

Abstract
The phosphorylation state of the C-terminal repeat domain (CTD) of the largest subunit of RNA polymerase II changes as polymerase transcribes a gene, and the distinct forms of the phospho-CTD (PCTD) recruit different nuclear factors to elongating polymerase. The Set2 histone methyltransferase from yeast was recently shown to bind the PCTD of elongating RNA polymerase II by means of a novel domain termed the Set2–Rpb1 interacting (SRI) domain. Here, we report the solution structure of the SRI domain in human Set2 (hSRI domain), which adopts a left-turned three-helix bundle distinctly different from other structurally characterized PCTD-interacting domains. NMR titration experiments mapped the binding surface of the hSRI domain to helices 1 and 2, and Biacore binding studies showed that the domain binds preferably to [Ser-2 + Ser-5]-phosphorylated CTD peptides containing two or more heptad repeats. Point-mutagenesis studies identified five residues critical for PCTD binding. In view of the differential effects of these point mutations on binding to different CTD phosphopeptides, we propose a model for the hSRI domain interaction with the PCTD.
Keywords: histone methylation, phospho-C-terminal-domain-interacting domain, RNA polymerase II, transcription
RNA polymerase II carries an intrinsically unstructured, flexible domain at the C terminus of its largest subunit. The principal function of this C-terminal repeat domain (CTD), which comprises multiple repeats of a consensus heptamer Y1S2P3T4S5P6S7, is to serve as a binding scaffold for various nuclear factors (reviewed in refs. 1–3). The CTD of preinitiating RNA polymerase II is mostly unphosphorylated, whereas after initiation and during elongation it is hyperphosphorylated, principally on Ser-5 and Ser-2 residues of the repeats (4–6); we refer to this form as the phospho-CTD (PCTD). Attendant with changes in patterns of CTD phosphorylation, the ensemble of CTD-bound proteins changes as RNA polymerase II progresses through the transcription cycle (5). Although knowledge of the number and types of PCTD-associating proteins (PCAPs) has expanded rapidly (7, 8), information about the molecular nature of PCAP–PCTD interactions remains quite limited; detailed binding properties and/or 3D structures are known for only a few PCTD-interacting domains (PCIDs) (9–14).
Describing in molecular detail the interactions between the PCTD and its binding partners is an important step in advancing our understanding of the PCTD and its manifold functions. One recently identified binding partner of the PCTD is the Saccharomyces cerevisiae Set2 (yeast Set2; ySet2), a histone methyltransferase that modifies K36 of histone H3 in nucleosomes of transcribed genes (15–19). The identification of ySet2 as a PCAP together with studies of transcription in set2 mutant strains suggests a role for the PCTD in chromatin structure modulation during elongation (20). Deletion studies of ySet2 mapped a novel PCID, termed the Set2–Rpb1 interacting (SRI) domain by Kizer et al. (20), to the C-terminal segment of ySet2. The yeast SRI domain binds preferentially to PCTD peptides with both Ser-2 and –5 phosphorylated; moreover, this domain is required for targeting ySet2 catalytic activity to the coding region of genes, thereby coupling H3 K36 methylation to transcription elongation in vivo (20). The yeast SRI domain shows significant sequence homology to the C-terminal regions of Set2-like proteins in different species but is not homologous to any other characterized PCIDs. Although ySet2 contains several domains that also are found in other SET proteins (AWS, SET, PostSET, and WW domains; see Fig. 5a, which is published as supporting information on the PNAS web site), the SRI domain is uniquely found in Set2; thus it was hypothesized to be a functional indicator for the Set2 family of histone methyltransferases. Sequence similarity searches uncovered SRI domain-homologous segments of proteins in several different species, including a single recognizably homologous segment in the human genome. That segment comprises the C terminus of the huntingtin yeast partner B (HYPB) protein, the presumptive human Set2 (hSet2) ortholog containing AWS, SET, PostSET, WW, and SRI domains (Fig. 5a) (20–22).
Here, we present the solution structure of the hSRI domain as solved by NMR spectroscopy. In addition we characterize its PCTD-interaction surface by NMR titration. We also show that the hSRI domain displays extreme specificity for contiguous Ser-2P/Ser-5P residues, requiring four such SerP residues in consecutive heptad repeats for maximal binding. Finally, we present results from site-directed mutagenesis and Biacore binding measurements that identify key hSRI domain residues essential for PCTD binding. The hSRI domain structure exemplifies a previously undescribed protein motif dedicated to linking histone methylation to transcription elongation.
Materials and Methods
DNA Constructs, Purification of Recombinant Proteins, and Initial Studies. A GST-fusion protein carrying residues 1,884–2,061 of hSet2/HYPB, containing a WW domain and a putative SRI domain, was expressed in and purified from bacterial cells (see Supporting Text, which is published as supporting information on the PNAS web site, for detailed procedures); far-Western blotting (performed as described in ref. 7) confirmed that this fusion protein binds the PCTD. Unexpectedly, thrombin cleavage released not only the intact hSet2 fragment but also a smaller piece that retained the ability to bind the PCTD (Fig. 5). This segment (identified as residues 1,954–2,061 by mass spectrometry), corresponding to the SRI homology region of hSet2/HYPB, was overexpressed as an N-terminal His-6-tagged protein and purified by a Ni-NTA column, then the N-terminal His-6-tag was removed by proteolytic cleavage. The resulting fragment, containing GSHM at the N terminus and residues 1,954–2,061 of hSet2 (“hSRI domain”), was renumbered 1–112, further purified by size-exclusion chromatography, and used for NMR studies.
Isotopically enriched hSRI domain was overexpressed in M9 minimal media with 15N-NH4Cl and 13C-glucose as the sole nitrogen and carbon sources (Cambridge Isotope Laboratories, Andover, MA). NMR samples were prepared with U-15N, U-13C/15N or 10% 13C labeling. Selective 15N-lysine labeling was achieved by overexpressing the hSRI domain in M9 minimal media supplemented with 15N-lysine during induction. All NMR samples were exchanged into a buffer containing 25 mM sodium phosphate, 100 mM KCl, 2 mM DTT, and 5% (vol/vol) D2O (pH 7.0) before experiments.
Site-Directed Mutagenesis. A series of single point mutations of hSRI domain were prepared by using the QuikChange site-directed mutagenesis kit (Stratagene) starting from a pET-15b vector containing the wild-type (WT) hSRI domain. The presence of the desired mutations was confirmed by DNA sequencing. All mutants were overexpressed by using BL21(DE3)STAR cells in LB and were purified by using the same procedures for the WT protein.
NMR Spectroscopy and Structure Calculation. All NMR experiments were conducted at 27°C using Varian INOVA 600 or 800 MHz spectrometers. Data were processed by using nmrpipe (23) and analyzed with xeasy/cara (24). Following standard protocols (see Supporting Text for details), we have obtained a final ensemble of 20 structures containing no Nuclear Overhauser Effect violations of >0.4 Å and no dihedral angle violations of >4°. The quality of these structures can be evaluated in Table 3, which is published as supporting information on the PNAS web site. To map its binding surface, we obtained a series of 1H-15N HSQC spectra of the hSRI domain in the presence of the 2,5,2,5,2,5 PCTD peptide at increasing molar ratios of 1:3, 2:3, 1:1, 2:1, and 3:1. Because of the technical difficulty of obtaining accurate chemical-shift values from the crowded resonances at the stoichiometric concentration and because of the concern of nonspecific interactions between the basic hSRI domain and the excess amount of negatively charged PCTD peptide, chemical shift perturbations from the first titration point are calculated as 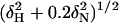 and plotted in Fig. 2a.
and plotted in Fig. 2a.
Fig. 2.

NMR titration maps the PCTD-binding surface of the hSRI domain to α1 and α2. (a) Chemical shift perturbations of the hSRI domain are calculated as 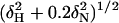 for each residue and plotted. Secondary structures are shown above the plot. (b) Resonances that experience chemical shift perturbation of >0.095 ppm or that are severely attenuated during titration (indicated as brown bars at full scale in a) are mapped on the surface of the hSRI domain. The orientation of the hSRI domain is identical to that in Fig. 1 a and b. b was generated by molmol (35).
for each residue and plotted. Secondary structures are shown above the plot. (b) Resonances that experience chemical shift perturbation of >0.095 ppm or that are severely attenuated during titration (indicated as brown bars at full scale in a) are mapped on the surface of the hSRI domain. The orientation of the hSRI domain is identical to that in Fig. 1 a and b. b was generated by molmol (35).
Binding (Biacore) Assay. Interaction analysis was performed essentially as described in ref. 7 by using a Biacore 3000 sensor. Detailed descriptions of the methods used are provided in Supporting Text.
Results
Identification of the Minimal hSRI Domain. A GST fusion protein carrying the C-terminal 178-aa segment (1,884–2,061) of hSet2/HYPB, which contains the SRI domain, interacts efficiently with the PCTD (Fig. 5). Thrombin digestion, which separates GST from the hSet2 fragment, also releases a smaller piece that retains the PCTD binding ability. This piece accumulates at the expense of the larger hSet2 fragment as cleavage times are increased and appears as a strong PCTD-interacting band after 6 days of thrombin treatment, even though it is stained only weakly by Ponceau S (Fig. 5). Mass spectrometric analysis revealed that this smaller-molecular-weight band in fact consists of two slightly different fragments, encompassing amino acids 1,948–2,061 and 1,954–2,061 of hSet2, i.e., the region of the protein analogous to the SRI of ySet2. Because both fragments bound the PCTD (data not shown), we used the smaller one for further studies.
To confirm that we had indeed identified the minimal hSRI domain, we expressed and purified this domain (residues 1,954–2,061 of hSet2) as a recombinant protein and examined its PCTD binding properties using surface plasmon resonance (Biacore). On the surface of a streptavidin sensor chip, we immobilized chemically synthesized, biotinylated three-repeat CTD peptides with phosphoserines at exactly known positions, as described in refs. 7 and 20. The peptides (see Table 1) are phosphorylated on either Ser-2 of each repeat (2,2,2 peptide), Ser-5 of each repeat (5,5,5 peptide), or both Ser-2 and -5 of each repeat (2,5,2,5,2,5 peptide), and they mimic PCTD forms likely encountered by Set2 in vivo. We found that, similar to the yeast SRI domain (20), the hSRI domain binds specifically to CTD repeats doubly phosphorylated on Ser-2 and -5 of each heptad (Fig. 5). As a charge control, we included the 6PC peptide (Table 1), which also contains six phosphoserines, but not in the context of the CTD heptad repeats. Thus, even though the hSRI domain is a basic protein with an abundance of Arg and Lys residues, its binding to the PCTD cannot be attributed solely to nonspecific charge-based interactions: the phosphoserines in the context of the CTD heptad sequence determine its binding specificity.
Table 1. Peptides used to study the hSRI domain–PCTD interactions.
| Synthetic CTD peptides
|
|
|---|---|
| Name | Sequence |
| NP | Y1 S2P3T4S5P6S7Y1S2P3T4S5P6S7Y1S2P3T4S5P6S7 |
| 5,5,5 | Y S P T S5 P S Y S P T S5 P S Y S P T S5 P S |
| 2,2,2 | Y S2 P T S P S Y S2 P T S P S Y S2 P T S P S |
| 2,5,2,5,2,5 | Y S2 P T S5 P S Y S2 P T S5 P S Y S2 P T S5 P S |
| 2,5,2,5 | T S P S Y S2 P T S5 P S Y S2 P T S5 P S Y S P T |
| 5,2,5,2 | S Y S P T S5 P S Y S2 P T S5 P S Y S2 P T S P S |
| 2,5,2 | T S P S Y S2 P T S5 P S Y S2 P T S P S |
| 5,2,5 | S Y S P T S5 P S Y S2 P T S5 P S Y S P T |
| 2,5 | T S P S Y S2 P T S5 P S Y S P T |
| 5,2 | S Y S P T S5 P S Y S2 P T S P S |
| 6PC | G S A P S S G S A P S P S G P S A S G P S G |
S = SerPO4.
hSRI Domain Possesses a Previously Undescribed Fold for PCTD Recognition. Recombinant hSRI domain was overexpressed, isotopically labeled, and extensively purified for structural studies by solution NMR. By analytical ultracentrifugation, we found the hSRI domain to be monomeric in solution (data not shown). By using multidimensional NMR spectroscopy, we obtained the complete assignment of the 1H, 13C, and 15N resonances of the hSRI domain, except for T50 (see Materials and Methods and Fig. 1c for numbering), which is exchange broadened. With the exception of a few residues at the N and C termini (residues 1–9 and 110–112), the protein is well structured. Twenty structures were calculated with 2,600 nuclear Overhauser effects and 178 dihedral angle constraints and further refined against 120 residual dipolar couplings by using a water refinement protocol (Table 3) (25, 26). The structural ensemble is presented in Fig. 1a, and the corresponding ribbon diagram is in Fig. 1b. The mean pairwise rms deviation for the backbone atoms of residues 10–109 was 0.43 Å.
Fig. 1.

Solution structure and sequence alignment of the hSRI domain. (a) Stereo view of backbone traces from 20 structures of the hSRI domain with helices colored in red and loops in gray. (b) Stereo view of the ribbon diagrams of the hSRI domain. Side chains of conserved hydrophobic residues important for the packing of the three-helix bundle are shown as stick models in green. a and b were prepared by using molmol (35). (c) Sequence alignment of SRI domains from different species (first amino acid of each sequence is numbered). Conserved hydrophobic residues are colored in yellow, basic residues are in blue, and acidic residues are in red. Residues important in maintaining the hydrophobic core of hSRI domain are denoted by green circles above the sequence. Residues important for PCTD interactions are denoted by asterisks. Secondary structures and residue numbers used in NMR studies are shown above the sequence alignment. See Supporting Text for listing of species and GenInfo Identifier (GI) accession numbers.
The hSRI domain forms a compact, closed three-helix bundle, with an up–down–up topology (Fig. 1 a and b). The first and second helices (α1 and α2) are antiparallel to each other and are of similar length, each containing ≈21–23 aa; the third helix (α3), which is packed across helices 1 and 2 at an ≈30° angle and is positioned in the back, is slightly shorter, consisting of only 15 aa. Most conserved hydrophobic residues in the SRI domain family (F22, M26, F29, I30, and L34 of α1; Y37, V44 of the α1–α2 loop; L56, A57, L60, T61, V64, M65, and L69 of α2; L78 of the α2–α3 loop; T88, Y91, I92, Y95, and M96 of α3; and F99 and Y103 of the C-terminal loop) are largely buried in the interior of the structure and form an extensive and contiguous hydrophobic core that stabilizes the packing of the three-helix bundle (Fig. 1 b and c). The packing and stability of these helices are augmented by an interhelix salt bridge involving highly conserved residues E68 in α2 and K87 in α3. Additional conserved residues are dominated by basic side chains, some of which are presumably involved in binding to highly negatively charged PCTD peptides (Fig. 1c).
The structure of the hSRI domain is very different from all of the characterized protein domains observed in complex with PCTD peptides (10, 11, 13, 14). Interestingly, the hSRI domain superficially resembles the FF domain from HYPA/FBP11, a compact PCID that also folds into a three-helix bundle (9). However, the topologies of these two proteins are quite distinct. In the hSRI domain, if helix 1 is positioned in the front with an “up” orientation, helices 2 and 3 are located to the left and right of helix 1, respectively. Borrowing the nomenclature of four-helix bundles (27, 28), the hSRI domain belongs to a family of “left-turned” three-helix bundles. In the FF domain, helices 2 and 3 are located to the right and left of helix 1, respectively; the FF domain thus belongs to a family of “right-turned” three-helix bundles. In addition, all three helices in the hSRI domain are roughly parallel or antiparallel, with a relatively small angle (≈30°) between any two helices. In contrast, the three helices in the FF domain are positioned in an almost orthogonal orientation. These differences are also reflected at the primary sequence level, because the two protein families do not appear to be related.
hSRI Domain Interacts with PCTD Predominantly Through α1 and α2. After establishing the hSRI domain as a previously undescribed PCID, we next began probing its binding surface by NMR titration, using the three-repeat CTD phosphopeptide (peptide 2,5,2,5,2,5) shown in Table 1. A series of 1H-15N heteronuclear single quantum correlation spectra of the hSRI domain were collected in the presence of increasing amounts of the 2,5,2,5,2,5 peptide. In the course of the titration, the 1H-15N resonances of K19, E20, M26, Q28, F29, H55, R58, M65, E68, D77, E79, K93, and K94 showed significant progressive perturbations (>2-fold above the average value), and the resonances of V31, A57, H62, G63, and E76 were severely attenuated, indicating that the hSRI domain–PCTD peptide interaction is in the fast-to-intermediate exchange regime on the NMR time scale. Most of the affected residues (K19, E20, M26, Q28, F29, V31, H55, A57, R58, H62, G63, M65, and E68) are distributed along helices 1 and 2 (Fig. 2). Surprisingly, the set of perturbed residues also includes some with negative charge, E76, D77, and E79, located in the loop connecting α2 and α3. These residues, which are not conserved within the SRI domain family, seem likely to cause repulsive electrostatic interactions with the highly negatively charged PCTD peptide (see below). Therefore, we expect that they do not contribute to PCTD binding directly. Taking all these considerations together, we propose that the primary PCTD docking site is situated in the concave surface between α1 and α2.
Substrate Specificity of hSRI Domain. Because of the circularly permuted nature of the CTD sequence, it is important to realize that the functional unit for recognition may span more than one canonical heptad repeat, and the boundary of this functional unit could start with any amino acid within the Y1S2P3T4S5P6S7 sequence (see, e.g., refs. 11 and 12). In addition, although the hSRI domain binds the three-repeat, Ser-2 + Ser-5 phosphorylated CTD peptide (2,5,2,5,2,5) with high specificity (Fig. 5), the small size and monomeric state in solution of the hSRI domain suggested that it would be unlikely to recognize all six Ser-2P + Ser-5P residues at once. To test this notion, and to define more narrowly both the number and arrangement of phosphoserine moieties that the hSRI domain requires for recognition (its “phosphoepitope”), we examined the hSRI domain binding to a series of peptide derivatives that differed in the disposition of their phosphoserine groups (Table 1). We used Biacore technology as before to obtain binding sensorgrams for each phosphopeptide, using the nonphosphorylated NP peptide as a control. Representative sensorgrams shown in Fig. 3a illustrate that the hSRI domain binds the 2,5,2,5,2,5 peptide with a high degree of specificity, as compared with controls (NP in this experiment, 6PC in Fig. 5c). Moreover, the hSRI domain binds as well to the 2,5,2,5 and the 5,2,5,2 peptides as it does to the 2,5,2,5,2,5 peptide, indicating that four contiguous Ser-2/Ser-5 phosphates are sufficient for maximal binding. Peptides with three contiguous Ser-2/Ser-5 phosphates (2,5,2 or 5,2,5 peptides) exhibit decreased binding (differences quantified by equilibrium binding analysis) (see Table 2; see also Fig. 6, which is published as supporting information on the PNAS web site). Importantly, the affinity of the hSRI domain for the 2,5,2 and the 5,2,5 peptides is orders of magnitude tighter than for either the 2,2,2 or the 5,5,5 peptides (binding too weak for accurate measurement; see Table 2), whose three Ser-phosphates are farther apart. These results demonstrate that the correct spacing of the phosphate groups on the peptide plays a vital role in determining the binding epitope and that the precise length of the peptide or its starting (N-terminal) amino acid appears to have minimal effects on binding. Finally, PCTD peptides carrying only two contiguous SerP residues (2,5 or 5,2 peptide) show virtually no binding to the hSRI domain, indicating that three or more contiguous SerPs are required for significant binding. In summary, specific binding of the hSRI domain to phosphorylated CTD repeats depends critically both on the number of SerP residues and on their spatial arrangement.
Fig. 3.

The hSRI domain–PCTD interaction. (a) Biacore sensorgrams showing the interaction of the hSRI domain with different PCTD peptides. The hSRI domain interacts best with [Ser-2 + Ser-5]-phosphorylated PCTDs containing at least two complete repeats (2,5,2,5,2,5 peptide, 2,5,2,5 peptide, and 5,2,5,2 peptide), with severalfold weaker affinity toward 2,5,2 and 5,2,5 peptides (see Table 2) and with extremely weak affinity for other PCTD peptides. (b) Equilibrium binding curves of WT hSRI domain and five single-point mutations that diminish the binding affinity of the hSRI domain toward the 2,5,2,5 PCTD peptide. (c) Surface mapping of the five residues in the hSRI domain important for the PCTD interactions. Orientation of the hSRI domain is identical to that in Fig. 1 a and b. c is generated by molmol (35).
Table 2. Binding affinities of hSRI mutant proteins toward PCTD peptides.
| Phosphopeptide
|
|||
|---|---|---|---|
| Protein | 2,5,2,5 | 2,5,2 | 5,2,5 |
| E76A + E79A | 1.3 ± 0.3 | 3.9 ± 0.5 | 3.7 ± 0.5 |
| E76A | 2.0 ± 0.1 | 5.7 ± 0.1 | 5.5 ± 0.1 |
| E79A | 2.3 ± 0.1 | 6.2 ± 0.3 | 5.5 ± 0.3 |
| WT | 5.4 ± 0.5 | 17.8 ± 0.9 | 12.5 ± 1.5 |
| V31A | 23.1 ± 2.3 | 90.8 ± 12.7 | 59.2 ± 5.3 |
| H62A | 37.6 ± 4.9 | 164.9 ± 35.9 | 80.4 ± 5.4 |
| K54A | 60.2 ± 11.0 | * | 76.1 ± 5.9 |
| F53L | 75.5 ± 18.0 | 279.6 ± 111.7 | 108.7 ± 15.4 |
| R58A | 132.5 ± 62.8 | * | 210.6 ± 71.1 |
KD values are in μM. Asterisks indicate binding too weak to measure reliably.
Mutagenesis Studies. The majority of the residues in the hSRI domain that experience backbone resonance perturbations upon titrating in CTD phosphopeptides are located in helices 1 and 2. Although some of these residues contribute side chains that form part of the hydrophobic core of the three-helix bundle, a significant number of them map onto one face of the protein; thus, their side chains could potentially contribute to PCTD binding. Residues in close proximity to a binding interface can display resonance perturbations even though they may not contribute significantly to the binding energy. To identify side chains actually involved in binding, we generated a series of site-specific mutations in α1 (R23, K24, F29, and V31) and α2 (F53, K54, R58, H62, and G63), expressed and purified the mutant proteins, and compared them with the WT hSRI domain in terms of PCTD peptide binding. We also included E76 and E79 for investigation, because they experienced some of the largest resonance perturbations during NMR titration. Circular dichroism spectra of these mutant proteins showed that none of these point mutations disrupts the folding of the protein (data not shown).
Point mutations of these residues differed significantly in the extent to which they affected binding. Some, like R23A, K24A, Q28A, F29A, G63T, and G63E, had little if any effect (data not shown), whereas others, like E76A/E79A appeared to actually increase binding, presumably by removing negative side chains that might be repulsive to the phosphates (Table 2). Most significantly, five single mutations considerably diminished the hSRI domain binding affinity. Equilibrium binding curves for the interactions between these mutant hSRI domains (V31A, F53L, K54A, R58A, and H62A) and the 2,5,2,5 peptide are shown in Fig. 3b. Interestingly, four of the five residues (F53, K54, R58, and H62) map on the same face of helix 2, and the fifth, V31, is located at the C terminus of helix 1 and is close to F53, a critical residue from helix 2 (Fig. 3c). The most deleterious mutations involve the loss of either a positive charge (R58A and K54A) or an aromatic group (the relatively conservative F53L); each of these changes raises the KD at least 10-fold. In addition, mutating these residues also appears to differentially affect binding to the two three-phosphate peptides: e.g., the H62A and F53L mutants bind the 2,5,2 peptide with 2- to 3-fold higher KD values than the 5,2,5 peptide, whereas R58A or K54A mutants bind so weakly to the 2,5,2 peptide that KD values could not be reliably measured (Table 2). Together, these observations suggest that V31, F53, K54, R58, and H62 play a major role in the interaction between the hSRI domain and the PCTD, most likely by forming part of the binding interface. The results also suggest that the surfaces on the hSRI domain that bind to the 2,5,2 and 5,2,5 peptides partially overlap and that the complete binding surface of the hSRI domain is most suitable for recognizing PCTD repeats containing more than three contiguous Ser-2/5P residues (see Discussion and Fig. 4; see also Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 4.

Speculative model of the hSRI domain interaction with PCTD peptides. Within the rectangle representing the hSRI domain, amino acids found to be critical for binding (H62, R58, V31, F53, and K54) are shown with brown arrows pointing to residues in the CTD heptad repeats with which they might interact (for Ser, when phosphorylated). Potential participation of K19 and R38 in binding is based on their positions in the 3D structure and the equivalent binding affinities of 5,2,5,2 and 2,5,2,5 peptides (see Discussion). Red dots in peptides indicate SerP.
Discussion
Model of the hSRI Domain–PCTD Interaction. The binding site on the hSRI domain, into which the PCTD docks, was first roughly localized by mapping NMR perturbations resulting from phosphopeptide binding. Contributions to binding of individual residues in the CTD docking site were then more precisely defined by using a combination of site-directed mutagenesis and phosphopeptide binding experiments. This approach identified three positive side chains from helix 2 (K54, R58, and H62) that contribute significantly to binding; we think it plausible that each interacts with a Ser-2P or Ser-5P residue in the preferred phosphoepitope (see Fig. 4). Two uncharged residues, F53 (from α2) and V31 (from α1), are also very important for binding; they appear to participate in forming a hydrophobic patch in a groove running roughly in parallel with and between helices 1 and 2. It seems credible that the aromatic ring of F53 may provide an interaction surface for the side chain of a Y1 residue in the PCTD repeats. This overall picture is reminiscent of a pattern seen in the structure of the capping enzyme in complex with a four-repeat phosphopeptide, in which ionic interactions (involving Ser-5P residues of the repeats) alternate with hydrophobic interactions (involving Tyr and Pro residues of the repeats); the binding surface on the capping enzyme is some 40-Å long, accommodating a fairly stretched-out, sparsely phosphorylated CTD (11). In the hSRI domain, binding surface features are arrayed analogously but with positively charged residues closer together to accommodate CTD repeats with more closely spaced SerP residues (positions 2 and 5 of consecutive repeats).
SRI domain-PCTD interactions such as those suggested in the speculative Fig. 4 may explain these results. If PCTD peptides bind in the orientation and register shown, for example, the 5,2,5,2 and 2,5,2,5 peptides could both use three SerPs, and residues between them, to interact with the “core” CTD docking site residues, those demonstrated by mutagenesis to be very important for peptide binding (H62, R58, V31, F53, and K54). The fourth SerP in these peptides (N-terminal in 5,2,5,2 and C-terminal in 2,5,2,5) could potentially interact with additional positive residues that appear to be appropriately situated in the hSRI domain structure (e.g., K19 and R38 for 5,2,5,2 peptide and 2,5,2,5 peptide, respectively). In this way “core” interactions plus an additional end-specific interaction could result in very similar KD values for the two different peptides. Continuing with this hypothetical scheme (Fig. 4), we would expect mutation K54A to affect binding of the 2,5,2 peptide more than that of the 5,2,5 peptide, because the former makes a contact with K54 whereas the latter does not; the data support this expectation. In contrast, for peptides with more widely spaced SerPs (e.g., 2,2,2 or 5,5,5), it is apparently not possible for the peptide to adopt a conformation that produces enough productive interactions for significant binding. Further structural studies will be needed to test the validity of the proposed interactions.
Conservation of SRI Domain and PCTD-Binding Interface. How well conserved is the SRI domain? The sequence alignment of putative SRI domains from diverse eukaryotes indicates that, although the α1–α2 loop may be longer in vertebrates, the hydrophobic residues that comprise the structural core of the three-helix bundle are highly conserved, as is the salt bridge between helices 2 and 3 (E68 and K87, respectively). Such conservation of core structural components argues for a similar tertiary organization of all SRI domains. In addition, three of the five amino acids believed to form part of the binding interface in the hSRI domain also appear to be well conserved; the Lys at position 54 is, in fact, one of two invariant residues (K87 is the other). The residue at position 58 is nearly always Arg or Lys, and the residue at position 62 is, in most cases, either His or Lys. The notion that these amino acids may be involved in recognizing the closely spaced negative charges on the CTD phosphopeptide is consistent with the shared phosphoepitope binding specificity of the yeast and human SRI domains. In contrast to these (largely) positively charged amino acids, two hydrophobic residues that contribute to the binding properties of the hSRI domain, V31 and F53, display a greater degree of evolutionary variability. Residue 31 at the end of α1 is often Pro (instead of Val) in many nonanimal eukaryotes, whereas position 53 at the beginning of α2 is occupied by an aromatic residue only in about half the sequences examined. We suggest that the functionality provided by this pair of closely apposed residues in the human protein may be contributed by other chemically similar amino acid pairs in other organisms. Although specific details of such compensatory interactions await additional experiments, this idea is consistent with the structural properties of the PCTD: in addition to being flexible enough to dock into binding domains with similar tertiary structures but nonidentical sequences, it also can accommodate several structurally different binding partners (13, 14, 29, 30).
Concluding Remarks. The hSRI domain is the founding member of a PCID family with a left-turned three-helix bundle. The primary CTD-docking site is located between helices 1 and 2 and employs side chains emanating from both helices. The hSRI domain recognizes a phosphoepitope comprising at least four contiguous SerP residues in consensus heptad repeat positions 2,5,2,5 or 5,2,5,2.
It is interesting to consider our results in the context of the finding that heptad repeat pairs (di-heptads) comprise the minimal “functional unit” in the CTD, in terms of cell viability (31): a binding requirement for four contiguous Ser-2/5P residues indicates an epitope length of just about one diheptad. In addition, our findings, like previous results (see introduction), support a role for Set2 during the elongation phase of transcription because RNA polymerase II actively traversing a transcription unit, after leaving the immediate vicinity of the promoter and before approaching the polyA and termination sites, is likely to carry doubly phosphorylated CTD repeats (6, 20, 32–34).
Supplementary Material
Acknowledgments
We thank Dr. Marcy MacDonald (Harvard Medical School, Boston) for providing the GST-HYPB construct and Dr. Munir Alam (Duke Human Vaccine Institute) for assistance with the Biacore analysis. This work was supported by National Institutes of Health Grant GM40505 (to A.L.G.) and the Whitehead Institute (to P.Z.).
Author contributions: M.L., H.P.P., A.L.G., and P.Z. designed research; M.L., H.P.P., Z.G., and H.S. performed research; M.L., H.P.P., Z.G., H.S., A.L.G., and P.Z. analyzed data; and H.P.P., A.L.G., and P.Z. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CTD, C-terminal repeat domain; PCTD, hyperphosphorylated CTD; PCID, PCTD-interacting domain; HYPB, huntingtin yeast partner B; SRI, Set2–Rpb1 interacting; hSRI, human SRI; ySet2, yeast Set2; hSet2, human Set2.
Data deposition: Atomic coordinates for the structural ensemble of hSRI domain have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2A7O).
References
- 1.Bentley, D. (2002) Curr. Opin. Cell. Biol. 14, 336–342. [DOI] [PubMed] [Google Scholar]
- 2.Howe, K. J. (2002) Biochim. Biophys. Acta 1577, 308–324. [DOI] [PubMed] [Google Scholar]
- 3.Proudfoot, N. J., Furger, A. & Dye, M. J. (2002) Cell 108, 501–512. [DOI] [PubMed] [Google Scholar]
- 4.Dahmus, M. (1996) J. Biol. Chem. 271, 19009–19012. [DOI] [PubMed] [Google Scholar]
- 5.Sims, R. J., III, Belotserkovskaya, R. & Reinberg, D. (2004) Genes Dev. 18, 2437–2468. [DOI] [PubMed] [Google Scholar]
- 6.Jones, J. C., Phatnani, H. P., Haystead, T. A., MacDonald, J. A., Alam, S. M. & Greenleaf, A. L. (2004) J. Biol. Chem. 279, 24957–24964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phatnani, H. P., Jones, J. C. & Greenleaf, A. L. (2004) Biochemistry 43, 15702–15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carty, S. M. & Greenleaf, A. L. (2002) Mol. Cell. Proteomics 1, 598–610. [DOI] [PubMed] [Google Scholar]
- 9.Allen, M., Friedler, A., Schon, O. & Bycroft, M. (2002) J. Mol. Biol. 323, 411–416. [DOI] [PubMed] [Google Scholar]
- 10.Verdecia, M. A., Bowman, M. E., Lu, K. P., Hunter, T. & Noel, J. P. (2000) Nat. Struct. Biol. 7, 639–643. [DOI] [PubMed] [Google Scholar]
- 11.Fabrega, C., Shen, V., Shuman, S. & Lima, C. D. (2003) Mol. Cell 11, 1549–1561. [DOI] [PubMed] [Google Scholar]
- 12.Greenleaf, A. (2003) Structure (London) 11, 900–902. [DOI] [PubMed] [Google Scholar]
- 13.Meinhart, A. & Cramer, P. (2004) Nature 430, 223–226. [DOI] [PubMed] [Google Scholar]
- 14.Noble, C. G., Hollingworth, D., Martin, S. R., Ennis-Adeniran, V., Smerdon, S. J., Kelly, G., Taylor, I. A. & Ramos, A. (2005) Nat. Struct. Mol. Biol. 12, 144–151. [DOI] [PubMed] [Google Scholar]
- 15.Li, J., Moazed, D. & Gygi, S. P. (2002) J. Biol. Chem. 277, 49383–49388. [DOI] [PubMed] [Google Scholar]
- 16.Xiao, T., Hall, H., Kizer, K. O., Shibata, Y., Hall, M. C., Borchers, C. H. & Strahl, B. D. (2003) Genes Dev. 17, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaft, D., Roguev, A., Kotovic, K. M., Shevchenko, A., Sarov, M., Shevchenko, A., Neugebauer, K. M. & Stewart, A. F. (2003) Nucleic Acids Res. 31, 2475–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, B., Howe, L., Anderson, S., Yates, J. R., III, & Workman, J. L. (2003) J. Biol. Chem. 278, 8897–8903. [DOI] [PubMed] [Google Scholar]
- 19.Krogan, N. J., Kim, M., Tong, A., Golshani, A., Cagney, G., Canadien, V., Richards, D. P., Beattie, B. K., Emili, A., Boone, C., et al. (2003) Mol. Cell. Biol. 23, 4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kizer, K. O., Phatnani, H. P., Shibata, Y., Hall, H., Greenleaf, A. L. & Strahl, B. D. (2005) Mol. Cell. Biol. 25, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Passani, L. A., Bedford, M. T., Faber, P. W., McGinnis, K. M., Sharp, A. H., Gusella, J. F., Vonsattel, J. P. & MacDonald, M. E. (2000) Hum. Mol. Genet. 9, 2175–2182. [DOI] [PubMed] [Google Scholar]
- 22.Faber, P. W., Barnes, G. T., Srinidhi, J., Chen, J., Gusella, J. F. & MacDonald, M. E. (1998) Hum. Mol. Genet. 7, 1463–1474. [DOI] [PubMed] [Google Scholar]
- 23.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 24.Bartels, C., Xia, T., Billeter, M., Güntert, P. & Wüthrich, K. (1995) J. Biol. NMR 6, 1–10. [DOI] [PubMed] [Google Scholar]
- 25.Schwieters, C. D., Kuszewski, J. J., Tjandra, N. & Clore, G. M. (2003) J. Magn. Reson. 160, 65–73. [DOI] [PubMed] [Google Scholar]
- 26.Nabuurs, S. B., Nederveen, A. J., Vranken, W., Doreleijers, J. F., Bonvin, A. M., Vuister, G. W., Vriend, G. & Spronk, C. A. (2004) Proteins 55, 483–486. [DOI] [PubMed] [Google Scholar]
- 27.Hecht, M. H., Richardson, J. S., Richardson, D. C. & Ogden, R. C. (1990) Science 249, 884–891. [DOI] [PubMed] [Google Scholar]
- 28.Kamtekar, S. & Hecht, M. H. (1995) FASEB J. 9, 1013–1022. [DOI] [PubMed] [Google Scholar]
- 29.Guo, Z. & Stiller, J. W. (2005) Mol. Biol. Evol. 22, 2166–2178. [DOI] [PubMed] [Google Scholar]
- 30.Meinhart, A., Kamenski, T., Hoeppner, S., Baumli, S. & Cramer, P. (2005) Genes Dev. 19, 1401–1415. [DOI] [PubMed] [Google Scholar]
- 31.Stiller, J. W. & Cook, M. S. (2004) Eukaryotic Cell 3, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boehm, A. K., Saunders, A., Werner, J. & Lis, J. T. (2003) Mol. Cell. Biol. 23, 7628–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahn, S. H., Kim, M. & Buratowski, S. (2004) Mol. Cell 13, 67–76. [DOI] [PubMed] [Google Scholar]
- 34.Phatnani, H. P. & Greenleaf, A. L. (2004) Methods Mol. Biol. 257, 17–28. [DOI] [PubMed] [Google Scholar]
- 35.Koradi, R., Billeter, M. & Wüthrich, K. (1996) J. Mol. Graphics 14, 51–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.