

Abstract
We describe the use of the matrix eigenvalue decomposition (EVD) and pseudoinverse projection and a tensor higher-order EVD (HOEVD) in reconstructing the pathways that compose a cellular system from genome-scale nondirectional networks of correlations among the genes of the system. The EVD formulates a genes × genes network as a linear superposition of genes × genes decorrelated and decoupled rank-1 subnetworks, which can be associated with functionally independent pathways. The integrative pseudoinverse projection of a network computed from a “data” signal onto a designated “basis” signal approximates the network as a linear superposition of only the subnetworks that are common to both signals and simulates observation of only the pathways that are manifest in both experiments. We define a comparative HOEVD that formulates a series of networks as linear superpositions of decorrelated rank-1 subnetworks and the rank-2 couplings among these subnetworks, which can be associated with independent pathways and the transitions among them common to all networks in the series or exclusive to a subset of the networks. Boolean functions of the discretized subnetworks and couplings highlight differential, i.e., pathway-dependent, relations among genes. We illustrate the EVD, pseudoinverse projection, and HOEVD of genome-scale networks with analyses of yeast DNA microarray data.
Keywords: DNA microarrays, eigenvalue decomposition, higher-order eigenvalue decomposition, pseudoinverse projection, yeast Saccharomyces cerevisiae cell cycle and mating
DNA microarrays make it possible to record the complete genomic signals, such as mRNA expression (e.g., refs. 1 and 2) and DNA-bound proteins' occupancy levels (e.g., ref. 3), that are generated and sensed by cellular systems. The underlying genome-scale networks of relations among all genes of the cellular systems can be computed from these signals (e.g., refs. 4–6). These relations among the activities of genes, not only the activities of the genes alone, are known to be pathway-dependent, i.e., conditioned by the biological and experimental settings in which they are observed (e.g., ref. 7). For example, the mRNA expression patterns of the yeast Saccharomyces cerevisiae genes KAR4 and CIK1 are correlated during mating yet anticorrelated during cell-cycle progression (8). A single genome-scale nondirectional network of correlations cannot describe the pathway-dependent differences in relations, such as those between the expression patterns of KAR4 and CIK1.
Recently, we showed that the matrix singular-value decomposition (SVD), generalized SVD, and pseudoinverse projection separate genome-scale signals, i.e., gene and array patterns of, e.g., mRNA expression and proteins' DNA binding, into mathematically defined patterns that correlate with the independent biological and experimental processes and cellular states that compose the signals (9–12). For example, the comparative generalized SVD of yeast and human mRNA expression during their cell cycles formulates the yeast expression as a linear superposition of cell-cycle oscillations, which are common to the yeast and human, and response to synchronization by the mating pheromone, which is exclusive to the yeast, and describes a differential relation in the expression of genes such as KAR4 and CIK1 that is in agreement with their pathway-dependent activities (11).
Now, we describe the use of the matrix eigenvalue decomposition (EVD) and pseudoinverse projection and a tensor higher-order EVD (HOEVD) in reconstructing the pathways, or genome-scale pathway-dependent relations among the genes of a cellular system, from nondirectional networks of correlations, which are computed from measured genomic signals and tabulated in symmetric matrices. The EVD formulates a genes × genes network, which is computed from a “data” signal, as a linear superposition of genes × genes decorrelated and decoupled rank-1 subnetworks. We show that significant EVD subnetworks might represent functionally independent pathways that are manifest in the data signal. The integrative pseudoinverse projection of a network, computed from a data signal, onto a designated “basis” signal approximates the network as a linear superposition of only the subnetworks that are common to both signals, i.e., pseudoinverse projection filters off the network the subnetworks that are exclusive to the data signal. We show that the pseudoinverse-projected network simulates observation of only the pathways that are manifest under both sets of the biological and experimental conditions where the data and basis signals are measured. We define a comparative HOEVD that formulates a series of networks computed from a series of signals as linear superpositions of decorrelated rank-1 subnetworks and the rank-2 couplings among these subnetworks. We show that significant HOEVD subnetworks and couplings might represent independent pathways or transitions among them common to all or exclusive to a subset of the signals. Boolean functions of the discretized subnetworks and couplings highlight known as well as previously unknown differential, i.e., pathway-dependent relations between genes. We illustrate the EVD, pseudoinverse projection, and HOEVD of genome-scale networks with analyses of mRNA expression data from the yeast Saccharomyces cerevisiae during its cell cycle (1) and DNA-binding data of yeast transcription factors that are involved in cell-cycle, development, and biosynthesis programs (3).
Mathematical Methods: EVD, Pseudoinverse Projection, and HOEVD of Networks
Eigenvalue Decomposition. Let the symmetric matrix â1 of size N-genes × N-genes tabulate the genome-scale nondirectional network of correlations among the genes of a cellular system.¶ The network â1 is computed from a genome-scale signal, designated the data signal, of, e.g., mRNA expression levels measured in a set of M1 samples of the cellular system using M1 DNA microarrays and tabulated in the N-genes × M1-arrays matrix ê1, such that 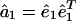 . We compute the EVD of the network â1,
. We compute the EVD of the network â1,
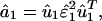 |
[1] |
from the SVD of the data signal 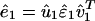 (9, 10, 13). The M1-“eigenarrays” × M1-“eigengenes” diagonal matrix
(9, 10, 13). The M1-“eigenarrays” × M1-“eigengenes” diagonal matrix 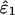 defines the M1 nonnegative “eigenexpression” levels, such that the expression of the mth eigengene in the mth eigenarray is the mth eigenexpression level of ê1,
defines the M1 nonnegative “eigenexpression” levels, such that the expression of the mth eigengene in the mth eigenarray is the mth eigenexpression level of ê1, 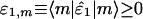 . The orthogonal transformation matrices û1 and
. The orthogonal transformation matrices û1 and 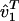 define the N-genes × M1-eigenarrays and the M1-eigengenes × M1-arrays subspaces, respectively. The mth column of û1, |α1,m 〉 ≡ û1|m 〉, lists the genome-scale expression of the mth eigenarray of ê1. The nth row of
define the N-genes × M1-eigenarrays and the M1-eigengenes × M1-arrays subspaces, respectively. The mth column of û1, |α1,m 〉 ≡ û1|m 〉, lists the genome-scale expression of the mth eigenarray of ê1. The nth row of 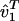 ,
, 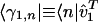 , lists the expression of the nth eigengene.
, lists the expression of the nth eigengene.
EVD formulates the network â1 as a linear superposition of a series of M1 rank-1 symmetric “subnetworks” of size N-genes × N-genes each, where the mth subnetwork is the outer product of the mth eigenarray with its transpose |α1,m 〉 〈α1,m| (Fig. 5 in Supporting Appendix, which is published as supporting information on the PNAS web site),
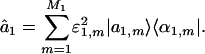 |
[2] |
The significance of the mth subnetwork is indicated by the mth “fraction of eigenexpression” 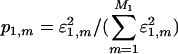 , i.e., the expression correlation captured by the mth subnetwork relative to that captured by all subnetworks. Each subnetwork is decorrelated of all other subnetworks, i.e., |α1,m 〉 〈α1,m|α1,n 〉 〈α1,n| = 0 for all m ≠ n, since û1 is orthogonal. Each subnetwork is also decoupled of all other subnetworks, such that there are no contributions to the network â1 from the M1(M1 – 1)/2 rank-2 symmetric “couplings” among the subnetworks, i.e., |α1,m 〉 〈α1,n| + |α1,n 〉 〈α1,m| for all m ≠ n, since
, i.e., the expression correlation captured by the mth subnetwork relative to that captured by all subnetworks. Each subnetwork is decorrelated of all other subnetworks, i.e., |α1,m 〉 〈α1,m|α1,n 〉 〈α1,n| = 0 for all m ≠ n, since û1 is orthogonal. Each subnetwork is also decoupled of all other subnetworks, such that there are no contributions to the network â1 from the M1(M1 – 1)/2 rank-2 symmetric “couplings” among the subnetworks, i.e., |α1,m 〉 〈α1,n| + |α1,n 〉 〈α1,m| for all m ≠ n, since 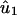 is diagonal. For a real data signal ê1, the eigenarrays are unique up to phase factors of ±1, and therefore the subnetworks are also unique, i.e., data-driven, except in degenerate subspaces defined by subsets of equal eigenexpression levels.
is diagonal. For a real data signal ê1, the eigenarrays are unique up to phase factors of ±1, and therefore the subnetworks are also unique, i.e., data-driven, except in degenerate subspaces defined by subsets of equal eigenexpression levels.
Pseudoinverse Projection. Let the matrix b̂ of size N-genes × L-arrays tabulate the genome-scale signal, designated the “basis” signal, of, e.g., proteins' DNA-binding occupancy levels measured in a set of L samples of the cellular system using L arrays. We compute the pseudoinverse projection (12, 13) of the network â1 onto the basis signal b̂,
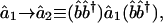 |
[3] |
from the projection of the data ê1 onto the basis b̂, ê1 → ê2 = b̂b̂†ê1, using the SVD of the basis 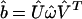 to compute its pseudoinverse
to compute its pseudoinverse 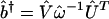 . The lth column of Û, |βl 〉≡ Û|l 〉, lists the genome-scale binding of the lth eigenarray of b̂. The pseudoinverse-projected network â2 is unique, i.e., data-driven. For a real basis signal b̂, b̂b̂† is an orthogonal projection matrix, and the projected network â2 is symmetric.
. The lth column of Û, |βl 〉≡ Û|l 〉, lists the genome-scale binding of the lth eigenarray of b̂. The pseudoinverse-projected network â2 is unique, i.e., data-driven. For a real basis signal b̂, b̂b̂† is an orthogonal projection matrix, and the projected network â2 is symmetric.
We compute the EVD of the projected network â2,
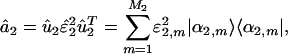 |
[4] |
where M2 = min{L, M1}, from the SVD of the projected signal 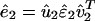 , where the mth column of û2, |α2,m 〉≡ û2|m 〉, lists the genome-scale expression of the mth eigenarray of ê2. In reconstructing â2, the pseudoinverse projection filters out of â1 each of its subnetworks |α1,m 〉 〈α1,m|, which is decorrelated of the series of L rank-1 symmetric subnetworks |βl 〉 〈βl| that compose the network b̂b̂T computed from the basis signal b̂, such that |βl 〉 〈βl|α1,m 〉 〈α1,m| = 0 for all l = 1, 2,..., L (Fig. 6 in Supporting Appendix).
, where the mth column of û2, |α2,m 〉≡ û2|m 〉, lists the genome-scale expression of the mth eigenarray of ê2. In reconstructing â2, the pseudoinverse projection filters out of â1 each of its subnetworks |α1,m 〉 〈α1,m|, which is decorrelated of the series of L rank-1 symmetric subnetworks |βl 〉 〈βl| that compose the network b̂b̂T computed from the basis signal b̂, such that |βl 〉 〈βl|α1,m 〉 〈α1,m| = 0 for all l = 1, 2,..., L (Fig. 6 in Supporting Appendix).
Higher-Order EVD (HOEVD). Let the third-order tensor {âk} of size K-networks × N-genes × N-genes tabulate a series of K genome-scale networks computed from a series of K genome-scale signals {êk}, of size N-genes × Mk-arrays each, such that 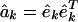 for all k = 1, 2,..., K. We define and compute a HOEVD of the tensor of networks {âk},
for all k = 1, 2,..., K. We define and compute a HOEVD of the tensor of networks {âk},
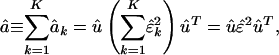 |
[5] |
using the SVD of the appended signals 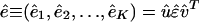 , where the mth column of û, |αm 〉 ≡ û|m 〉, lists the genome-scale expression of the mth eigenarray of ê. Whereas the matrix EVD is equivalent to the matrix SVD for a symmetric nonnegative matrix, this tensor HOEVD is different from the tensor higher-order SVD (14–16) for the series of symmetric nonnegative matrices {âk}, where the higher-order SVD is computed from the SVD of the appended networks (â1, â2,..., âK) rather than the appended signals. This HOEVD formulates the overall network computed from the appended signals â = êêT as a linear superposition of a series of
, where the mth column of û, |αm 〉 ≡ û|m 〉, lists the genome-scale expression of the mth eigenarray of ê. Whereas the matrix EVD is equivalent to the matrix SVD for a symmetric nonnegative matrix, this tensor HOEVD is different from the tensor higher-order SVD (14–16) for the series of symmetric nonnegative matrices {âk}, where the higher-order SVD is computed from the SVD of the appended networks (â1, â2,..., âK) rather than the appended signals. This HOEVD formulates the overall network computed from the appended signals â = êêT as a linear superposition of a series of 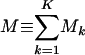 rank-1 symmetric “subnetworks” that are decorrelated of each other
rank-1 symmetric “subnetworks” that are decorrelated of each other 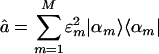 . Each subnetwork is also decoupled of all other subnetworks in the overall network â, since
. Each subnetwork is also decoupled of all other subnetworks in the overall network â, since 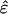 is diagonal.
is diagonal.
This HOEVD formulates each individual network in the tensor {âk} as a linear superposition of this series of M rank-1 symmetric decorrelated subnetworks and the series of M(M-1)/2 rank-2 symmetric couplings among these subnetworks (Fig. 7 in Supporting Appendix), such that
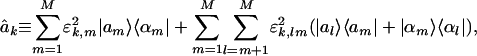 |
[6] |
for all k = 1, 2,..., K. The subnetworks are not decoupled in any one of the networks {âk}, since, in general, 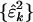 are symmetric but not diagonal, such that
are symmetric but not diagonal, such that 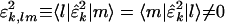 . The significance of the mth subnetwork in the kth network is indicated by the mth fraction of eigenexpression of the kth network
. The significance of the mth subnetwork in the kth network is indicated by the mth fraction of eigenexpression of the kth network 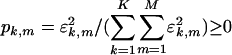 , i.e., the expression correlation captured by the mth subnetwork in the kth network relative to that captured by all subnetworks (and all couplings among them, where
, i.e., the expression correlation captured by the mth subnetwork in the kth network relative to that captured by all subnetworks (and all couplings among them, where 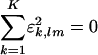 for all l ≠ m) in all networks. Similarly, the amplitude of the fraction
for all l ≠ m) in all networks. Similarly, the amplitude of the fraction 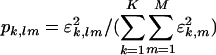 indicates the significance of the coupling between thelth and mth subnetworks in the kth network. The sign of this fraction indicates the direction of the coupling, such that pk,lm > 0 corresponds to a transition from the lth to the mth subnetwork and pk,lm < 0 corresponds to the transition from the mth to the lth. For real signals {êk}, the subnetworks are unique, and the couplings among them are unique up to phase factors of ±1, except in degenerate subspaces of
indicates the significance of the coupling between thelth and mth subnetworks in the kth network. The sign of this fraction indicates the direction of the coupling, such that pk,lm > 0 corresponds to a transition from the lth to the mth subnetwork and pk,lm < 0 corresponds to the transition from the mth to the lth. For real signals {êk}, the subnetworks are unique, and the couplings among them are unique up to phase factors of ±1, except in degenerate subspaces of 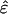 .
.
Interpretation of the Subnetworks and Their Couplings. We parallel- and antiparallel-associate each subnetwork or coupling with most likely expression correlations, or none thereof, according to the annotations of the two groups of x pairs of genes each, with largest and smallest levels of correlations in this subnetwork or coupling among all X = N(N – 1)/2 pairs of genes, respectively. The P value of a given association by annotation is calculated by using combinatorics and assuming hypergeometric probability distribution of the Y pairs of annotations among the X pairs of genes, and of the subset of y ⊆ Y pairs of annotations among the subset of x ⊆ X pairs of genes, 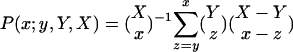 , where
, where 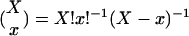 is the binomial coefficient (17). The most likely association of a subnetwork with a pathway or of a coupling between two subnetworks with a transition between two pathways is that which corresponds to the smallest P value. Independently, we also parallel- and antiparallel-associate each eigenarray with most likely cellular states, or none thereof, assuming hypergeometric distribution of the annotations among the N-genes and the subsets of n ⊆ N genes with largest and smallest levels of expression in this eigenarray. The corresponding eigengene might be inferred to represent the corresponding biological process from its pattern of expression.
is the binomial coefficient (17). The most likely association of a subnetwork with a pathway or of a coupling between two subnetworks with a transition between two pathways is that which corresponds to the smallest P value. Independently, we also parallel- and antiparallel-associate each eigenarray with most likely cellular states, or none thereof, assuming hypergeometric distribution of the annotations among the N-genes and the subsets of n ⊆ N genes with largest and smallest levels of expression in this eigenarray. The corresponding eigengene might be inferred to represent the corresponding biological process from its pattern of expression.
For visualization, we set the x correlations among the X pairs of genes largest in amplitude in each subnetwork and coupling equal to ±1, i.e., correlated or anticorrelated, respectively, according to their signs. The remaining correlations are set equal to 0, i.e., decorrelated. We compare the discretized subnetworks and couplings using Boolean functions (6).
Biological Results: Yeast Pathways from mRNA Expression and Proteins' DNA-Binding Signals
Significant EVD Subnetworks Are Associated with Functionally Independent Pathways. We compute the network â1 from the data signal ê1, which tabulates relative mRNA expression levels of n = 4,153 yeast genes with valid data in at least 15 of the M = 18 samples of a cell cycle time course of a culture synchronized by the mating pheromone α factor (1). The relative expression level of the nth gene in the mth sample is presumed valid when the ratio of the measured expression to the background signal is >1.5 for both the synchronized culture and asynchronous reference. Before computing â1, we use SVD to estimate the missing data in ê1 (10, 18) and to approximately center the expression pattern of each gene in ê1 at its time-invariant level (Supporting Appendix).
EVD of the network â1 uncovers four significant subnetworks, which capture >60%, 10%, 5%, and 5%, respectively, of the expression correlation of â1. These subnetworks are associated with the independent pathways manifest in the data signal ê1, following the P values for the distribution of the Y = 1,035 pairs of the 46 genes that were microarray-classified as pheromoneregulated (2) among all X = 2,926 pairs of the 77 genes that were traditionally classified as cell-cycle-regulated (1), and among each of the subsets of x = 150 pairs of genes with largest and smallest levels, respectively, of expression correlation (Table 2 in Supporting Appendix). The associations of the EVD subnetworks of â1 are consistent with those of the corresponding SVD eigenarrays of ê1 following the P values for the distribution of the 284 pheromone-regulated genes and that of the 574 genes, which were traditionally or microarray-classified as cell-cycle-regulated, among all 4,153 genes and among each of the subsets of 150 genes with largest and smallest levels, respectively, of expression (Table 1 in Supporting Appendix). The associations of the EVD subnetworks of â1 are also consistent with the patterns of expressions of the corresponding SVD eigengenes of ê1 (Fig. 8 in Supporting Appendix). We visualize the discretized four subnetworks and their Boolean functions in the subset of 70 genes that constitute the x = 150 correlations in each subnetwork that are largest in amplitude among the X = 2,926 pairs of traditionally classified cell-cycle-regulated genes.
The first and most significant subnetwork is associated with the α factor signal-transduction pathway, where the relations among the genes depend only on their pheromone-response classifications. Genes that are up-regulated in response to pheromone, and separately also genes that are down-regulated, are correlated, even when these genes are classified into antipodal cell-cycle stages. Genes that are up-regulated in response to pheromone are anticorrelated with genes that are down-regulated, even when these genes are classified into the same cell-cycle stages. For example, KAR4, which is up-regulated in response to pheromone, is correlated with CIK1, which is also up-regulated, and anticorrelated with CLN2, which is down-regulated (Fig. 1a), even though the expression of both KAR4 and CLN2 peaks at the cell-cycle stage G1 while the expression of CIK1 peaks at the antipodal stage S/G2. In the second subnetwork, which is associated with the exit from the α factor-induced cell-cycle arrest in M/G1 and the entry into cell-cycle progression at G1, genes that are up-regulated in response to pheromone are correlated, independent of their cell-cycle classification. The relations among genes that are down-regulated, however, depend on their cell-cycle, rather than their pheromone-response, classification. For example, CLN2 and CLB2, which encode cyclins of the antipodal stages G1 and G2/M, respectively, are anticorrelated, even though both are down-regulated in response to pheromone; and SWI4, which encodes a G1 transcription factor, is correlated with CLN2 and anticorrelated with CLB2 (Fig. 1b). In the third and fourth subnetworks, which are associated with the two pathways of antipodal cell-cycle-expression oscillations that are orthogonal, i.e., π /2 out of phase relative to one another, the relations among genes depend only on their cell-cycle classifications. For example, in the third subnetwork, which is associated with the cell-cycle-expression oscillations at S vs. those at M, KAR4 is anticorrelated with CIK1, where KAR4 is correlated, and CIK1 is anticorrelated with ASH1 (Fig. 1c). In the fourth subnetwork, which is associated with expression at G1 vs. that at G2, KAR4 is correlated with CLN2 (Fig. 1d).
Fig. 1.

Discretized significant EVD subnetworks of the network â1 in the subsets of 150 correlations (red) and anticorrelations (green) largest in amplitude among all traditionally classified cell-cycle genes of â1, color-coded according to their cell-cycle classifications, M/G1 (yellow), G1 (green), S (blue), S/G2 (red), and G2/M (orange), and separately also according to their pheromone-response classifications, up-regulated (black) and down-regulated (gray). (a) The first subnetwork shows pheromone-response-dependent and cell-cycle-independent relations among the genes. (b) The second subnetwork shows pheromone-response- and cell-cycle-dependent relations. (c and d) The third and fourth subnetworks show cell-cycle-dependent relations that are orthogonal to each other.
Boolean functions of the discretized subnetworks highlight known pathway-dependent relations among genes, common to a subset of the subnetworks or antipodal across the subnetworks (Fig. 9 in Supporting Appendix).
Integrative Pseudoinverse-Projected Networks Simulate Observation of only the Pathways Manifest in both the Data and Basis Signals. We compute the network â2 by pseudoinverse-projecting the network â1 onto the basis signal, which tabulates the relative DNA-bound protein occupancy levels of the 2,120 genes with at least one valid data point in any one of L = 12 samples that correspond to 12 yeast-cell-cycle transcription factors (3). The relative binding occupancy level of the nth gene in the lth sample is presumed valid when the associated P value is <0.1. Similarly, â3 is computed by projecting â1 onto the basis signal, which tabulates the occupancy levels of 2,476 genes in 12 samples of transcription factors involved in developmental programs, such as mating; and â4 is computed by projecting â1 onto the basis signal, which tabulates the occupancy levels of 2,943 genes in eight samples of factors involved in biosynthesis, such as DNA replication. Before computing â2, â3, and â4 for the 1,588, 1,827, and 2,254 genes at the intersections of â1 and the proteins' DNA-binding basis signals, we divide each gene measurement in each basis signal by the arithmetic mean of the measurements for that gene in that signal, thus converting the signals to DNA-binding levels of each transcription factor relative to those of all other factors. We also approximately center the binding pattern of each gene at its transcription factor-invariant level using SVD (Supporting Appendix).
EVD of the cell-cycle-projected network â2 uncovers only two significant subnetworks, which capture ≈55% and 30% of the expression correlation of â2, respectively, and are associated with the two pathways of antipodal cell-cycle-expression oscillations at G1 vs. those at G2 and at S vs. M, respectively [Table 4 (row a) in Supporting Appendix]. Boolean AND intersection of the discretized first subnetwork of â2, in the subset of 200 correlations largest in amplitude among all traditionally classified cell-cycle genes of â2, with the discretized fourth subnetwork of â1 highlights correlations among traditionally classified M/G1,G1, and S genes, and anticorrelations among these genes and G2/M genes, independent of their responses to pheromone (Fig. 2a). Boolean AND of the second subnetwork of â2 with the third subnetwork of â1 highlights correlations among M/G1 genes and their anticorrelations with S and S/G2 genes (Fig. 2b). The α factor signal-transduction pathway that is manifest in the data but not in the basis signal is not associated with either one of the subnetworks of â2. Similarly, EVD of the development-projected network â3 uncovers only one significant subnetwork, which captures >90% of the expression correlation of â3 and is associated with the α factor signal-transduction pathway [Table 4 (row b) in Supporting Appendix]. Boolean AND of the subnetwork of â3 with the first subnetwork of â1 highlights correlations among genes that are up-regulated in response to pheromone and their anticorrelations with down-regulated genes, independent of their cell-cycle classifications (Fig. 2c). The cell-cycle-expression oscillation pathways that are manifest in the data but not in the basis signal are not associated with either one of the subnetworks of â3. EVD of the biosynthesis-projected network â4 uncovers three significant subnetworks, which capture together >90% of the expression correlation of â4, all of which are associated with the activity of histones that peaks during DNA replication at the cell-cycle stage S [Table 4 (row c) and Fig. 13 in Supporting Appendix].
Fig. 2.

Boolean AND intersections of the discretized EVD subnetworks of the pseudoinverse-projected â2 and â3, in the subsets of 200 correlations largest in amplitude among all traditionally classified cell-cycle genes of â2 and â3, respectively, with these of â1.(a) The first subnetwork of â2 AND fourth subnetwork of â1.(b) The second subnetwork of â2 AND third subnetwork of â1.(c) The subnetwork of â3 AND first subnetwork of â1.
The associations of the EVD subnetworks of the projected networks â2, â3, and â4 are consistent with the associations of the corresponding SVD eigenarrays (Table 3 in Supporting Appendix) and eigengenes (Figs. 10–12 in Supporting Appendix) of the projected signals ê2, ê3, and ê4, respectively.
Comparative HOEVD Subnetworks and Their Couplings Are Associated with Pathways and the Transitions Among Them Common to the Series or Exclusive to a Subset of Networks. HOEVD of the series of networks {â1, â2, â3} uncovers three significant subnetworks, which capture ≈40%, 15%, and 9% of the expression correlation of the overall network â ≡ â1 + â2 + â3, respectively, and the three couplings among these subnetworks, which capture expression correlations only in the individual networks. The associations of the HOEVD subnetworks and couplings of {â1, â2, â3} (Table 6 in Supporting Appendix) are consistent with the associations of the corresponding SVD eigenarrays (Table 5 in Supporting Appendix) and eigengenes (Fig. 14 in Supporting Appendix) of the appended signals ê ≡ (ê1, ê2, ê3), computed for the 868 genes at the intersection of ê1, ê2, and ê3.
The subnetworks are associated with the independent pathways that are manifest in the overall network as well as the individual networks. The first subnetwork, which is associated with the α factor signal-transduction pathway (Fig. 3a), contributes to the expression correlations of the network â1 as well as to the development-projected network â3, but its contribution to the cell-cycle-projected network â2 is negligible (Fig. 4a). The second and third subnetworks, which are associated with the two pathways of antipodal cell-cycle-expression oscillations at G1 vs. that at G2 and at S vs. that at M, respectively (Fig. 3 b and c), contribute to â1 and â2 but not to â3. The couplings are associated with the transitions among these independent pathways that are manifest in the individual networks only. The coupling between the first and second subnetworks is associated with the transition between the two pathways of response to pheromone and cell-cycle expression at G1 vs. that at G2, i.e., the exit from pheromone-induced arrest and entry into cell-cycle progression (Fig. 3d). The coupling between the first and third subnetworks is associated with cell-cycle expression at G1/S vs. that at M (Fig. 3e). The coupling between the second and third subnetworks is associated with cell-cycle-expression oscillations at the two antipodal cell-cycle checkpoints of G1/S vs. G2/M (Fig. 3f). All these couplings contribute to the expression correlation of â2. Their contributions to the expression correlations of â1 and â3 are negligible (Fig. 4b).
Fig. 3.

Discretized significant HOEVD subnetworks of the series of networks {â1, â2, â3} and their couplings, in the subsets of 100 correlations largest in amplitude among all traditionally classified cell-cycle genes of {â1, â2, â3}. (a) The first subnetwork shows pheromone-response-dependent only relations among the genes. (b and c) The second and third subnetworks show orthogonal cell-cycle-dependent relations. (d and e) The couplings between the first and second, and first and third subnetworks, respectively, both show pheromone-response- and cell-cycle-dependent relations. (f) The coupling between the second and third subnetworks shows cell-cycle-dependent only relations.
Fig. 4.

Fractions of eigenexpression of the HOEVD subnetworks (a) and their couplings (b) in the individual networks â1 (red), â2 (blue), and â3 (green). The contributions of each coupling in each individual network cancel out in the overall network â ≡ â1 + â2 + â3.
Boolean functions of the discretized subnetworks and couplings highlight known as well as previously unknown pathway-dependent relations among genes that are in agreement with current understanding of the cellular system of yeast (Fig. 15 in Supporting Appendix) (19).
Discussion
We have shown that the matrix EVD and pseudoinverse projection and a tensor HOEVD can separate genome-scale nondirectional networks of, e.g., mRNA expression and proteins' DNA-binding relations among genes into mathematically defined subnetworks and their couplings that can be associated with functionally independent pathways and the transitions among them. In analyses of genome-scale yeast networks, these subnetworks and couplings uncover coordinated differential relations among cell-cycle- and pheromone-regulated genes that are in agreement with reported pathway-dependent activities of these genes. Possible additional applications of EVD, pseudoinverse projection, and HOEVD include reconstruction of pathways and transitions among these pathways from nondirectional networks of correlations among sets of orthologous genes, which are computed from genome-scale signals of different types and from different organisms to elucidate organism, as well as pathway, dependence of relations among genes (e.g., refs. 6, 11, 20, and 21).
Supplementary Material
Acknowledgments
We thank T. G. Kolda and T. O. Yeates for thoughtful reviews of this manuscript; J. F. X. Diffley, V. R. Iyer, E. M. Marcotte, and B. K. Tye for helpful comments; and the American Institute of Mathematics in Palo Alto for hosting the 2004 Workshop on Tensor Decompositions where some of this work was done. This work was supported by National Science Foundation Grant CCR-0430617 (to G.H.G.) and National Human Genome Research Institute Individual Mentored Research Scientist Development Award in Genomic Research and Analysis 5 K01 HG00038 (to O.A.).
Author contributions: O.A. and G.H.G. designed research; O.A. performed research; O.A. analyzed data; and O.A. and G.H.G. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: EVD, eigenvalue decomposition; HOEVD, higher-order EVD; SVD, singular-value decomposition.
Footnotes
In this article, m̂ denotes a matrix, |v 〉 denotes a column vector, and 〈u| denotes a row vector, such that m̂|v 〉, 〈u|m̂, and 〈u|v 〉 all denote inner products, and |v 〉 〈u| denotes an outer product.
References
- 1.Spellman, P. T., Sherlock, G., Zhang, M. Q., Iyer, V. R., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher, B. (1998) Mol. Biol. Cell 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts, C. J., Nelson, B., Marton, M. J., Stoughton, R., Meyer, M. R., Bennett, H. A., He, Y. D., Dai, H., Walker, W. L., Hughes, T. R., et al. (2000) Science 287, 873–880. [DOI] [PubMed] [Google Scholar]
- 3.Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., Hannett, N. M., Harbison, C. T., Thompson, C. M., Simon, I., et al. (2002) Science 298, 799–804. [DOI] [PubMed] [Google Scholar]
- 4.Ihmels, J., Levy, R. & Barkai, N. (2002) Nat. Biotechnol. 22, 86–92. [DOI] [PubMed] [Google Scholar]
- 5.Balazsi, G., Barabasi, A. L. & Oltvai, Z. N. (2005) Proc. Natl. Acad. Sci. USA 102, 7841–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowers, P. M., Cokus, S. J., Eisenberg, D. & Yeates, T. O. (2004) Science 306, 2246–2249. [DOI] [PubMed] [Google Scholar]
- 7.Braun, E. & Brenner, N. (2004) Phys. Biol. 1, 67–76. [DOI] [PubMed] [Google Scholar]
- 8.Kurihara, L. J., Stewart, B. G., Gammie, A. E. & Rose, M. D. (1996) Mol. Cell. Biol. 16, 3990–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alter, O., Brown, P. O. & Botstein, D. (2000) Proc. Natl. Acad. Sci. USA 97, 10101–10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alter, O., Brown, P. O. & Botstein, D. (2001) in Microarrays: Optical Technologies and Informatics, eds. Bittner, M. L., Chen, Y., Dorsel, A. N. & Dougherty, E. R. (Int. Soc. Optical Eng., Bellingham, WA), Vol. 4266, pp. 171–186. [Google Scholar]
- 11.Alter, O., Brown, P. O. & Botstein, D. (2003) Proc. Natl. Acad. Sci. USA 100, 3351–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alter, O. & Golub, G. H. (2004) Proc. Natl. Acad. Sci. USA 101, 16577–16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golub, G. H. & Van Loan, C. F. (1996) Matrix Computation (Johns Hopkins Univ. Press, Baltimore), 3rd Ed.
- 14.De Lathauwer, L., De Moor, B. & Vandewalle, J. (2000) SIAM J. Matrix Anal. Appl. 21, 1253–1278. [Google Scholar]
- 15.Kolda, T. G. (2001) SIAM J. Matrix Anal. Appl. 23, 243–255. [Google Scholar]
- 16.Zhang, T. & Golub, G. H. (2000) SIAM J. Matrix Anal. Appl. 23, 534–550. [Google Scholar]
- 17.Tavazoie, S., Hughes, J. D., Campbell, M. J., Cho, R. J. & Church, G. M. (1999) Nat. Genet. 22, 281–285. [DOI] [PubMed] [Google Scholar]
- 18.Kim, H., Golub, G. H. & Park, H. (2005) Bioinformatics 21, 187–198. [DOI] [PubMed] [Google Scholar]
- 19.Caro, L. H., Smits, G. J., van Egmond, P., Chapman, J. W. & Klis, F. M. (1998) FEMS Microbiol. Lett. 161, 345–349. [DOI] [PubMed] [Google Scholar]
- 20.Stuart, J. M., Segal, E., Koller, D. & Kim, S. K. (2003) Science 302, 249–255. [DOI] [PubMed] [Google Scholar]
- 21.Bergmann, S., Ihmels, J. & Barkai, N. (2004) PLoS Biol. 2, E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.