
Figure 1.
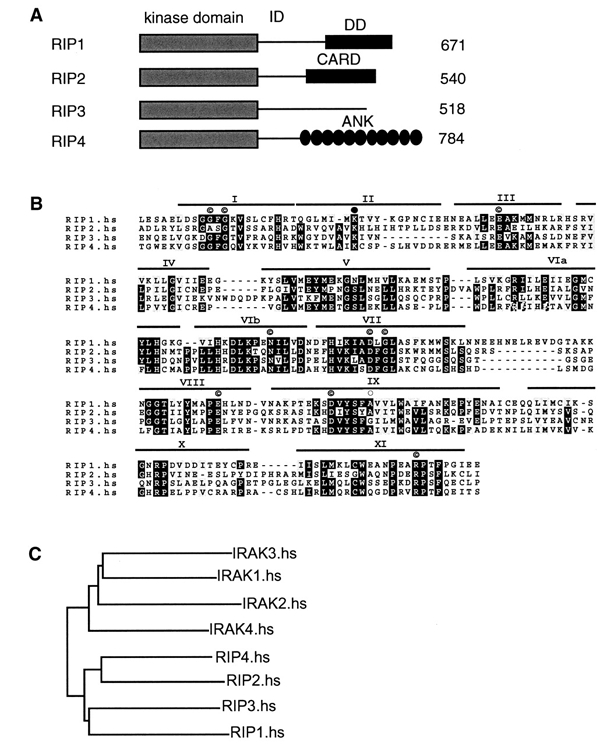
RIP4 is a novel member of the RIP family of kinases. (A) Domain organization of RIP family members. All members share a homologous N-terminal kinase domain. RIP1 and RIP2 have C-terminal DD and CARD motifs, respectively, whereas the C-terminus of RIP3 lacks obvious sequence homology to known proteins in public databases. RIP4 has C-terminal ankyrin repeats. (B) Sequence alignment of the kinase domains of RIPs. Black and gray shading indicate 75% amino acid sequence identity and similarity, respectively. I–XI represent conserved modules (Hanks and Hunter, 1995). Indicated are residues conserved in 95% of the kinases (©), residues important for ATP binding (filled circle) and the Gly residue present in RIP3 (open circle) that is conserved in 95% of all kinases but is substituted into Ala in RIP1, RIP2 and RIP4. (C) Phylogenic tree of the kinase domain of human RIPs and IRAKs.
The physiological function of RIP3 is less well understood (Sun et al., 1999, 2002; Kasof et al., 2000). In contrast to RIP1 and RIP2, RIP3 has no DD or CARD motif at its C-terminus. RIP3 interacts with and phosphorylates RIP1 to inhibit RIP1- and TNFR1-induced NF-κB activation (Sun et al., 2002).
Here, we report the identification and characterization of DIK/PKK (Bähr et al., 2000; Chen et al., 2001) as a novel member of the RIP family of kinases that we refer to as RIP4.
Results and discussion
We screened public databases with a generalized profile of the RIP-like kinase motif based on the RIP1, RIP2 and RIP3 kinase sequences (Bucher et al., 1996). Using this approach, we found a kinase that was previously identified as PKC-δ interacting protein kinase (DIK) (Bähr et al., 2000). The probable mouse ortholog has been described previously and given the name PKK (protein kinase C-associated kinase) (Chen et al., 2001). Because of its close similarity to the other members of the RIP family of kinases, we will refer to DIK and PKK as human and murine RIP4, respectively. Human RIP4 has 784 amino acids (aa). Its predicted structural organization comprises an N-terminal RIP-like kinase domain, a Ser/Thr-rich intermediate domain (ID) and a C-terminal region containing 11 ankyrin repeats (Figure 1A). The kinase domain of RIP4 shares 32, 45 and 31% of amino acid identity with RIP3, RIP2 and RIP1, respectively, and contains highly conserved key residues important for kinase activity (Hanks and Hunter, 1995) (Figure 1B). Sequence similarity comparisons with the kinase domains of other kinases in the human genome clearly identified RIP4 as a member of the RIP family of kinases, which is closely related but distinct from the IRAK family of kinases (Figure 1C). It has been demonstrated that murine RIP4 exhibits protein kinase activity in vitro and that it associates with cellular membranes (Bähr et al., 2000). The physiological function of RIP4 and the role of its kinase activity, however, are not known.
In view of its apparent similarity to the NF-κB-activating RIP kinases, we overexpressed RIP4 in 293T cells to test its capacity to activate an NF-κB luciferase reporter plasmid. Expression of RIP4 in 293T cells resulted in a dose-dependent activation of the reporter gene (Figure 2A). NF-κB transcriptional activation correlated with induction of IκB phosphorylation, to an extent comparable to that seen with the Toll receptor adaptor protein, MyD88 (Figure 2B). Next, we analyzed whether the kinase activity of RIP4 was necessary for the observed NF-κB activation. To this purpose, we generated a point mutant of RIP4 (K51R) predicted to result in reduced ATP binding and catalytic activity of the kinase domain (Hanks and Hunter, 1995). In comparison to the wild-type (wt) form, the K51R mutant showed dramatically reduced in vitro kinase activity (Figure 2C). Inactivation of the kinase domain also led to the complete loss of the NF-κB activation capacity (Figure 2D), demonstrating that RIP4-mediated NF-κB signals require the active kinase domain. Moreover, the study of a series of deletion mutants of RIP4 indicated that the kinase domain alone was capable of activating the NF-κB reporter plasmid, whereas mutants containing the ID or the ankyrin repeats were inactive (Figure 2D and E).
Figure 2.
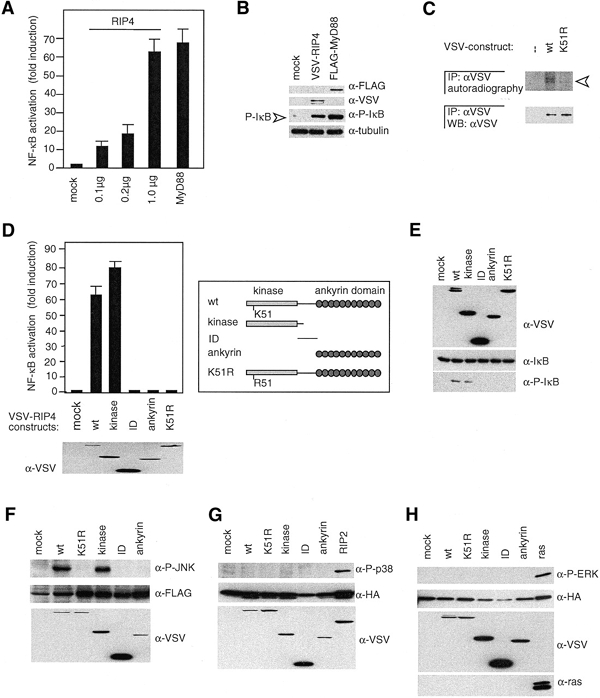
The kinase activity of RIP4 is critical for NF-κB and JNK activation. (A) 293T cells were transfected with an NF-κB reporter plasmid, together with mock or MyD88 plasmids (1 μg) or the indicated amounts of a RIP4 construct, and analyzed for NF-κB-dependent luciferase activity. Data shown are mean values ± standard deviations from one representative out of three independent experiments, each done in triplicate. (B) 293T cells were co-transfected with IκB and mock, RIP4 or MyD88 constructs, as indicated, and cell lysates were analyzed for the presence of phosphorylated IκB (P-IκB) by western blot. (C) 293T cells were transfected with the indicated RIP4 constructs, and anti-VSV immunoprecipitates (IP) were analyzed by in vitro kinase assay and autoradiography (upper) or by anti-VSV western blot (lower) The arrowhead indicates phosphorylated RIP4. (D) 293T cells transfected with the indicated RIP4 constructs were analyzed as in (A). The structure and expression level of the various RIP4 constructs used is shown. (E) 293T cells transfected with IκB and the indicated RIP4 constructs were analyzed for IκB phosphorylation as in (B). (F–H) 293T cells were co-transfected with FLAG-JNK, HA-p38 or HA-ERK constructs, respectively, together with the indicated RIP4 constructs, and cell lysates were assessed for MAPK activation by western blot.
We next tested whether RIP4 was able to stimulate the activation of the JNK, p38 or ERK MAPK pathways by co-expressing RIP4 with these MAPKs in 293T cells. Compared to the vector control, RIP4 potently induced JNK activation (Figure 2F). Similar to the requirement for NF-κB activation, the initiation of JNK signals by RIP4 was dependent on the presence of an intact kinase domain, since overexpression of the kinase-dead version of RIP4 did not result in increased JNK activation. In contrast, overexpression of the various RIP4 constructs did not lead to p38 or ERK activation (Figure 2G and H).
Activation of NF-κB is dependent on the formation of a multiprotein complex that comprises TNF-receptor-associated factors (TRAFs), IKKs and NEMO, leading to the subsequent phosphorylation and degradation of IκB (Karin and Delhase, 2000). Dominant negative (DN) versions of some of these proteins can block NF-κB-activating signals triggered by upstream proteins. Indeed, DN-IκBα potently inhibited RIP4-mediated NF-κB activation, whereas DN-IKKβ had a partial inhibitory effect. These findings indicate that overexpression of RIP4 initiated the NF-κB signal cascade upstream of IKKβ and IκB (Figure 3A).
Figure 3.
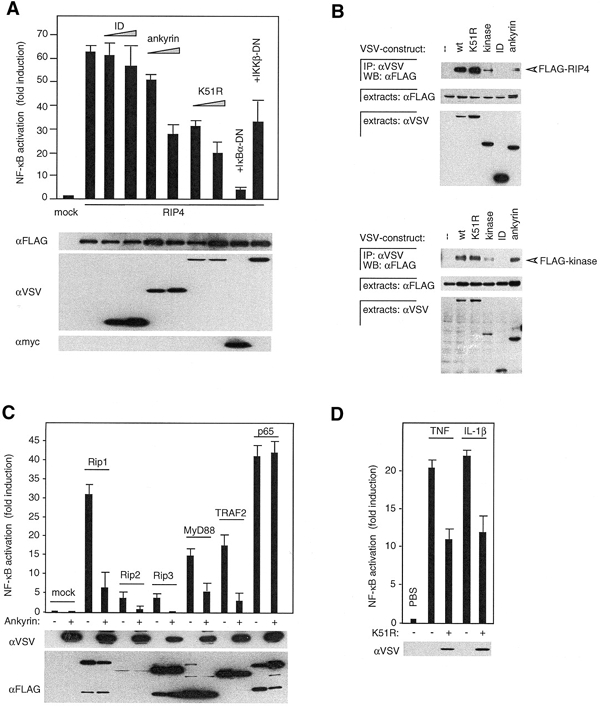
DN versions of RIP4 inhibit NF-κB activation by multiple pathways. (A) 293T cells were transfected with an NF-κB reporter plasmid and RIP4, in combination with mock plasmid (1 μg) or the indicated RIP4 constructs, DN-IKKβ or DN-IκBα, and analyzed for NF-κB-dependent luciferase activity. The expression level of the various transfected constructs is shown. (B) 293T cells were transfected with the indicated RIP4 constructs, and anti-VSV immunoprecipitates (IP) and cell extracts were analyzed by western blot. (C) 293T cells were transfected with an NF-κB reporter plasmid and the indicated constructs, together with the RIP4 ankyrin domain (+) or mock plasmid (−). (D) HeLa cells were transfected with an NF-κB reporter plasmid together with RIP4 K51R (+) or mock plasmid (−) and analyzed for TNF- and IL-1β-induced NF-κB activation.
At similar expression levels, the kinase domain alone appeared to be at least as efficient as full-length RIP4 at activating NF-κB (Figure 2D), suggesting that the ankyrin or ID may act as auto-inhibitory domain. Therefore, we next analyzed the effect of co-expression of either the ID or the ankyrin domain on RIP4-induced NF-κB reporter gene activation. Expression of the ankyrin repeats caused a concentration-dependent decrease in RIP4-triggered NF-κB activation, whereas no effect was seen with the ID (Figure 3A). The kinase mutant version of RIP4, K51R, also acted as an inhibitory molecule. These findings support the idea that the RIP4 kinase activity may be negatively regulated by an intramolecular interaction of the kinase domain with the ankyrin repeats. Overexpression of wt RIP4 may overcome this inhibition and induce the formation of RIP4 kinase homodimers and subsequent activation by autophosphorylation. To further investigate these potential regulatory interactions, various RIP4 constructs were analyzed for their association with wt RIP4 by co-immunoprecipitation studies (Figure 3B). Indeed, full-length RIP4 and its kinase domain interacted with the wt and K51R versions of RIP4. Moreover, a small but significant proportion of the ankyrin and N-terminal kinase domain constructs was found to associate with full-length RIP4 (Figure 3B, upper), but also with the kinase domain alone (Figure 3B, lower). Together, these data demonstrate that RIP4 can homodimerize via the kinase fold and the ankyrin domains and suggest that the RIP4 kinase activity may be negatively regulated by an intra- and/or inter-molecular interaction between the kinase domain and the ankyrin repeats.
Next, we took advantage of the two DN-RIP4 versions and co-expressed them along with various inducers of NF-κB activation. NF-κB activation induced by overexpression of RIP1, RIP2, RIP3, MyD88 or TRAF2 was partially or fully inhibited by the ankyrin construct (Figure 3C). In contrast, p65-induced NF-κB activation was not affected. Similar results were obtained using the K51R mutant (data not shown). Moreover, NF-κB activation induced by TNF or Il-1β treatment was clearly inhibited by the K51R construct (Figure 3D). Together with the results shown in Figure 3A, these findings suggest that RIP4 is implicated in the regulation of various NF-κB-inducing signaling pathways upstream or at the level of the IKK complex.
A characteristic feature of RIP family members is their capacity to bind to TRAF molecules. In vitro studies have shown that RIP1 interacts with TRAF1, TRAF2 and TRAF3 (Hsu et al., 1996), whereas RIP2 associates with TRAF1, TRAF2, TRAF5 and TRAF6 (McCarthy et al., 1998; Thome et al., 1998). Co-expression of RIP4 with the individual six TRAF family members indicated that TRAF1, TRAF2, TRAF3 and TRAF5, but not TRAF6 bound RIP4 (Figure 4A). Moreover, no binding to TRAF4 was detected (data not shown). We next tested the effect of DN forms of several TRAF members on RIP4-induced NF-κB activation. DN-TRAF1 efficiently blocked RIP4-induced NF-κB activation, whereas DN-TRAF3 and DN-TRAF6 had only a moderate effect and no significant inhibition was observed using DN-TRAF2, DN-TRAF5 or a combination of both, despite their binding to RIP4 (Figure 4B; data not shown). Thus, RIP4 seems to physically and functionally interact with multiple TRAF-dependent pathways leading to NF-κB activation.
Figure 4.

RIP4 interacts with TRAF proteins. (A) 293T cells were transfected with a VSV-tagged RIP4 construct together with the indicated TRAF constructs, and anti-FLAG immunoprecipitates (IP) and cell extracts were analyzed by western blot. (B) 293T cells were transfected with an NF-κB reporter plasmid together with RIP4 and the indicated DN-TRAF constructs, and lysates were analyzed for NF-κB-dependent luciferase activity.
Yet another feature of RIP kinases is their processing during apoptosis. RIP1 is cleaved in the ID, after Asp324, by caspase-8 during Fas ligand (FasL)-induced apoptosis (Lin et al., 1999; Martinon et al., 2000). This proteolytic cleavage separates the DD from the kinase domain, generating a DN version that efficiently blocks NF-κB anti-apoptotic signals (Martinon et al., 2000). RIP3 undergoes similar processing (our unpublished data). Therefore, we analyzed proteolytic processing of RIP4 upon FasL treatment of RIP4-transfected 293T cells by western blotting with an antibody directed against the N-terminal tag or an anti-RIP4 antibody directed against the C-terminal ankyrin domains (Figure 5B). In non-treated cells, a small proportion of RIP4 is constitutively cleaved into an N-terminal, VSV-tagged 37 kDa fragment and a C-terminal 44 kDa fragment. FasL treatment strongly increased the generation of these fragments. Processing of RIP4 was caspase dependent, since pre-incubation of the cells with the caspase inhibitor zVAD completely blocked RIP4 processing. In the ID, we identified two candidate caspase cleavage sites, SQLD340 and SSVD378 (Figure 5A). Individual mutation of these sites identified both sites as targets of Fas-induced cleavage of RIP4, since more slowly migrating N-terminal or C-terminal fragments were detected for the D340E and D378E, respectively (Figure 5B). In contrast, mutation of both sites led to complete resistance of the mutated RIP4 against processing, indicating that both sites are involved in caspase-dependent processing (Figure 5C). Together with the observed DN effect of the ankyrin construct (Figure 3A and C), these data suggest that pro-apoptotic stimuli may lead to the generation, via caspase-dependent cleavage, of a C-terminal RIP4 fragment that inhibits NF-κB-dependent gene transcription. Consistent with this hypothesis, the non-cleavable Asp/Glu 340/378 double mutant, but not the corresponding Asp340 or Asp378 single mutants, shows increased NF-κB activation (Figure 5D).
Figure 5.
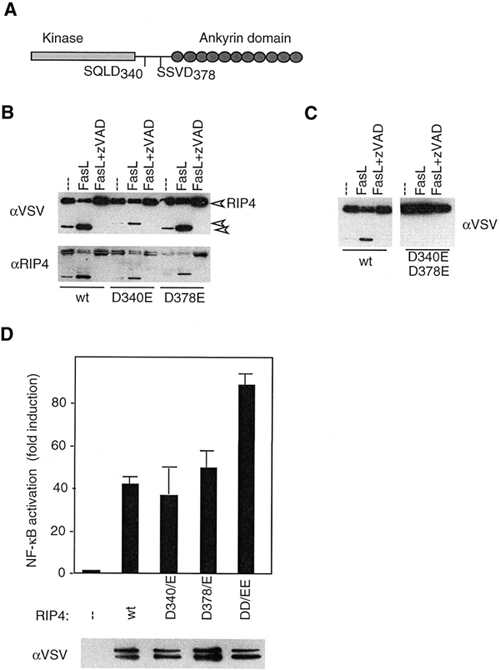
RIP4 is cleaved after Asp340 and Asp378 during FasL-dependent induction of apoptosis. (A) The ID of RIP4 contains two consensus caspase cleavage sites, SQLD340 and SSVD378. (B and C) 293T cells were transfected with the indicated N-terminally VSV-tagged RIP4 constructs. Twenty-four hours after transfection, cells were treated with recombinant FasL in the presence or absence of the caspase inhibitor zVAD, and cell lysates were analyzed for the presence of RIP4 cleavage products by western blot using anti-VSV and anti-RIP4 antibodies, as indicated. The arrowheads in (B) indicate full-length and cleaved forms of RIP4. (D) 293T cells were transfected with wild-type (wt) RIP4, the double D340/D378-Glu (DD/EE) or the respective single mutants, together with an NF-κB reporter plasmid, and NF-κB activation was analyzed by luciferase assay.
The RIP-family kinases have been shown to affect NF-κB and caspase pathways, but little is known about the precise molecular mechanisms involved. Our data demonstrate that the RIP4 kinase activity is necessary for NF-κB and JNK activation. Overexpression of RIP1 and RIP2 has been shown to induce NF-κB activation independently of their kinase activity (Ting et al., 1996; Thome et al., 1998). However, an important role for the RIP1 and RIP2 kinase activities was revealed under conditions of limited oligomerization, suggesting that the respective kinase activities may be necessary for efficient NF-κB activation under conditions of limiting upstream signals (Inohara et al., 2000). The substrates of RIP4 and the related RIP kinases are not known, but are likely to include the RIP kinases themselves and the kinases directly involved in the NF-κB and JNK pathways.
The generation of mice deficient for RIP4 and the identification of novel RIP4-interacting proteins will help to further elucidate the precise physiological role of RIP4 in signaling pathways, leading to NF-κB and JNK activation and to apoptosis.
Methods
Expression vectors.
A VSV-RIP4 expression construct was obtained by subcloning from a pFlag-CMV vector (Bähr et al., 2000) into a derivative of pCR3 (Invitrogen), in frame with an N-terminal VSV tag. RIP4 kinase (aa 1–340), ID (aa 277–438) and ankyrin (aa 430–784) constructs were amplified by PCR using Pwo polymerase (Boehringer). Mutation of Asp340 or Asp378 into Glu (D340E and D378E, respectively) and of Lys51 into Arg to generate a kinase-dead RIP4 (K51R) was achieved by a site-directed mutagenesis with two sequential rounds of PCR. The double DD/EE mutant was generated from the two single mutants by double PCR.
Antibodies.
Polyclonal anti-RIP4 has been described previously (Bähr et al., 2000). Antibodies used include anti-FLAG M2 and anti-VSV P5D4 (Sigma); anti-HA (BabCo), anti-phospho-IκB, anti-phospho-JNK, anti-phospho-p38 and anti-phospho-ERK (Cell Signaling); anti-IκB (Santa-Cruz); anti-tubulin (a gift of S. Lens, Amsterdam, The Netherlands); and anti-ras (Transduction Laboratories). Secondary peroxidase-conjugated antibodies were from Jackson ImmunoResearch.
Transfection, immunoprecipitation, in vitro kinase assay and western blotting.
These assays were performed as described previously (Thome et al., 1998). HeLa cells were transfected using lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Supplementary Material
Additional material and methods
Acknowledgments
The authors would like to thank K. Burns and M. Thurau for critical review of the manuscript. This study was supported by grants from the Swiss National Science Foundation and the CTI (to J.T.) and the Swiss Cancer Ligue (to M.T. and J.T.).
References
- Bähr C., Rohwer A., Stempka L., Rincke G., Marks F. and Gschwendt M. (2000) DIK, a novel protein kinase that interacts with protein kinase Cδ. Cloning, characterization, and gene analysis. J. Biol. Chem., 275, 36350–36357. [DOI] [PubMed] [Google Scholar]
- Bucher P., Karplus K., Moeri N. and Hofmann K. (1996) A flexible search technique based on generalized profiles. Comp. Chem., 20, 3–24. [DOI] [PubMed] [Google Scholar]
- Chen L., Haider K., Ponda M., Cariappa A., Rowitch D. and Pillai S. (2001) Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J. Biol. Chem., 276, 21737–21744. [DOI] [PubMed] [Google Scholar]
- Chin A.I., Dempsey P.W., Bruhn K., Miller J.F., Xu Y. and Cheng G. (2002) Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature, 416, 190–194. [DOI] [PubMed] [Google Scholar]
- Hanks S.K. and Hunter T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J., 9, 576–596. [PubMed] [Google Scholar]
- Hofmann K. (1999) The modular nature of apoptotic signaling proteins. Cell. Mol. Life Sci., 55, 1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H., Huang J., Shu H.B., Baichwal V. and Goeddel D.V. (1996) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity, 4, 387–396. [DOI] [PubMed] [Google Scholar]
- Inohara N., del Peso L., Koseki T., Chen S. and Nunez G. (1998) RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J. Biol. Chem., 273, 12296–12300. [DOI] [PubMed] [Google Scholar]
- Inohara N., Koseki T., Lin J., del Peso L., Lucas P.C., Chen F.F., Ogura Y. and Nunez G. (2000) An induced proximity model for NF-κB activation in the Nod1/RICK and RIP signaling pathways. J. Biol. Chem., 275, 27823–27831. [DOI] [PubMed] [Google Scholar]
- Karin M. and Delhase M. (2000) The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin. Immunol., 12, 85–98. [DOI] [PubMed] [Google Scholar]
- Karin M. and Lin A. (2002) NF-κB at the crossroads of life and death. Nat. Immunol., 3, 221–227. [DOI] [PubMed] [Google Scholar]
- Kasof G.M., Prosser J.C., Liu D., Lorenzi M.V. and Gomes B.C. (2000) The RIP-like kinase, RIP3, induces apoptosis and NF-κB nuclear translocation and localizes to mitochondria. FEBS Lett., 473, 285–291. [DOI] [PubMed] [Google Scholar]
- Kelliher M.A., Grimm S., Ishida Y., Kuo F., Stanger B.Z. and Leder P. (1998) The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity, 8, 297–303. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Inohara N., Hernandez L.D., Galan J.E., Nunez G., Janeway C.A., Medzhitov R. and Flavell R.A. (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature, 416, 194–199. [DOI] [PubMed] [Google Scholar]
- Lin Y., Devin A., Rodriguez Y. and Liu Z. (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev., 13, 2514–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F., Holler N., Richard C. and Tschopp J. (2000) Activation of a pro-apoptotic amplification loop through inhibition of NF-κB-dependent survival signals by caspase-mediated inactivation of RIP. FEBS Lett., 468, 134–136. [DOI] [PubMed] [Google Scholar]
- McCarthy J.V., Ni J. and Dixit V.M. (1998) RIP2 is a novel NF-κB-activating and cell death-inducing kinase. J. Biol. Chem., 273, 16968–16975. [DOI] [PubMed] [Google Scholar]
- Sun X., Lee J., Navas T., Baldwin D.T., Stewart T.A. and Dixit V.M. (1999) RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem., 274, 16871–16875. [DOI] [PubMed] [Google Scholar]
- Sun X., Yin J., Starovasnik M.A., Fairbrother W.J. and Dixit V.M. (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem., 277, 9505–9511. [DOI] [PubMed] [Google Scholar]
- Thome M., Hofmann K., Burns K., Martinon F., Bodmer J.L., Mattmann C. and Tschopp J. (1998) Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol., 8, 885–888. [DOI] [PubMed] [Google Scholar]
- Ting A.T., Pimentel-Muinos F.X. and Seed B. (1996) RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J., 15, 6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Yu P.W., Huang B.C., Shen M., Quast J., Chan E., Xu X., Nolan G.P., Payan D.G. and Luo Y. (1999) Identification of RIP3, a RIP-like kinase that activates apoptosis and NFκB. Curr. Biol., 9, 539–542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional material and methods