

Abstract
Signal transducer and activator of transcription 5 (STAT5) is constitutively activated by BCR/ABL, the oncogenic tyrosine kinase responsible for chronic myelogenous leukemia. The mechanism of BCR/ABL-mediated STAT5 activation is unknown. We show here that the BCR/ABL SH3 and SH2 domains interact with hematopoietic cell kinase (Hck), leading to the stimulation of Hck catalytic activity. Active Hck phosphorylated STAT5B on Tyr699, which represents an essential step in STAT5B stimulation. Moreover, a kinase-dead Hck mutant and Hck inhibitor PP2 abrogated BCR/ABL-dependent activation of STAT5 and elevation of expression of STAT5 downstream effectors A1 and pim-1. These data identify a novel BCR/ABL–Hck–STAT5 signaling pathway, which plays an important role in BCR/ABL-mediated transformation of myeloid cells.
Keywords: activation/BCR/ABL/Hck/pathway/STAT5
Introduction
BCR/ABL is derived from translocation of the c-ABL gene on chromosome 9 to the BCR locus on chromosome 22 [t(9;22), Philadelphia chromosome], and is present in essentially all cases of chronic myelogenous leukemia (CML) and a cohort of acute lymphocytic leukemia (ALL) patients (Shtivelman et al., 1986; Clark et al., 1988). BCR/ABL hybrid genes produce p230, p210 and p185 fusion proteins with constitutive tyrosine kinase activity that transform hematopoietic cells in vitro, and cause CML- or ALL-like syndromes in mice (Daley et al., 1990; Heisterkamp et al., 1990; Gishizky and Witte, 1992).
BCR/ABL activates multiple signaling pathways responsible for the protection from apoptosis, stimulation of growth factor-independent proliferation, modulation of adhesion/invasion ability and induction of resistance to genotoxic drugs and γ-radiation (Raitano et al., 1997; Zou and Calame, 1999). Previous reports, including those from our laboratory, revealed that activation of signal transducer and activator of transcription 5 (STAT5) contributed to BCR/ABL-dependent changes in the phenotype of transformed cells (Ilaria et al., 1999; Nosaka et al., 1999; Kieslinger et al., 2000; Slupianek et al., 2001). STAT5A and STAT5B proteins belong to the family of STATs, which are latent transcription factors that become activated upon phosphorylation on tyrosine and also on serine (Horvath and Darnell, 1997). Phosphorylation of Tyr694 (Y694) in STAT5A and Tyr699 (Y699) in STAT5B is essential for dimerization and subsequent translocation to the nucleus (Gouilleux et al., 1994), resulting in the transactivation of several STAT5-inducible genes (Mui et al., 1996). Protein products of these genes are involved in regulation of growth factor independence, differentiation, adhesion/invasion and DNA repair/drug resistance (Ilaria et al., 1999; Nosaka et al., 1999; Kieslinger et al., 2000; Slupianek et al., 2001).
The mechanism(s) of STAT5 activation by BCR/ABL remains unknown. Myeloid cells expressing the BCR/ABL ΔSH3 + ΔSH2 mutant (BCR/ABLΔΔ mutant, which lacks both SH3 and SH2 domains) failed to exhibit constitutive activation of STAT5, implicating the SH3 + SH2 region of BCR/ABL in recruiting STAT5 (Nieborowska-Skorska et al., 1999). In addition, BCR/ABL signaling from SH3 and SH2 domains to STAT5 was required for the leukemogenic activity of BCR/ABL (Nieborowska-Skorska et al., 1999). Therefore, it was reasonable to speculate that the SH3 and SH2 domains of BCR/ABL are directly responsible for recruitment of STAT5, especially because these domains are involved in the communication between proteins, specifically between activated tyrosine kinases and their downstream effectors (Koch et al., 1991; Pawson and Gish, 1992). SH2 domains bind to peptide motifs containing phosphorylated tyrosine residues (Waksman et al., 1993), while SH3 domains interact with proline-rich peptides that form polyproline type II helices (Weng et al., 1995). However, the immunoprecipitation experiments revealed that only a small portion (if any) of STAT5 could be detected in complex with BCR/ABL (Ilaria and Van Etten, 1996; Nieborowska-Skorska et al., 1999), which does not favor a direct activation mechanism. On the other hand, STAT5A and STAT5B displayed pronounced phosphorylation on tyrosine residue(s) in BCR/ABL-positive cells, suggesting interaction with an activated tyrosine kinase (Carlesso et al., 1996; Ilaria and Van Etten, 1996; Shuai et al., 1996; Chai et al., 1997). BCR/ABL-mediated phosphorylation of STAT5 may not depend on Jak2 (Ilaria and Van Etten, 1996; Chai et al., 1997; Xie et al., 2001), strongly suggesting that another tyrosine kinase(s) is involved.
Cytoplasmic tyrosine kinases belonging to the Src family (e.g. Hck and Lyn) can activate STAT5 independently of Jak2 in order to transduce signals from receptors activated by interleukin-3 (IL-3), erythropoietin and epidermal growth factor (EGF) (Chaturvedi et al., 1998; Chin et al., 1998; Olayioye et al., 1999; Reddy et al., 2000; Okutani et al., 2001). Hck and Lyn are also stimulated by BCR/ABL in myeloid cells (Danhauser-Riedl et al., 1996; Lionberger et al., 2000). In addition, studies with dominant-negative mutants have demonstrated that Hck is required for BCR/ABL-mediated transformation of myeloid leukemia cells to cytokine independence (Lionberger et al., 2000). Thus, we sought to determine if Hck could act as an intermediate in BCR/ABL-dependent activation of STAT5.
Results
STAT5 is phosphorylated by BCR/ABL immunoprecipitates
STAT5 plays an important role in BCR/ABL-mediated leukemogenesis, and its tyrosine phosphorylation is easily detectable in BCR/ABL-positive cells (de Groot et al., 1999; Nieborowska-Skorska et al., 1999; Horita et al., 2000; Sillaber et al., 2000; Sonoyama et al., 2002). Since phosphorylation of the C-terminal portion of STAT5 (e.g. Y694 and S725 in STAT5A, and Y699 and S730 of STAT5B) is an essential step for its dimerization and activation of transcriptional activity (Gouilleux et al., 1994), we examined the role of BCR/ABL in STAT5 phosphorylation. An in vitro kinase reaction was performed using anti-ABL immunoprecipitates from cells expressing BCR/ABL or its kinase-dead K1172R mutant. GST–STAT5B fusion protein was phosphorylated by immunoprecipitates containing BCR/ABL kinase, but not the K1172R mutant protein (Figure 1A, upper box, arrow 1). Moreover, only the C-terminal fragment (C5), not the N-terminal fragment (N5), of STAT5B was phosphorylated in the presence of BCR/ABL kinase (Figure 1A, upper panel, arrow 3 and arrow 2, respectively).

Fig. 1. STAT5 is phosphorylated by BCR/ABL immunoprecipitates. (A) STAT5 phosphorylation was examined in anti-ABL immunoprecipitates obtained from IL-3- and serum-starved 32Dcl3 cells expressing BCR/ABL wild-type (WT) or the kinase-defective mutant (K1172R), using full-length STAT5 or the C5 (amino acids 547–787) and N5 (amino acids 1–546) fragments conjugated to GST as substrates and [γ-32P]ATP (upper box). GST alone was not phosphorylated (data not shown). GST–STAT5 proteins loaded onto the gel were detected by western blot using anti-STAT5B antibodies (C-17 recognizes the C- terminus and N-20 recognizes the N-terminus) (middle box). Arrows 1, 2 and 3 indicate the position of full-length GST–STAT5, GST–N5 and GST–C5, respectively. GST was detected by Ponceau red staining (not shown). Immunoprecipitated BCR/ABL proteins (arrow 4) were visualized by western analysis using anti-ABL antibody (bottom box). (B) Phosphorylation of GST–C5 (C5) and enolase (Eno) was examined in anti-ABL immunoprecipitates obtained from IL-3- and serum-starved 32Dcl3 cells (Parental) and cells expressing BCR/ABL kinase in the absence (–) or presence (+) of 1 µM STI571. C5 and enolase substrates were detected by Coomassie Blue staining of the gel. Results represent three independent experiments.
Interestingly, addition of STI571 (imatinib mesylate), a selective inhibitor of ABL kinase (Druker et al., 1996), did not significantly affect the in vitro phosphorylation of the C5 fragment (Figure 1B, upper panel), whereas phosphorylation of the generic tyrosine kinase substrate enolase was completely blocked (Figure 1B, lower panel). This result suggests that a kinase other than BCR/ABL was present in the anti-ABL immunoprecipitate, which was able to phosphorylate STAT5 when BCR/ABL kinase was inhibited.
Hck is an intermediate in BCR/ABL-dependent phosphorylation of STAT5B on Y699
As shown above, another kinase associated with BCR/ABL may be responsible for STAT5 phosphorylation in vitro. In order to search for this kinase, the anti-STAT5 immunoprecipitates were examined for the presence of phospho-proteins, possibly also tyrosine kinases. The immunoprecipitates were obtained from the growth factor-starved parental cells (STAT5 inactive), cells expressing BCR/ABL (STAT5 active) or BCR/ABLΔΔ cells (STAT5 inactive) (Nieborowska-Skorska et al., 1999). As expected, western analysis revealed lack of STAT5 phosphorylation in the immunoprecipitates from parental and BCR/ABLΔΔ cells (Figure 2, upper box). Conversely, STAT5 was tyrosine phosphorylated in the immunoprecipitate from BCR/ABL cells. In addition, bands of ∼200 and 55–60 kDa were present in the immunoprecipitate from BCR/ABL cells, but absent or less evident in the immunoprecipitates from parental and BCR/ABLΔΔ cells (Figure 2, upper box). Subsequent western analyses showed that the former band represents BCR/ABL and the latter Hck (as marked on Figure 2, upper box). This finding is important, because previous studies have shown that Src family kinases associate with BCR/ABL (Danhauser-Riedl et al., 1996; Lionberger et al., 2000), suggesting that these additional activities may contribute to STAT5 activation as well (Chaturvedi et al., 1998; Chin et al., 1998; Olayioye et al., 1999; Reddy et al., 2000; Okutani et al., 2001).
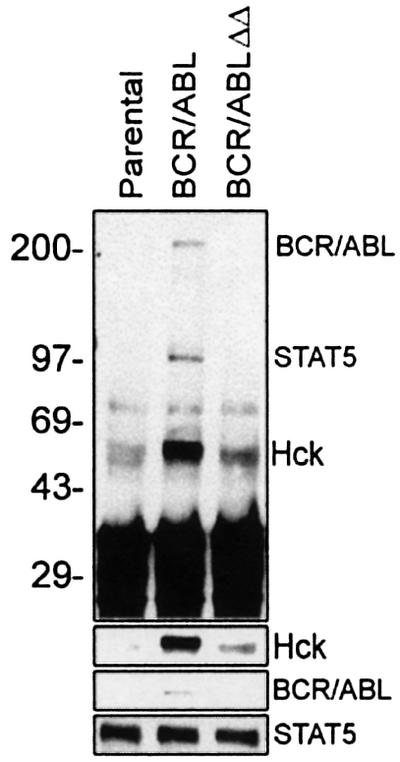
Fig. 2. Pattern of phosphotyrosine proteins co-immunoprecipitating with STAT5. STAT5 was immunoprecipitated from the lysates of 32Dcl3 cells (Parental) and cells expressing BCR/ABL or the BCR/ABLΔΔ mutant, after being starved of IL-3 for 12 h. The immunoprecipitates were analyzed by western analysis with anti-P.Tyr antibodies (upper panel). Subsequent blotting with anti-STAT5, anti-ABL and anti-Hck confirmed the localization of the indicated proteins (lower panels). An equal amount of STAT5 was immunoprecipitated in each sample. Results represent two independent experiments.
Next, a series of experiments was performed to provide more evidence about the interactions between BCR/ABL, STAT5 and Hck. Anti-STAT5 and anti-Hck immunoprecipitates were analyzed for the presence and phosphorylation status of BCR/ABL, Hck and STAT5. Hck was readily detectable in anti-STAT5 immunoprecipitates, and a small portion of BCR/ABL, but not BCR/ABLΔΔ mutant, was also present (Figure 3A). STAT5 and Hck present in the immunoprecipitate from BCR/ABL, but not BCR/ABLΔΔ cells, were tyrosine phosphorylated. Although Hck was detected in all anti-STAT5 immunoprecipitates, more protein was present in those obtained from BCR/ABL cells (Figure 3A, compare lane BCR/ABL with lanes ‘Parental’ and BCR/ABLΔΔ). STAT5 was detectable in anti-Hck immunoprecipitates from parental, BCR/ABL and BCR/ABLΔΔ cell lysates, but the phosphorylated form was present only in BCR/ABL cell lysates (Figure 3B). In addition, BCR/ABL, but not the BCR/ABLΔΔ mutant, was readily detectable in complex with Hck. BCR/ABL–Hck complex formation was associated with tyrosine phosphorylation of Hck (Figure 3B). In conclusion, these experiments suggest the existence of a BCR/ABL–Hck–STAT5 pathway and implicate Hck as an intermediate in BCR/ABL-dependent tyrosine phosphorylation of STAT5. This hypothesis is supported by the results of GST pull-down experiments showing that BCR/ABL could interact directly with Hck, but not with STAT5, and that Hck could bind directly to STAT5 (Lionberger et al., 2000; and data not shown).

Fig. 3. The BCR/ABL SH3 + SH2 region interacts with the Hck–STAT5 complex. 32Dcl3 cells (Parental) and cells expressing BCR/ABL, or the indicated BCR/ABL mutants, were starved of IL-3 for 5 h. STAT5 immunoprecipitates (A) and Hck immunoprecipitates (B) were examined by SDS–PAGE followed by western analysis with anti-STAT5, anti-Hck, anti-P.Tyr and anti-ABL antibodies. Hck immuno precipitation and kinase assay (C) were performed to detect its association with BCR/ABL and to measure its catalytic activity. Hck immunoprecipitates were examined by SDS–PAGE followed by western analysis with anti-ABL (upper panel) and anti-anti-Hck (bottom panel) antibody. An in vitro kinase reaction was performed in these immunoprecipitates using [γ-32P]ATP and 5 µg of the standard substrate for the Src-related family of tyrosine kinases, Sam68 (middle panel). Results represent two or three independent experiments.
Deletion of the SH3 and SH2 domains from BCR/ABL abrogated its ability not only to bind Hck but also to stimulate Hck catalytic activity, as determined by co-immunoprecipitation and western analysis, and by in vitro kinase assay using a standard Hck substrate, Sam68 (Figure 3C). To provide more detailed information about the recruitment and activation of Hck by the SH3 + SH2 region of BCR/ABL, several smaller mutants have been employed (Nieborowska-Skorska et al., 1999). Deletion of either the SH3 or SH2 domain from BCR/ABL (ΔSH3 or ΔSH2, respectively) did not prevent the association and activation of Hck; a similar effect was obtained after introduction of the P1013L single amino acid mutation disrupting the ability of the BCR/ABL SH3 domain to interact with a proline-rich region, and the R1053L mutation abrogating the ability of the BCR/ABL SH2 domain to bind phosphotyrosine (P1013L + R1053L mutant) (Figure 3C). In addition, a BCR/ABL mutant lacking the SH3 domain and containing an R1053L point mutation (ΔSH3 + R1053L) recruited and activated Hck; however, the mutant containing the P1013L mutation and an SH2 deletion (P1013L + ΔSH2) did not. Thus, the mutation pattern in the BCR/ABL SH3 + SH2 region, which abrogated the recruitment and activation of Hck (Figure 3C), is identical to that preventing stimulation of STAT5 (Nieborowska-Skorska et al., 1999).
To confirm the hypothesis that the BCR/ABL SH3 + SH2 region activates the Hck–STAT5 pathway, the Hck kinase assay was performed using Sam68 (positive control for the reaction) and the C-terminal STAT5 fragment (C5) fused to GST as substrates. Anti-Hck immunoprecipitates from BCR/ABL cells, but not from the parental or BCR/ABLΔΔ cells, phosphorylated Sam68 and C5 (Figure 4A). This observation implicates Hck interaction with the BCR/ABL SH3 + SH2 region resulting in Hck activation and subsequent phosphorylation of STAT5. In accordance with previous studies (see Figure 1B), addition of STI571 to the in vitro kinase reaction mixture did not significantly affect the tyrosine phosphorylation of the STAT5 C5 fragment by anti-Hck immunoprecipitates from BCR/ABL-positive cells (data not shown). To determine if Hck can phosphorylate Tyr699 of STAT5B, this tyrosine was replaced by phenylalanine in the C5 fragment (C5-YF mutant). This mutation completely abolished phosphorylation of the fragment (Figure 4B), indicating that Y699 is the primary phosphorylation site for Hck activated by BCR/ABL. To confirm that this phenomenon is dependent on Hck and not on BCR/ABL, which may be present in anti-Hck immunoprecipitates (see Figure 3B), the kinase reaction was performed in anti-Hck immunoprecipitates obtained from Sf9 cells expressing either catalytically active Hck (Hck-WT) or a kinase-defective mutant (Hck-KE). In concordance with previous experiments, catalytically active Hck phosphorylated the C5 wild-type fragment, but not the C5-YF mutant (Figure 4C). Altogether, these results strongly suggest that Hck activated by BCR/ABL could phosphorylate STAT5B on Y699, which is essential for its activation.
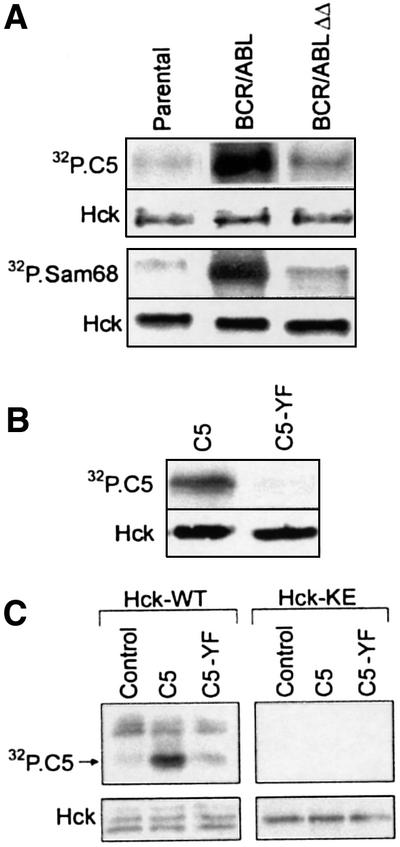
Fig. 4. The BCR/ABL SH3 + SH2 region is required for activation of Hck and phosphorylation of STAT5B on Y699. (A) Hck was immunoprecipitated from cell lysates obtained from 32Dcl3 parental cells (Parental) and from cells expressing BCR/ABL or the BCR/ABLΔΔ mutant after being starved of IL-3 for 5 h. An in vitro kinase reaction was performed using [γ-32P]ATP and 5 µg of the substrates: Sam68 (lower panel) or GST–C5 (upper panel). (B) Hck was immunoprecipitated from BCR/ABL cells. GST–C5 (C5) or GST–C5[Y699F] mutant (C5-YF) proteins were added as substrates along with [γ-32P]ATP. (C) Wild-type Hck and Hck-KE were immunopurified from infected Sf9 cell lysates and incubated in vitro with [γ-32P]ATP alone (Control) or together with the C5 or C5-YF proteins. The kinase reactions were resolved by SDS–PAGE. Phosphorylation of GST fusion proteins was assessed by autoradiography (32P.C5 and 32P.Sam68 boxes). The presence of Hck in each reaction was verified by immunoblotting (Hck panel). Results represent three independent experiments.
To provide additional evidence that Hck can directly phosphorylate and activate STAT5, the Hck–STAT5 interaction was examined in Sf9 insect cells. When Hck and STAT5A proteins were co-expressed in Sf9 cells, STAT5A became tyrosine phosphorylated (Figure 5A). In contrast, kinase-dead Hck did not phosphorylate STAT5A, indicating that STAT5A phosphorylation depends on the catalytic activity of Hck. Hck-mediated tyrosine phosphorylation of STAT5A in Sf9 cells was associated with activation of its DNA binding ability as measured by electrophoretic mobility shift assay (EMSA) using STAT5-responsive mammary gland factor element (MGFE) as a probe (Figure 5B).
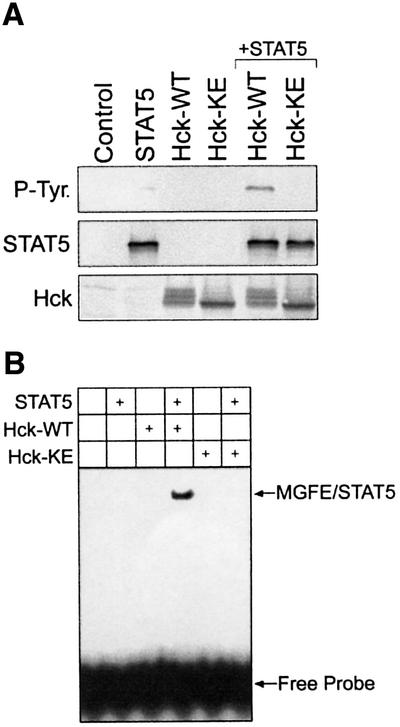
Fig. 5. Analysis of STAT5 activation by Hck in Sf9 insect cells. (A) STAT5, wild-type Hck (Hck-WT) and kinase-defective Hck (Hck-KE) were expressed in Sf9 insect cells. Uninfected Sf9 cells were included as a negative control (Control). STAT5 was immunoprecipitated from infected cell lysates and analyzed for phosphotyrosine content by immunoblotting with anti-phosphotyrosine antibodies (P-Tyr. panel). Equivalent recovery of STAT5 was verified by immunoblotting an aliquot of each immunoprecipitate with anti-STAT5 antibodies (STAT5 panel). Hck protein expression was verified by immunoblotting of the cell lysates (bottom). (B) Sf-9 cells were infected with recombinant baculoviruses carrying the indicated cDNAs. Aliquots of the nuclear extracts were tested for STAT5–DNA binding activity by EMSA using a [γ-32P]ATP-labeled MGFE probe. Results are representative of three independent experiments.
Hck is an intermediate in BCR/ABL-dependent activation of STAT5
To obtain more evidence about functional aspects of the BCR/ABL–Hck–STAT5 pathway, a kinase-dead dominant-negative mutant of Hck (Hck-KE) was transiently expressed in BCR/ABL-positive cells. As a control, cells were infected with the empty construct. Expression of Hck-KE strongly reduced the total cellular Hck catalytic activity (Figure 6A, bottom box), which was associated with down-modulation of BCR/ABL-dependent STAT5 tyrosine phosphorylation (Figure 6A, middle box) and DNA binding ability (Figure 6A, upper box). STAT5 was responsible for the majority of FcγRI bandshift in BCR/ABL-positive 32Dcl3 cells, as confirmed by the supershift assay using anti-STAT5 antibody (Nieborowska-Skorska et al., 1999). In addition, transient expression of the Hck-KE mutant completely inhibited the BCR/ABL-dependent STAT5-mediated transactivation of the β-casein promoter (Figure 6B) (Nieborowska-Skorska et al., 1999).

Fig. 6. Hck is a required intermediate in STAT5 activation by BCR/ABL. (A) Parental 32Dcl3 cells and BCR/ABL-positive cells were transfected with IRES–GFP (E), [Hck-YF]-IRES–GFP or [Hck-KE]-IRES–GFP retroviral particles or incubated with STI571, PP2 and PP3. GFP-positive cells and inhibitor-treated cells were examined for STAT5 binding ability by EMSA using a [γ-32P]ATP-labeled fragment of the FcγRI probe (upper large boxes). STAT5 tyrosine phosphorylation (P-Tyr.STAT5 boxes) was analyzed by immunoprecipitation followed by western analysis using anti-P.Tyr antibodies. Hck and BCR/ABL kinase activities were assayed as described in Figures 4A and 1B, respectively (*P.Sam68 and 32P.Eno boxes, respectively). STAT5, Hck and BCR/ABL proteins were detected in the immunoprecipitates by western analysis (STAT5, Hck and BCR/ABL boxes, respectively). (B) A transactivation assay was performed in Tk–ts13 cells transfected with the β-casein–luciferase reporter construct and the indicated constructs, and treated with the inhibitors. Activation of the luciferase gene is shown in arbitrary units in comparison with the control group transfected with the reporter plasmid. (C) Western analysis of the levels of A1 and pim-1 proteins in 32Dcl3 parental cells and cells expressing the indicated BCR/ABL and/or Hck proteins. The cells eventually were treated with the inhibitors as indicated. Actin was detected as a loading control. Results represent the mean ± SD from two or three independent experiments.
Expression of a constitutively active form of Hck (Hck-YF) in the parental cells resulted in elevation of cellular STAT5 tyrosine phosphorylation (Figure 6A, middle box) and DNA binding activity (Figure 6A, upper box). Moreover, transient overexpression of Hck or expression of the Hck-YF mutant (kinase-active), but not of the Hck-KE (kinase-dead) mutant, caused transactivation of the STAT5-responsive β-casein promoter (Figure 6B).
To prove the biological importance of the BCR/ABL–Hck–STAT5 pathway, expression of STAT5 downstream effectors A1 and pim-1 (Nieborowska-Skorska et al., 2002) was examined after modification of the Hck activity (Figure 6C). These effectors were described recently to play an essential collaborative role in BCR/ABL leukemogenesis (Nieborowska-Skorska et al., 2002). Induction of Hck activity in the parental 32Dcl3 cells by expression of the Hck-YF mutant was associated with up-regulation of the expression of A1 and pim-1 (Figure 6C). Conversely, the Hck-KE mutant diminished the levels of A1 and pim-1 in BCR/ABL cells.
In addition to the mutant/transfection strategy, the inhibitors of ABL kinase (STI571) and Src family kinases (PP2) were employed to confirm the existence of the BCR/ABL–Hck–STAT5 pathway. As expected, incubation of BCR/ABL-positive cells with STI571 abrogated BCR/ABL kinase activity, Hck kinase activity and STAT5 activation (tyrosine phosphorylation and DNA binding ability) (Figure 6A). PP2, however, did not inhibit BCR/ABL kinase, but abrogated Hck catalytic activity and STAT5 activation. PP3 compound, a negative control for PP2, did not exert these effects. Moreover, both STI571 and PP2 (but not PP3) reduced the BCR/ABL-dependent STAT5-mediated transactivation of the β-casein promoter, but only PP2 and not STI571 inhibited Hck-dependent STAT5-mediated transactivation of the promoter (Figure 6B). In addition, treatment with STI571 and PP2 (but not PP3) diminished the expression of A1 and pim-1 proteins in BCR/ABL-positive cells (Figure 6C). As expected, PP2 (but not STI571 and PP3) inhibited the biological effect of the expression of the Hck-YF mutant in parental 32Dcl cells (data not shown).
Discussion
The oncogenic BCR/ABL tyrosine kinase regulates multiple signaling proteins (Maru, 2001; Sattler and Griffin, 2001). In addition, BCR/ABL activates other cytoplasmic tyrosine kinases including the Src family members Hck and Lyn, which in turn can stimulate their own signaling pathways (Danhauser-Riedl et al., 1996). The Src family of non-receptor tyrosine kinases consists of eight members (Src, Lck, Hck, Fyn, Yes, Blk, Lyn and Fgr), and has been implicated in a wide variety of intracellular signaling pathways in hematopoietic cells (Corey and Anderson, 1999). Each hematopoietic cell lineage may express more than one member of the Src family preferentially, for example myeloid cells express Hck and Lyn, T lymphocytes express Lck and Fyn, and B lymphocytes express Blk and Lyn (Corey and Anderson, 1999).
BCR/ABL expressed in myeloid cells activated both Hck and Lyn (Danhauser-Riedl et al., 1996), suggesting that these kinases might play a role in CML. However, in Philadelphia chromosome-positive (Ph+) ALL cells, BCR/ABL may stimulate different Src family kinases such as Blk, Lck and/or Fyn. The binding and activation of Hck, Lyn and Fyn involved the SH3 + SH2 region of BCR/ABL (Lionberger et al., 2000). More precisely, the ability of BCR/ABL to bind and activate Hck depended on the proline-rich motif binding ability of the SH3 domain and the entire SH2 domain (but not its phosphotyrosine-binding sequence FLVRES) of BCR/ABL (this work). One possibility is that the BCR/ABL SH3 domain recognizes a proline-rich motif of Hck, and the BCR/ABL SH2 domain interacts with Hck in a non-phosphotyrosine-dependent manner (Pendergast et al., 1991). The BCR/ABL SH3 and SH2 domains may also create a ‘pocket’ required for the recruitment of Hck. There is evidence to suggest intramolecular contact between the BCR/ABL SH3 and SH2 domains, collaboration between these domains, and mutual functional influence of one domain on the other (Nam et al., 1996). The same region of the BCR/ABL SH3 and SH2 domains was also engaged in activation of STAT5 (Nieborowska-Skorska et al., 1999). This observation suggested that Src kinases might serve as intermediates linking BCR/ABL to STAT5 activation and downstream signaling. The BCR/ABL region involved in recruitment of Hck was not necessary for activation of RAS and binding of the p85 subunit of phosphatidylinositol-3 kinase (PI-3K), c-Cbl and Shc (Skorski et al., 1997; Nieborowska-Skorska et al., 1999), implicating specificity in the BCR/ABL–Hck–STAT5 interaction.
Although protein kinases such as PDGFβR and Bmx can directly activate STAT5 proteins (Saharinen et al., 1997; Paukku et al., 2000), BCR/ABL seems not to be a good candidate for the kinase activating STAT5 in vivo, because it probably does not associate with STAT5 or the association is very weak (Ilaria and Van Etten, 1996; Nieborowska-Skorska et al., 1999). In addition, BCR/ABL-induced stimulation of STAT5 was not dependent on Jak2 (Ilaria and Van Etten, 1996; Xie et al., 2001), indicating that STAT5 activation by BCR/ABL proceeds via a mechanism distinct from the classical Jak-dependent pathway described for STAT activation in cytokine receptor signaling (Okuda et al., 1999). Interestingly, Jak-independent activation of STAT5 has been reported for Src family kinases (Kazansky et al., 1999; Olayioye et al., 1999; Okutani et al., 2001).
Here we showed that BCR/ABL-mediated activation of Hck represents a major signaling pathway to activate STAT5. Hck associated with STAT5, which did not depend on the Hck catalytic activity or STAT5 phosphorylation because the complex was detectable in the immunoprecipitates from BCR/ABL cells (Hck activated and STAT5 phosphorylated) as well as from parental and BCR/ABLΔΔ cells (Hck inactive and STAT5 not phosphorylated). In addition, it seems that the BCR/ABL SH3 + SH2 region preferentially bound the inactive form of Hck by ABL kinase-independent mechanisms (Warmuth et al., 1997; Lionberger et al., 2000) and that more STAT5 was associated with the kinase-dead Hck in a pull-down assay (data not shown). The latter observation is in agreement with the report that Src formed a kinase-independent complex with STAT5 (Kazansky et al., 1999), which probably depends on an ‘open yet inactive’ configuration of Src (the SH2 domain does not interact with Y509 due to its dephosphorylation, but the positive phosphorylation of Y397 is not yet achieved) allowing the SH2 and SH3 domains of the protein to interact with cellular substrates (Chaturvedi et al., 1998). In fact, BCR/ABL may enhance the pool of Hck molecules exhibiting an ‘open yet inactive’ configuration due to the activation of phosphatases such as Syp (Tauchi et al., 1994; Peng and Cartwright, 1995). Taking all these findings together, the following model for the BCR/ABL–Hck–STAT5 signaling pathway could be proposed: (i) BCR/ABL elevates the pool of Hck molecules in the ‘open yet inactive’ conformation, which bind STAT5; (ii) the Hck (open yet inactive)–STAT5 (inactive) complex is recruited by the BCR/ABL SH3 + SH2 region; (iii) BCR/ABL kinase phosphorylates and activates Hck; (iv) Hck phosphorylates STAT5 on Tyr699 (STAT5B) or Tyr694 (STAT5A); and (v) activation of Hck and subsequent phosphorylation of STAT5 weakens the BCR/ABL–Hck (active)–STAT5 (phosphorylated) complex and causes its dissociation. Our finding that Hck phosphorylated STAT5B on Tyr699 is supported by previous reports indicating that Src kinase phosphorylated STAT5A on Tyr694 (Olayioye et al., 1999; Okutani et al., 2001). Phosphorylation of these essential tyrosine residues of STAT5A and STAT5B triggers their dimerization and transactivation ability (Gouilleux et al., 1994; Yamashita et al., 1998), or allows association with other signaling proteins such as PI-3K (Corey et al., 1993).
That more Hck was immunoprecipitated with tyrosine-phosphorylated STAT5 detected in cell lysates from BCR/ABL cells in comparison with parental and BCR/ABLΔΔ cells may be due to the additional complexes formed by phospho-STAT5 with the p85 subunit of PI-3K associated with activated Hck (Corey et al., 1993). This speculation is supported by the observation that IL-3- or EGF-induced stimulation of Src activity was associated with enhanced interaction with STAT3 or STAT1, respectively (Chaturvedi et al., 1998; Olayioye et al., 1999). In addition, higher levels of Hck were detected in the former cells after they had been starved of IL-3 (data not shown).
Several different Src family kinases often associate with the same cell surface receptor in the same cell type, suggesting that the kinases may have overlapping functions. The redundancy between Src family members was confirmed using knockout mice because the single knockout mice had a very subtle defect, while more pronounced defects were unmasked by cross-breeding to create multiple knockouts (Lowell et al., 1994, 1996; Stein et al., 1994). Therefore, to obtain more information about the role of Hck in BCR/ABL leukemogenesis, the dominant-negative mutant of Hck instead of the Hck knockout cells was applied. Expression of a kinase-defective Hck mutant suppressed BCR/ABL-dependent transformation of myeloid cells to growth factor independence (Lionberger et al., 2000) and activation of STAT5 (this work), implicating Hck as a major signaling molecule in BCR/ABL-induced pathways. Although another member of the Src family, the Lyn kinase, is highly expressed in myeloid cells (Corey and Anderson, 1999) and activated by BCR/ABL (Danhauser-Riedl et al., 1996), the Hck kinase seems to play a primary role downstream of BCR/ABL. There are several observations that may explain this phenomenon. Purified Hck (this work), but not Lyn (Gouilleux et al., 1994), was able to activate STAT5 in vitro, demonstrating that either Lyn requires an intermediating protein to bind and/or activate STAT5, or that the binding exhibits low affinity. In accordance with this speculation, STAT5 was readily detectable in anti-Hck, but not anti-Lyn immunoprecipitates, even if both kinases were able to phosphorylate STAT5 in Sf9 cells (data not shown). In addition to Hck and Lyn, Jak2 can also activate STAT5 (Schindler and Darnell, 1995). However, a Hck kinase-defective mutant inhibited BCR/ABL-mediated STAT5 phosphorylation and activation (this work). Thus, Hck is most likely to be the primary kinase in BCR/ABL–STAT5 signaling, and Lyn and Jak2 are the secondary kinases. Hck, Lyn and Jak2 can bind to the C-terminal and kinase regions of BCR/ABL; in addition, Hck and Lyn may bind to the SH3 + SH2 region of BCR/ABL (Lionberger et al., 2000; Xie et al., 2001). Thus, Src family kinases and Jak kinases probably compete for the binding site(s) on BCR/ABL, and Hck outcompetes the others. In support of this speculation, the BCR/ABL–Hck complex was readily detectable by simple immunoprecipitation, whereas BCR/ABL–Lyn association was demonstrated only after applying a more sensitive radioisotope technique (Danhauser-Riedl et al., 1996). Moreover, Jak kinases (Jak1, Jak2, Jak3 and Tyk2) were not activated consistently in BCR/ABL-transformed cells (Carlesso et al., 1996; Ilaria and Van Etten, 1996; Chai et al., 1997).
The biological effect of a kinase-defective Hck mutant is probably due to its strong interaction with BCR/ABL (Lionberger et al., 2000), which not only blocks activation of the endogenous Hck, but also prevents the potential interaction of BCR/ABL with Lyn and Jak2, resulting in pronounced inhibition of STAT5 activation. However, Lyn and Jak2 may replace Hck in the knockout cells (Hck–/–) and activate STAT5. In fact, the activity of Lyn protein kinase is increased in Hck–/– cells, implying that Lyn may compensate for a deficiency in Hck (Lowell et al., 1994). In addition, Hck–/– mice did not display hematological abnormalities characteristic of STAT5A + B–/– mice, supporting the redundancy among Src-like kinases and/or Jak kinases (Lowell et al., 1994; Bunting et al., 2002).
Better understanding of the biological role of the BCR/ABL–Hck–STAT5 signaling pathway seems to be very important, because: (i) the pathway is essential for leukemogenesis; and (ii) Hck could be considered as a potential selective target for anti-leukemia treatment. The first statement is supported by the reports indicating that BCR/ABL SH3 + SH2 mutants unable to activate STAT5 and Hck did not transform hematopoietic cells (Nieborowska-Skorska et al., 1999), and that expression of the dominant-negative mutants of either STAT5 or Hck blocked growth factor independence and leukemogenic potential of BCR/ABL-transformed myeloid cells (Nieborowska-Skorska et al., 1999; Lionberger et al., 2000). Application of Hck inhibitors (Src kinase family inhibitors) confirmed that Hck is essential for growth factor independence of Ph+ leukemia cells (Wilson et al., 2002). In addition, expression of the two prominent STAT5 downstream effectors A1 and pim-1, which are important for growth factor independence of BCR/ABL-transformed cells (Nieborowska-Skorska et al., 2002), was dependent on Hck activation (this work). Thus, the BCR/ABL–Hck–STAT5 pathway seems to play an essential role in Ph+ myeloid leukemias. Interestingly, the Hck dominant-negative mutant inhibited growth factor-independent proliferation of BCR/ABL-transformed hematopoietic cells, but did not affect cytokine-dependent proliferation of non-transformed cells (Lionberger et al., 2000). Moreover, inhibitors of the Src kinase family members exerted a selective inhibitory effect on Ph+ cells, but not on Ph– cells (Wilson et al., 2002). Altogether, Hck may be considered as a potential selective target for anti-CML treatment. Importantly, combination of an ABL kinase inhibitor (STI571) and an inhibitor of a BCR/ABL downstream effector (e.g. Hck) may exert synergistic anti-leukemia effects, as shown for the combination of STI571 and wortmannin or LY294002 (PI-3K inhibitors) (Klejman et al., 2002).
In summary, we have identified Hck as a primary protein kinase responsible for BCR/ABL-mediated activation of STAT5 in myeloid cells. However, due to a high level of redundancy and selectivity of expression in different cell lineages, we cannot exclude the possibility that other Src family members and Jak kinases may become important for BCR/ABL signaling in the absence of Hck or in non-myeloid cells. Thus, BCR/ABL may switch between Src and Jak kinases dependent on the conditions and cell lineage.
Materials and methods
Cells
Murine growth factor-dependent 32Dcl3 myeloid cells as well as BCR/ABL-transfected counterparts (Nieborowska-Skorska et al., 1999) were cultured in Iscove’s medium (IMDM) supplemented with 10% fetal bovine serum (FBS) and IL-3. Tk–ts13 hamster fibroblast were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS. Sf9 insect cells were cultured in Grace’s complete insect cell medium supplemented with 10% FBS.
Plasmids and constructs
Full-length human STAT5B and STAT5B[Y699] mutant were obtained from Dr Hallgeir Rui (Department of Pathology, Uniformed Services University of the Health Sciences, Bethesda, MD). Full-length STAT5B, STAT5B[1–546] (N5B), STAT5B[547–787] (C5B) and STAT5B[547–787] [Y699F mutant] (C5B-YF) were cloned in-frame into a pGEX-2TK vector in order to express the appropriate GST fusion proteins. Wild-type, kinase-defective (K269E = KE) and activated (Y501F = YF) forms of human Hck, described elsewhere (Briggs et al., 1997), were subcloned into the retroviral vector pMigR1 as a Hck-IRES–green fluorescent protein (GFP) sequence (Briggs et al., 1997).
Kinase inhibitors
STI571 (imatinib mesylate) was a generous gift of Novartis Pharma AG, Basel, Switzerland. STI571 was dissolved in phosphate-buffered saline (PBS) and stored as a 1 mM stock solution. PP2, a potent and selective inhibitor of the Src family of protein tyrosine kinases, and PP3, a negative control for PP2, were from Calbiochem-Novabiochem Co. (San Diego, CA). PP2 and PP3 were dissolved in dimethylsulfoxide (DMSO) and stored as 5 mM stock solutions at –20°C. Cells (106/ml) were treated for 24 h with 1 µM STI571, 10 µM PP2 or PP3, or with the solvent only.
Expression of Hck and STAT5 in Sf9 cells
Generation of recombinant Hck and STAT5 baculoviruses as well as expression of recombinant Hck and STAT5 proteins in Sf-9 cells has been described elsewhere (Lerner and Smithgall, 2002).
Immunoprecipitations and western analysis
STAT5A + B and Hck were immunoprecipitated from cell lysates as described (Skorski et al., 1995), using anti-STAT5 (C-17, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or anti-Hck (Upstate Biotechnology, Lake Placid, NY) antibodies, respectively. Immunoprecipitates and cell lysates were analyzed by SDS–PAGE followed by western analysis using anti-STAT5 (C-17, Santa Cruz), anti-Hck (M-28, Santa Cruz), anti-phosphotyrosine (4G10 from Upstate Biotechnology, and PY20 from Oncogene Research Products, Cambridge, MA), anti-ABL (Ab-3, Oncogene), anti-A1 (T-18 + C-19, Santa Cruz), anti-pim-1 (C-20 + N-16, Santa Cruz) and anti-actin (C-11, Santa Cruz) antibodies.
In vitro kinase reactions
Substrates for the kinase reactions: GST–STAT5 (full-length or fragments) fusion proteins were purified from Escherichia coli using Bulk and RediPack GST Purification Modules (Amersham Pharmacia Biotech, Inc., Piscataway, NJ), enolase (Sigma Chemical Co., St Louis, MO) and GST–Sam68[331–443] (Sam68) protein was purchased from Santa Cruz Biotechnology, Inc. 32Dcl3 cells and clones expressing BCR/ABL proteins were starved of growth factor and serum for 5 h. Kinase reactions were performed as described (Nieborowska-Skorska et al., 1999), with modifications. Briefly, anti-ABL or anti-Hck immunoprecipitates were incubated with 5 µg of the substrate protein in the presence of 1 µM [γ-32P]ATP (Perkin-Elmer). When indicated, 1 µM STI571 or 10 µM PP2 or PP3 were added to the reaction. Reactions were resolved by SDS–PAGE, transferred to nitrocellulose and exposed to Kodak film.
STAT5 functional assays
The DNA binding activity of STAT5 was examined by EMSA using the FcγRI fragment or MGFE probe, as described (Nelson et al., 1998; Skorski et al., 1998). STAT5-dependent transactivation was examined by luciferase assay (Nieborowska-Skorska et al., 1999) with modifications. Briefly, Tk–ts13 hamster fibroblasts were co-transfected with the expression vectors containing BCR/ABL or Hck (wild-type or mutants), or with the insert-less vector along with the STAT5-responsive luciferase reporter construct (β-casein-luc) and the expression plasmid for β-galactosidase (β-gal). At 36 h post-transfection, cells were starved of serum [0.1% bovine serum albumin (BSA), in the presence or absence of the inhibitors] for 12 h and harvested for the luciferase assay using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. For each transfection, luciferase activity was normalized using β-gal activity as an internal control.
Expression of Hck mutants in hematopoietic cells
32Dcl2 cells and BCR/ABL-positive counterparts were infected with pMigR1 retroviral vector particles encoding Hck(YF)-IRES–GFP, Hck(KE)-IRES–GFP or IRES–GFP, as described (Nieborowska-Skorska et al., 1999). Cells were collected after 72 h of co-cultivation with the packaging cell line. GFP-positive cells were isolated by fluorescence-activated cell sorting (FACS).
Acknowledgments
Acknowledgements
This work was supported by American Cancer Society grants RPG9834801LBC, RSG9834804LIB (to T.S.), RPG9605204-TBE and NIH CA81398 (to T.E.S.). T.S. is a Scholar of the Leukemia and Lymphoma Society. A.S. was a Fellow of the Leukemia Research Foundation and is sponsored by an Elsa U.Pardee grant.
References
- Briggs S.D., Sharkey,M., Stevenson,M. and Smithgall,T.E. (1997) SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem., 272, 17899–17902. [DOI] [PubMed] [Google Scholar]
- Bunting K.D., Bradley,H.L., Hawley,T.S., Moriggl,R., Sorrentino,B.P. and Ihle,J.N. (2002) Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood, 99, 479–487. [DOI] [PubMed] [Google Scholar]
- Carlesso N., Frank,D.A. and Griffin,J.D. (1996) Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J. Exp. Med., 183, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai S.K., Nichols,G.L. and Rothman,P. (1997) Constitutive activation of JAKs and STATs in BCR–Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol., 159, 4720–4728. [PubMed] [Google Scholar]
- Chaturvedi P., Reddy,M.V. and Reddy,E.P. (1998) Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene, 16, 1749–1758. [DOI] [PubMed] [Google Scholar]
- Chin H., Arai,A., Wakao,H., Kamiyama,R., Miyasaka,N. and Miura,O. (1998) Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood, 91, 3734–3745. [PubMed] [Google Scholar]
- Clark S.S. et al. (1988) Expression of a distinctive BCR–ABL oncogene in Ph1-positive acute lymphocytic leukemia (ALL). Science, 239, 775–777. [DOI] [PubMed] [Google Scholar]
- Corey S.J. and Anderson,S.M. (1999) Src-related protein tyrosine kinases in hematopoiesis. Blood, 93, 1–14. [PubMed] [Google Scholar]
- Corey S., Eguinoa,A., Puyana-Theall,K., Bolen,J.B., Cantley,L., Mollinedo,F., Jackson,T.R., Hawkins,P.T. and Stephens,L.R. (1993) Granulocyte macrophage-colony stimulating factor stimulates both association and activation of phosphoinositide 3OH-kinase and src-related tyrosine kinase(s) in human myeloid derived cells. EMBO J., 12, 2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley G.Q., Van Etten,R.A. and Baltimore,D. (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science, 247, 824–830. [DOI] [PubMed] [Google Scholar]
- Danhauser-Riedl S., Warmuth,M., Druker,B.J., Emmerich,B. and Hallek,M. (1996) Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res., 56, 3589–3596. [PubMed] [Google Scholar]
- de Groot R.P., Raaijmakers,J.A., Lammers,J.W., Jove,R. and Koenderman,L. (1999) STAT5 activation by BCR–Abl contributes to transformation of K562 leukemia cells. Blood, 94, 1108–1112. [PubMed] [Google Scholar]
- Druker B.J., Tamura,S., Buchdunger,E., Ohno,S., Segal,G.M., Fanning,S., Zimmermann,J. and Lydon,N.B. (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med., 2, 561–566. [DOI] [PubMed] [Google Scholar]
- Gishizky M.L. and Witte,O.N. (1992) Initiation of deregulated growth of multipotent progenitor cells by bcr–abl in vitro. Science, 256, 836–839. [DOI] [PubMed] [Google Scholar]
- Gouilleux F., Wakao,H., Mundt,M. and Groner,B. (1994) Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J., 13, 4361–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisterkamp N., Jenster,G., ten Hoeve,J., Zovich,D., Pattengale,P.K. and Groffen,J. (1990) Acute leukaemia in bcr/abl transgenic mice. Nature, 344, 251–253. [DOI] [PubMed] [Google Scholar]
- Horita M., Andreu,E.J., Benito,A., Arbona,C., Sanz,C., Benet,I., Prosper,F. and Fernandez-Luna,J.L. (2000) Blockade of the Bcr–Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J. Exp. Med., 191, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath C.M. and Darnell,J.E. (1997) The state of the STATs: recent developments in the study of signal transduction to the nucleus. Curr. Opin. Cell Biol., 9, 233–239. [DOI] [PubMed] [Google Scholar]
- Ilaria R.L. Jr, and Van Etten,R.A. (1996) P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem., 271, 31704–31710. [DOI] [PubMed] [Google Scholar]
- Ilaria R.L., Hawley,R.G. and Van Etten,R.A. (1999) Dominant negative mutants implicate STAT5 in myeloid cell proliferation and neutrophil differentiation. Blood, 93, 4154–4166. [PubMed] [Google Scholar]
- Kazansky A.V., Kabotyanski,E.B., Wyszomierski,S.L., Mancini,M.A. and Rosen,J.M. (1999) Differential effects of prolactin and src/abl kinases on the nuclear translocation of STAT5B and STAT5A. J. Biol. Chem., 274, 22484–22492. [DOI] [PubMed] [Google Scholar]
- Kieslinger M., Woldman,I., Moriggl,R., Hofmann,J., Marine,J.C., Ihle,J.N., Beug,H. and Decker,T. (2000) Antiapoptotic activity of Stat5 required during terminal stages of myeloid differentiation. Genes Dev., 14, 232–244. [PMC free article] [PubMed] [Google Scholar]
- Klejman A., Rushen,L., Morrione,A., Slupianek,A. and Skorski,T. (2002) Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene, 21, 5868–5876. [DOI] [PubMed] [Google Scholar]
- Koch C.A., Anderson,D., Moran,M.F., Ellis,C. and Pawson,T. (1991) SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science, 252, 668–674. [DOI] [PubMed] [Google Scholar]
- Lerner E.C. and Smithgall,T.E. (2002) SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat. Struct. Biol., 9, 365–369. [DOI] [PubMed] [Google Scholar]
- Lionberger J.M., Wilson,M.B. and Smithgall,T.E. (2000) Transformation of myeloid leukemia cells to cytokine independence by Bcr–Abl is suppressed by kinase-defective Hck. J. Biol. Chem., 275, 18581–18585. [DOI] [PubMed] [Google Scholar]
- Lowell C.A., Soriano,P. and Varmus,H.E. (1994) Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev., 8, 387–398. [DOI] [PubMed] [Google Scholar]
- Lowell C.A., Niwa,M., Soriano,P. and Varmus,H.E. (1996) Deficiency of the Hck and Src tyrosine kinases results in extreme levels of extramedullary hematopoiesis. Blood, 87, 1780–1792. [PubMed] [Google Scholar]
- Maru Y. (2001) Molecular biology of chronic myeloid leukemia. Int. J. Hematol., 73, 308–322. [DOI] [PubMed] [Google Scholar]
- Mui A.L., Wakao,H., Kinoshita,T., Kitamura,T. and Miyajima,A. (1996) Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J., 15, 2425–2433. [PMC free article] [PubMed] [Google Scholar]
- Nam H.J., Haser,W.G., Roberts,T.M. and Frederick,C.A. (1996) Intramolecular interactions of the regulatory domains of the Bcr–Abl kinase reveal a novel control mechanism. Structure, 4, 1105–1114. [DOI] [PubMed] [Google Scholar]
- Nelson K.L., Rogers,J.A., Bowman,T.L., Jove,R. and Smithgall,T.E. (1998) Activation of STAT3 by the c-Fes protein-tyrosine kinase. J. Biol. Chem., 273, 7072–7077. [DOI] [PubMed] [Google Scholar]
- Nieborowska-Skorska M., Wasik,M.A., Slupianek,A., Salomoni,P., Kitamura,T., Calabretta,B. and Skorski,T. (1999) Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med., 189, 1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieborowska-Skorska M., Hoser,G., Kossev,P., Wasik,M.A. and Skorski,T. (2002) Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase pim-1 in the BCR/ABL-mediated leukemogenesis. Blood, 99, 4531–4539. [DOI] [PubMed] [Google Scholar]
- Nosaka T., Kawashima,T., Misawa,K., Ikuta,K., Mui,A.L. and Kitamura,T. (1999) STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J., 18, 4754–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K., Foster,R. and Griffin,J.D. (1999) Signaling domains of the βc chain of the GM-CSF/IL-3/IL-5 receptor. Ann. NY Acad. Sci., 872, 305–313. [DOI] [PubMed] [Google Scholar]
- Okutani Y., Kitanaka,A., Tanaka,T., Kamano,H., Ohnishi,H., Kubota,Y., Ishida,T. and Takahara,J. (2001) Src directly tyrosine-phosphorylates STAT5 on its activation site and is involved in erythropoietin-induced signaling pathway. Oncogene, 20, 6643–6650. [DOI] [PubMed] [Google Scholar]
- Olayioye M.A., Beuvink,I., Horsch,K., Daly,J.M. and Hynes,N.E. (1999) ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem., 274, 17209–17218. [DOI] [PubMed] [Google Scholar]
- Paukku K., Valgeirsdottir,S., Saharinen,P., Bergman,M., Heldin,C.H. and Silvennoinen,O. (2000) Platelet-derived growth factor (PDGF)-induced activation of signal transducer and activator of transcription (Stat) 5 is mediated by PDGFβ-receptor and is not dependent on c-src, fyn, jak1 or jak2 kinases. Biochem. J., 345, 759–766. [PMC free article] [PubMed] [Google Scholar]
- Pawson T. and Gish,G.D. (1992) SH2 and SH3 domains: from structure to function. Cell, 71, 359–362. [DOI] [PubMed] [Google Scholar]
- Pendergast A.M., Muller,A.J., Havlik,M.H., Maru,Y. and Witte,O.N. (1991) BCR sequences essential for transformation by the BCR–ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell, 66, 161–171. [DOI] [PubMed] [Google Scholar]
- Peng Z.Y. and Cartwright,C.A. (1995) Regulation of the Src tyrosine kinase and Syp tyrosine phosphatase by their cellular association. Oncogene, 11, 1955–1962. [PubMed] [Google Scholar]
- Raitano A.B., Whang,Y.E. and Sawyers,C.L. (1997) Signal transduction by wild-type and leukemogenic Abl proteins. Biochim. Biophys. Acta, 1333, F201–F216. [DOI] [PubMed] [Google Scholar]
- Reddy E.P., Korapati,A., Chaturvedi,P. and Rane,S. (2000) IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene, 19, 2532–2547. [DOI] [PubMed] [Google Scholar]
- Saharinen P., Ekman,N., Sarvas,K., Parker,P., Alitalo,K. and Silvennoinen,O. (1997) The Bmx tyrosine kinase induces activation of the Stat signaling pathway, which is specifically inhibited by protein kinase Cδ. Blood, 90, 4341–4353. [PubMed] [Google Scholar]
- Sattler M. and Griffin,J.D. (2001) Mechanisms of transformation by the BCR/ABL oncogene. Int. J. Hematol., 73, 278–291. [DOI] [PubMed] [Google Scholar]
- Schindler C. and Darnell,J.E.,Jr (1995) Transcriptional responses to polypeptide ligands: the JAK–STAT pathway. Annu. Rev. Biochem., 64, 621–651. [DOI] [PubMed] [Google Scholar]
- Shtivelman E., Lifshitz,B., Gale,R.P., Roe,B.A. and Canaani,E. (1986) Alternative splicing of RNAs transcribed from the human abl gene and from the bcr–abl fused gene. Cell, 47, 277–284. [DOI] [PubMed] [Google Scholar]
- Shuai K., Halpern,J., ten Hoeve,J., Rao,X. and Sawyers,C.L. (1996) Constitutive activation of STAT5 by the BCR–ABL oncogene in chronic myelogenous leukemia. Oncogene, 13, 247–254. [PubMed] [Google Scholar]
- Sillaber C., Gesbert,F., Frank,D.A., Sattler,M. and Griffin,J.D. (2000) STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood, 95, 2118–2125. [PubMed] [Google Scholar]
- Skorski T., Kanakaraj,P., Nieborowska-Skorska,M., Ratajczak,M.Z., Wen,S.C., Zon,G., Gewirtz,A.M., Perussia,B. and Calabretta,B. (1995) Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood, 86, 726–736. [PubMed] [Google Scholar]
- Skorski T. et al. (1997) Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J., 16, 6151–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorski T. et al. (1998) The SH3 domain contributes to BCR/ABL-dependent leukemogenesis in vivo: role in adhesion, invasion and homing. Blood, 91, 406–418. [PubMed] [Google Scholar]
- Slupianek A., Schmutte,C., Tombline,G., Nieborowska-Skorska,M., Hoser,G., Nowicki,M.O., Pierce,A.J., Fishel,R. and Skorski,T. (2001) BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell, 8, 795–806. [DOI] [PubMed] [Google Scholar]
- Sonoyama J. et al. (2002) Functional cooperation among Ras, STAT5 and PI3-K is required for full oncogenic activities of BCR/ABL in K562 cells. J. Biol. Chem., 277, 8076–8082. [DOI] [PubMed] [Google Scholar]
- Stein P.L., Vogel,H. and Soriano,P. (1994) Combined deficiencies of Src, Fyn and Yes tyrosine kinases in mutant mice. Genes Dev., 8, 1999–2007. [DOI] [PubMed] [Google Scholar]
- Tauchi T., Feng,G.S., Shen,R., Song,H.Y., Donner,D., Pawson,T. and Broxmeyer,H.E. (1994) SH2-containing phosphotyrosine phosphatase Syp is a target of p210bcr–abl tyrosine kinase. J. Biol. Chem., 269, 15381–15387. [PubMed] [Google Scholar]
- Waksman G., Shoelson,S.E., Pant,N., Cowburn,D. and Kuriyan,J. (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell, 72, 779–790. [DOI] [PubMed] [Google Scholar]
- Warmuth M., Bergmann,M., Priess,A., Hauslmann,K., Emmerich,B. and Hallek,M. (1997) The Src family kinase Hck interacts with Bcr–Abl by a kinase-independent mechanism and phosphorylates the Grb2-binding site of Bcr. J. Biol. Chem., 272, 33260–33270. [DOI] [PubMed] [Google Scholar]
- Weng Z., Rickles,R.J., Feng,S., Richard,S., Shaw,A.S., Schreiber,S.L. and Brugge,J.S. (1995) Structure–function analysis of SH3 domains: SH3 binding specificity altered by single amino acid substitutions. Mol. Cell. Biol., 15, 5627–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.B., Schreiner,S.J., Choi,H.-J., Kamens,J. and Smithgall,T.E. (2002) Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene, in press. [DOI] [PubMed] [Google Scholar]
- Xie S., Wang,Y., Liu,J., Sun,T., Wilson,M.B., Smithgall,T.E. and Arlinghaus,R.B. (2001) Involvement of Jak2 tyrosine phosphorylation in Bcr–Abl transformation. Oncogene, 20, 6188–6195. [DOI] [PubMed] [Google Scholar]
- Yamashita H., Xu,J., Erwin,R.A., Farrar,W.L., Kirken,R.A. and Rui,H. (1998) Differential control of the phosphorylation state of proline-juxtaposed serine residues Ser725 of Stat5a and Ser730 of Stat5b in prolactin-sensitive cells. J. Biol. Chem., 273, 30218–30224. [DOI] [PubMed] [Google Scholar]
- Zou X. and Calame,K. (1999) Signaling pathways activated by oncogenic forms of Abl tyrosine kinase. J. Biol. Chem., 274, 18141–18144. [DOI] [PubMed] [Google Scholar]