

Abstract
Population genetic models play an important role in human genetic research, connecting empirical observations about sequence variation with hypotheses about underlying historical and biological causes. More specifically, models are used to compare empirical measures of sequence variation, linkage disequilibrium (LD), and selection to expectations under a “null” distribution. In the absence of detailed information about human demographic history, and about variation in mutation and recombination rates, simulations have of necessity used arbitrary models, usually simple ones. With the advent of large empirical data sets, it is now possible to calibrate population genetic models with genome-wide data, permitting for the first time the generation of data that are consistent with empirical data across a wide range of characteristics. We present here the first such calibrated model and show that, while still arbitrary, it successfully generates simulated data (for three populations) that closely resemble empirical data in allele frequency, linkage disequilibrium, and population differentiation. No assertion is made about the accuracy of the proposed historical and recombination model, but its ability to generate realistic data meets a long-standing need among geneticists. We anticipate that this model, for which software is publicly available, and others like it will have numerous applications in empirical studies of human genetics.
The search for inherited influences on human disease and the effort to understand the history of human populations rely on a detailed knowledge of human genome sequence variation. Population genetic models serve as important tools in this quest. By simulating variation under neutral evolution, they provide background expectations about genetic variation, for example, for the frequency distribution of disease alleles and for patterns of linkage disequilibrium. They also serve as a tool to evaluate evidence for evolutionary selection, and to infer the history of populations.
A primary difficulty in using such models lies in deciding which ones to employ. Given the complex history of human populations and the wide variety of plausible model parameters, as well as considerable uncertainty about how mutation and recombination rates vary, it is impossible rigorously to infer from data a single “correct” model: The problem is badly underdetermined. Until comparatively recently, in fact, genome-scale data were scarce enough that they provided few constraints on models. As a result, a common practice in simulating human variation has been to use a set of simple models that are easy to implement. The most basic model is the standard Wright-Fisher neutral model of a freely mixing, constant-sized population, with uniform rates of recombination and mutation across the genome. This model has been (and continues to be) widely used, as have other simple models (e.g., island models).
Simple models have been of enormous utility as heuristics. In some respects, they have also offered surprisingly good fits to empirical data (see, e.g., Fig. 1C,D). As empirical data have accumulated, however, disagreements between expectation and observation have become clear. Perhaps the most obvious discrepancy occurs in predictions of linkage disequilibrium, the nonrandom association of nearby alleles (Frisse et al. 2001; Pritchard and Przeworski 2001; Ardlie et al. 2002). Human heterozygosity (given measured mutation rates) suggests an effective population size (Ne) of 10,000 or perhaps 20,000. A simple version of the standard neutral model with this size population and with measured recombination rates, however, predicts far less LD than is observed (Fig. 1A,B) in most populations; the disagreement is particularly severe for non-African populations. Despite its other virtues, therefore, the standard neutral model is not a good model of human LD, and simulations based on that model are unlikely to have great utility for applications where LD is important.
Figure 1.

Fit of standard neutral model to empirical data. Comparison of simulated data under standard neutral model to empirical data on autosomes. Error bars represent one standard error. (A,B) Linkage disequilibrium (measured by r2 and D′) as a function of distance. (Solid line) Standard neutral model; (squares) West African data; (triangles) European data. (A) r2 as a function of physical distance. (B) D′ as a function of genetic distance. (C,D) Genetic distance (FST) and allele frequency spectrum for data and standard neutral model. (White) Data; (gray) model. (C) FST between European and West African populations. (D) European allele frequency spectrum.
Given the prevailing ignorance about the details of human demographic history, and because of the evident inconsistencies between simple models and empirical data, working models of human genetic variation are increasingly including a range of additional features (Wakeley et al. 2001; Reich et al. 2002; Sabeti et al. 2002; Wakeley and Lessard 2003; Wiuf and Posada 2003; Akey et al. 2004; Anderson and Slatkin 2004; Marth et al. 2004); the same is also true for studies of other species (Wall et al. 2002; Tenaillon et al. 2004). Commonly used features include recent population expansion, past bottlenecks, population structure, and “hotspots” of recombination.
What have not been published to date, however, are models that have been calibrated by comparison to many aspects of large, multilocus empirical data sets. As a result, despite all of the work done on modeling (much of it quite sophisticated), no tool currently exists for creating simulated human genetic data that is a good approximation to real, empirical data. It is to address this lack that we have carried out the model calibration described here. We have two goals in doing so: First, to create, for our and others' use, a simulation package that can produce realistic genetic data; and second, to encourage the development of similar (and better) calibrated models.
Results
In order to carry out our simulations, we implemented a coalescent population genetic model (Hudson 1990) in software. Conceptually, the program is similar to Richard Hudson's widely used program (Hudson 2002), but with one additional feature: The recombination rate can vary (to any extent the user cares to specify) within the locus being simulated. The simulation software was validated by comparison with standard predictions of the Wright-Fisher model. The program generates multimarker haplotype data for large chromosome segments (500,000 to 1 million base pairs, Mb) and permits a wide range of demographic histories for multiple populations (population splits, admixture, changes in size, bottlenecks, and migration); a simple model of gene conversion is also included. The simulation package can be obtained from http://www.broad.mit.edu/∼sfs/cosi.
As the basis for calibrating our model, we used a set of empirical observations from numerous genomic loci studied in population samples from three continental regions: West Africa, Europe, and East Asia (see Methods for details of the empirical data sets used). Since our goal was to create simulated data that simultaneously matches many aspects of empirical data, we compared the results of simulation to a broad set of empirical measures: (1) the frequency distribution of alleles, (2) the relationship between the frequency of an allele and the probability that it is the ancestral allele (estimated for empirical data from the chimpanzee allele), (3) a measure of the genetic distance between current populations (FST), (4 and 5) two measures of the extent of linkage disequilibrium (r2 and the fraction of marker pairs with D′ = 1), and (6) the total diversity (measured as heterozygosity, or π). We used data from autosomal loci to tune the model parameters, and also compared model predictions to a second data set of X-chromosome markers. To our knowledge, no previous study has attempted simultaneously to match this wide range of statistics.
Since our simulated data are intended for a variety of applications, there is no single natural choice of a metric for assessing how closely the simulated results match empirical observations. We therefore adopted a simple statistical framework that incorporated all of our statistical measures. For each measure (e.g., allele frequency, r2), we estimated the root-mean-square (RMS) deviation between simulated values and the mean empirical value, calculating it for each of the distributions shown in Figure 2 (see Methods). Four of the measures (minor allele frequency, ancestral/chimpanzee fraction, and the two LD measures) contribute three distributions each, one for each population; treating each of the pairwise FST values as a separate distribution, we have a total of 15 distributions. While the choice of this particular combination of measures was arbitrary, the goal of the procedure was to produce a model yielding adequate simulations of all measures, making it useful for a variety of applications. We compared the statistical uncertainty (standard error on the mean, see Methods for details) of our training data set to the distance between the mean values of the prediction and the empirical data. That is, our goal was to achieve an agreement between the prediction and the data that was comparable to the statistical uncertainty in a large empirical data set such as ours; for this purpose, we set our threshold for acceptable performance at 1.5 times the statistical uncertainty. As an overall composite measure of statistical fit, we calculated the RMS error (RMSE) for all 15 distributions together, normalizing each by its empirical statistical uncertainty.
Figure 2.

Comparison of best-fit model with empirical data, autosomes. Error bars represent one standard error. (A,B,C) Allele frequency spectrum. (White) Data; (black) model. (A) West African. (B) East Asian. (C) European sample. (D,E,F) Fraction of alleles that are ancestral/chimpanzee, binned by allele frequency. (White) Data; (black) model. (D) West African. (E) East Asian. (F) European. (G,H,I) Linkage disequilibrium (r2) versus physical distance. (Points) Data; (line) model. (G) West African. (H) East Asian. (I) European. (J,K,L) Fraction of marker pairs with perfect LD (D′ = 1.0) versus genetic distance. (J) West African. (K) East Asian. (L) European. (M) Genetic distance (FST). (White) Data; (black) model.
We started our calibrated model from what was essentially a standard neutral model, modified to incorporate an “Out of Africa” scenario for the origin of our three test populations. In the base model, each population was a Wright-Fisher population of effective size 10,000; dates for the splitting of the populations were set to 3500 generations (before the present) for the separation between the African and non-African populations, and to 2000 generations for the separation of the two non-African populations (see Fig. 3). Predictions for this simple model, assuming a constant recombination rate of 1.3 cM/Mb, matched the empirical data quite poorly (Fig. 2): the RMSE was 4.7, that is, the disagreement between empirical measures and model prediction was almost five times larger than the statistical uncertainty in our empirical data set.
Figure 3.

Demographic model. N1: ancestral population size. (N2) African population size. (N3) non-African population size. (Texp) Time of ancestral population expansion (if any). Bottlenecks are indicated by constrictions. (Not shown: recurring migration between African and European populations, and between Asian and African populations.)
To this base model we introduced additional parameters and tuned them until the overall match between simulation and empirical data met our criterion. Where possible, we tried to make choices that were demographically and biologically plausible. The resulting parameters, however, represent a mechanism for producing realistic-looking data, not an inference about the actual history of human populations or about details of recombination rate variation. Since the potential search space among many parameters is large, we took a stepwise approach: first choosing a set of parameters to add to the model, optimizing them by minimizing the RMSE, and then iterating the procedure by adding further parameters until the model fit was acceptable. At each step, fitting first used coarse step sizes (e.g., ±2000 for population sizes), and then finer ones, in an effort to avoid local minima.
Beginning with the base model described above, we first altered parameters that affect single-locus features of the data, which are influenced only by demography (not recombination): specifically, heterozygosity, allele frequency spectrum, fraction of ancestral/chimpanzee alleles, and FST. Of these, we first fit the West African allele frequency spectrum, since it is generally accepted that the human population originated in Africa, finding that models with a historical population expansion resulted in an improved fit to the data by increasing the fraction of low-frequency alleles. Next, we considered the remaining single-locus measures, and successively added parameters (primarily population bottlenecks, but also small amounts of continuing migration between populations, which served to reduce the genetic distances between populations) until the RMSE for the single-locus measures was 1.15. That is, based solely on the characteristics of single markers, the model fit the data nearly as well as the sampling error within the empirical observations, and much better than our base, standard neutral, model. Finally, the mutation rate in the model was tuned to match the observed heterozygosity (see Methods) (Sachidanandam et al. 2001).
We then turned to the recombination model. In order to generate the considerable extent of LD seen in empirical data, we held the demographic parameters fixed and introduced variation in recombination rates, first by including observed large-scale variation, as measured in the deCODE genetic map (Kong et al. 2002), and then by adding fine-scale variation, including localized hotspots of recombination. The need for non-uniform recombination to obtain sufficient extent of LD is consistent with a range of observations indicating non-uniform recombination in the human genome (Jeffreys et al. 2001; Cullen et al. 2002; May et al. 2002; Reich et al. 2002; Kauppi et al. 2003; Wall and Pritchard 2003a,b; Crawford et al. 2004; McVean et al. 2004).
In total, approximately 2 billion coalescent trees were generated in the fitting process. The best-fitting set of parameters (Table 1; Fig. 2) yielded good agreement with all aspects of the observed data listed above. Agreement was not perfect: The RMSE between predicted values and the mean empirical values was on average 1.35 (that is, the disagreement was 35% larger than what would be seen solely based on random sampling of the empirical data). The agreement was, however, far superior to the discrepancy of 4.7 found with the standard neutral model. We explored the effects of re-estimating the parameters once all had been added to the model (i.e., iterating the fit); we found further improvements of the fit to be negligible. To our knowledge, this is the first time population genetic simulations have produced data that agree with multiple aspects of empirical data from multiple parts of the genome and in more than one population sample.
Table 1.
Parameters of best-fitting model
| Variable parameters | Best-fit model |
| Ne (ancestral) | 12,500 |
| Ne (African) | 24,000 |
| Ne (non-African) | 7700 |
| T (African expansion) (gens) | 17,000 |
| OoA bottleneck (F) | 0.085 |
| Asian bottleneck (F) | 0.067 |
| European bottleneck (F) | 0.020 |
| African bottleneck (F) | 0.008 |
| Africa ↔ Europe migration rate (per chromosome) | 3.2 × 10-5 |
| Africa ↔ Asia migration rate (per chromosome) | 0.8 × 10-5 |
| Recombination hotspot spacing (bp) | 8500 |
| Hotspot spacing shape parameter | 0.35 |
| Fraction of recombination in hotspots | 88% |
| Gene conversion (initiation prob/bp) | 4.5 × 10-9 |
| Fixed parameters | |
| Mutation rate (per base pair per generation) | 1.5 × 10-8 |
| Ne (post-agriculture) | 100,000 |
| T (out of Africa) (gens) | 3500 |
| T (Eur/Asia split) (gens) | 2000 |
| T (Asian agriculture) (gens) | 400 |
| T (European agriculture) (gens) | 350 |
| T (African agriculture) (gens) | 200 |
Having tuned our simulation, we next examined how well it predicted aspects of the data not used in the tuning process. First, we used the model to generate predictions for the same measures as above, but for the X-chromosome instead of the autosomes. We compared the results to an X-chromosome data set, derived from the same population samples as the autosomal data. Results for both our best-fitting model and the standard neutral model are shown in Figure 4. The statistical power of this data set for evaluating simulations is limited, but it is sufficient to demonstrate that the calibrated model does perform visibly better than the standard neutral model (RMSE = 0.97 for the best-fit model vs. 1.51 for the standard neutral model). The smaller effective population size of the X-chromosome (three-quarters that of the autosomes) makes it a useful test of how well the simulation models genetic drift, which is dependent on population size: The effect of the smaller population can be clearly seen in the larger genetic distances and the high fraction of X-linked markers that are monomorphic. Second, we looked at how well the calibrated model simulated haplotype blocks, contiguous stretches of chromosome observed to have very low rates of historical recombination and low haplotype diversity (Fig. 4L,M). Again the model performed very well (within the statistical limits of our ability to measure it), and much better than the standard neutral model (Phillips et al. 2003).
Figure 4.

Comparison of best fit-model with empirical data, X-chromosome. Error bars represent one standard error. (A,B,C) Allele frequency spectrum. (White) Data; (black) best-fit model; (gray) standard neutral model. (A) West African. (B) East Asian. (C) European sample. (D,E,F) Fraction of alleles that are ancestral/chimpanzee, binned by allele frequency. (White) Data; (black) best-fit model; (gray) standard neutral model. (D) West African. (E) East Asian. (F) European. (G,H) Linkage disequilibrium (r2) versus physical distance. (Points) Data; (solid line) best-fit model; (dashed line) standard neutral model. (G) West African. (H) European. (East Asian omitted because of poor statistics.) (I,J) Fraction of marker pairs with perfect LD (D′ = 1.0) versus genetic distance. (I) West African. (J) European. (Points) Data; (solid line) best-fit model; (dashed line) standard neutral model. (East Asian omitted because of poor statistics.) (K) Genetic distance (FST). (White) Data; (black) best-fit model; (gray) standard neutral model. (L,M) Fraction of sequence in haplotype blocks of different sizes. (White) Data; (black) best-fit model; (gray) standard neutral model. (L) West African. (M) non-African (European + East Asian).
Finally, we show in Figure 5 an application of our calibrated model to a study of human genetic variation, a search for evidence of positive selection in a set of ∼100 genes (Walsh et al. 2005). The empirical data consisted of SNPs in and near genes with a density of 1 per 4 kb, genotyped in samples from three populations; since the three populations were similar to those used in calibrating our model, simulation results could be compared directly with data. Several statistics were calculated from the genotype data as possible indicators for the occurrence of selective sweeps around the genes; shown are the mean FST and the mean heterozygosity for each population. While the distributions differ between populations, in all three cases the agreement with simulation is excellent. Since this is a neutral simulation, there appears to be no need to invoke selective explanations even for the outliers, at least for these statistics.
Figure 5.

Comparison of best-fit model with data: 52 gene regions. Here 40 genes are genotyped in three populations; long genes were subdivided into smaller regions. The mean FST and heterozygosity are shown (black), and compared to the same measures for simulated data (gray); simulated regions were 120 kb long with 30 ± 10 SNPs per region. (A) Yoruba sample. (B) Chinese sample. (C) CEPH sample.
Discussion
We have described the development of a particular calibrated model. Since this was not an exhaustive survey, it is certain that better-fitting parameters can be found even within the same model framework, and it is likely that a broad array of different models would also perform acceptably. The model parameters, therefore, do not represent inferences about real-world processes, but values that happen to generate useful simulations. Nevertheless, our experience in developing our model suggests, but does not prove, that some of its features are likely to recur in any successful simulation. In particular, we found that substantially increased coalescence in our non-African lineages was a necessary component. Thus, our best-fitting model had a probability of European coalescence of 22% (equivalent to a bottleneck with an inbreeding coefficient of 0.22 relative to the source population) (Reich et al. 2001); we were unable to find models in which this parameter was outside the range of ∼0.20–0.25. More detailed features of the model (e.g., migration rates, or the attribution of inbreeding into population size and discrete bottleneck), on the other hand, are likely to be quite variable between successful models. Similarly, a substantial non-uniformity in recombination rate was required, but the specifics of how the non-uniformity was implemented are not likely to be very meaningful.
The justification for this kind of model, therefore, is not that it enables us to draw conclusions about human history. There are, instead, two reasons for developing it. First, the ability to produce realistic-looking data is itself useful, regardless of the historical accuracy of the underlying model. It is true that there are many applications for which simple models of human genetic variation serve admirably. For many other applications, however, it is important to be able to produce simulated data that bear a close resemblance to empirical data. For example, simulated data sets for comparing haplotype-phasing algorithms, or for assessing the density of markers needed to provide good coverage for disease studies, are only useful if they accurately reflect LD patterns in human populations. In these cases, a calibrated model is greatly preferable to something like the standard neutral model, and the model presented here is already in use for studying haplotype phasing. We anticipate that it (and similar models, when they appear) will continue to be used, and will continue to be refined as additional comparisons with empirical data are made.
Second, while the model is unlikely to reflect accurately the details of either demographic history or recombination, it does represent one of the many models that is consistent with what is currently known about human genetics. While it would be preferable to have the true model, for many purposes it is better to have one model that is consistent with data than to have none. For example, consider the X-chromosome data presented earlier (Fig. 4). In the absence of any model, or with only the standard neutral model as guidance, there is no way to interpret the differences between the X data and that from the autosomes. Are the increased monomorphism in non-African populations and the increased FST values to be expected from the smaller effective population of the X, or are they suggestive of positive selection acting on these loci? Given that our model, a randomly selected instance drawn from the space of consistent neutral models, predicts such features, there seems little need to invoke selection. Clearly, for more robust inference, especially in the case that model and data disagree, a broad range of calibrated models would be preferable, and we anticipate that such additional models will be developed in the near future.
Methods
Data sets
The autosomal data set (previously described in Gabriel et al. 2002) contained 3738 markers distributed in 54 regions across the genome. The X-chromosome set contained 250 markers in 16 roughly equidistant regions; it excluded regions homologous to the Y pseudoautosomal regions. For both sets, SNPs were selected from TSC (The SNP Consortium) SNPs in dbSNP, the public SNP database, without regard to genes or other genomic features. The autosomal regions averaged 250 kb in length. Two of the X regions were of comparable size, and the remainder were 80 kb in length; LD measurements for the X sample were therefore confined to distances <80 kb. All SNPs were genotyped in 31 parent–offspring trios (93 individuals) of European ancestry from the CEPH pedigrees, 42 unrelated individuals of Japanese and Chinese origin, and 30 parent–offspring trios from Nigeria (Yoruba). Gender was determined for Asian individuals by genotyping a Y-linked SNP. Ancestral allele status was taken to be that found by genotyping one chimpanzee (Pan troglodytes); chimpanzee alleles were not available for the X markers. Genotyping for the X markers was as described in Gabriel et al. (2002).
Samples and genotyping for the genes shown in Figure 5 are described in Walsh et al. (2005), as are the genes studied. Long genes were broken into ∼120-kb regions, and the regions were treated as independent; regions with <10 working SNPs were dropped. The remaining set consisted of 52 regions in 40 genes. Simulated regions were 120 kb in length and contained 30 ± 10 markers.
Empirical measures
Allele frequencies and the fraction of ancestral/chimpanzee markers were calculated only for markers for which both alleles appeared somewhere in the three data samples. LD measures (D′ and r2) were calculated only for markers with a minor allele frequency (MAF) >0.20. Statistical uncertainty for all autosomal measures were estimated by bootstrap (random sampling of the available regions, with replacement). Because two of the X regions had a disproportionate number of markers, where possible (allele frequencies and FST), X-based measures were calculated separately for each region, and then means and variances were calculated over the set of regions; for measures with too little data in each region (ancestral/chimpanzee fraction and LD), bootstrap estimates were used.
Simulation software
The simulation program implements a coalescent model, loosely based on Hudson's program (Hudson 1990), written in C. It generates an ancestral tree for a hypothetical data sample and randomly places mutation on the branches. The coalescent simulation was validated by comparison with standard predictions for a constant-sized, neutral population (distribution of times to the most recent common ancestor, allele frequency distribution, ratio of external to total branch length). Two versions of the simulation were written. A fast, two-locus version was used to optimize model parameters, and the full-sequence version was used for studying haplotype blocks and simulating gene loci; the latter is being made publicly available. The demographic model for the coalescent simulator is defined at run time via a parameter file; any number of populations can be simulated, and any combination of population size, bottlenecks, splits or extinctions, admixture, continuing migration between populations, and instantaneous or exponential changes in population size can be specified by parameters. The local recombination map is defined in an input file; our recombination model is implemented in a second program, which is also driven by a parameter file.
Demographic model
All models were based on an ancestral population that split to form a modern West African population and a Eurasian population, which subsequently split into European and East Asian populations. Dates of the splits were left fixed. All non-African populations were given the same effective population size. Population bottlenecks and changes in size were treated as instantaneous. Late expansion to a large size, roughly coincident with the advent of agriculture, was included for the sampled populations in all calibrated models, but had little effect on results; see Table 1 for the parameters used. X-chromosome simulations used the same parameter values as autosomal simulations, except for parameters that are intrinsically different on the X-chromosome: Population sizes, bottleneck intensities, and migration rates were scaled for the smaller effective population size of the X-chromosome, while regional recombination rates were drawn from an X-based distribution, and were corrected for the lack of recombination in males.
Recombination model
The recombination model was hierarchical. First, a regional rate was chosen from the observed distribution of rates for these regions, based on the deCODE genetic map (Kong et al. 2002). A variable fraction of this recombination rate was distributed uniformly (the background fraction listed in Table 1); the rest was clustered into hotspots. A local rate was drawn randomly from a gamma distribution with a fixed shape parameter of 0.3, with a mean equal to the regional rate; this additional variation is included to model changes in recombination rate at scales of hundreds of kilobases, which are smoothed out by the ∼2-Mb resolution of the genetic map. The local rate was then used to create individual hotspots. These had random spacing (gamma distribution with shape and mean as variable parameters, listed in Table 1) and random intensity (gamma distribution with fixed shape parameter of 0.3 and with mean determined by the local rate). Note that, in this model, most “hotspots” are weak and closely spaced; only the high end of the distribution corresponds to reported hotspots observed in human data. Gene conversion was treated as a double recombinant, with a tract length of 0.5 kb, parameterized by a (uniform) probability of initiation.
SNP ascertainment
When simulating genotype data, accurate modeling of the ascertainment process for the markers is essential. The SNPs in our data sets (all from TSC) were ascertained (Sachidanandam et al. 2001) by two methods, alignment of whole-genome shotgun (WGS) reads and alignment of reduced-representation (RRS) reads, both with the public genome sequence; in each case reads were drawn from an ethnically mixed pool of 48 chromosomes (Collins et al. 1998). WGS ascertainment (60% of all SNPs) was modeled as the comparison of a single pair of chromosomes, while RRS ascertainment (40%), which involved multiple overlapping reads at the same locus, was modeled by comparison of four chromosomes to a single reference chromosome. In practice, the only difference created by the two ascertainment schemes was a modest shift toward lower allele frequency for the RRS set. A pool of ascertainment chromosomes was included in the coalescent simulation, but withheld from the simulated output data. Ascertainment chromosomes were selected at random from the sampled populations, with the probability of selection based on the ethnic composition of the discovery resources (known in the case of the NIH panel, estimated based on allele frequencies in the case of the public genome); the fraction of chromosomes identified by the NIH as Native American was treated for this purpose as Asian (the effect of this approximation was tested by explicitly modeling a Native American population, and was found to be negligible). Different ascertainment chromosomes were selected every 500 bp (the approximate length of a TSC sequence read) for one chromosome and every 150,000 bp (the approximate length of a Human Genome Project clone insert). All sites that differed between the ascertainment chromosomes were defined to be identified polymorphisms. These comparisons also provided a measurement of heterozygosity, which was used to set the overall mutation rate in the model; the rate was set so that the simulated heterozygosity matched that measured by the TSC project using the same methodology (Sachidanandam et al. 2001). When simulating our empirical data sets, markers were randomly dropped from the simulation to match the mean marker density and the distribution of marker spacing in the empirical data.
Sensitivity to details of ascertainment modeling was tested by (1) varying the source-population composition of the public genome model by ±20% for each population, and (2) carrying out the simulation with only 1× or only 4× coverage. Variation in the source population, and keeping all model parameters fixed, resulted in RMS deviations of 1.36–1.40 (vs. 1.35 for the original model), and incorrectly specifying the coverage resulted in deviations of 1.39 and 1.45. Such modest changes—all results met our threshold requirement for acceptable results—suggest that details of the ascertainment model are not of great importance.
Parameter fitting
Model parameters were optimized by grid searches over two to four parameters at a time. Goodness of fit was evaluated by calculating the total RMS discrepancy, Δtot, between predicted values and the empirical means:
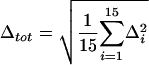 |
where Δi is the RMSE for one of the 15 distributions (1 per population for r2, D′, allele frequency spectrum, and fraction ancestral/chimpanzee; and 1 for each of the three genetic distances). Δi is defined as
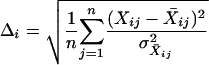 |
where Xij is the model's predicted value for the j-th bin of the i-th distribution, X̄ij is the empirical mean for the same value, 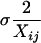 is the standard error on the empirical mean, and the sum runs over the n bins in the distribution i. Note that, with these definitions, Δtot calculated from a random resampling (rather than from model predictions) of the empirical data is 1.0.
is the standard error on the empirical mean, and the sum runs over the n bins in the distribution i. Note that, with these definitions, Δtot calculated from a random resampling (rather than from model predictions) of the empirical data is 1.0.
Not all parameters were of equal importance in the model. Setting the three least important parameters (the migration rates and the African bottleneck) to zero yielded agreement only slightly worse than our threshold value (1.55 times the standard error). In contrast, eliminating bottlenecks in the non-African lineages (whether supplied by an explicit bottleneck or by a small population size) produced discrepancies at least a factor of 2 larger than those seen in the full model. In the recombination model, eliminating either the regional variation or the discrete hotspots produced roughly the same loss of performance (discrepancy ∼2.7).
Acknowledgments
We thank David Cutler for helpful comments, members of the Broad Institute's Program in Medical and Population Genetics for useful discussions, and the International Haplotype Map Consortium for financial support.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3709305. Freely available online through the Genome Research Immediate Open Access option.
References
- Akey, J.M., Eberle, M.A., Rieder, M.J., Carlson, C.S., Shriver, M.D., Nickerson, D.A., and Kruglyak, L. 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E.C. and Slatkin, M. 2004. Population-genetic basis of haplotype blocks in the 5q31 region. Am. J. Hum. Genet. 74: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardlie, K.G., Kruglyak, L., and Seielstad, M. 2002. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 3: 299–309. [DOI] [PubMed] [Google Scholar]
- Collins, F.S., Brooks, L.D., and Chakravarti, A. 1998. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 8: 1229–1231. [DOI] [PubMed] [Google Scholar]
- Crawford, D.C., Bhangale, T., Li, N., Hellenthal, G., Rieder, M.J., Nickerson, D.A., and Stephens, M. 2004. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 36: 700–706. [DOI] [PubMed] [Google Scholar]
- Cullen, M., Perfetto, S.P., Klitz, W., Nelson, G., and Carrington, M. 2002. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am. J. Hum. Genet. 71: 759–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse, L., Hudson, R.R., Bartoszewicz, A., Wall, J.D., Donfack, J., and Di Rienzo, A. 2001. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am. J. Hum. Genet. 69: 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, S.B., Schaffner, S.F., Nguyen, H., Moore, J.M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M., Lochner, A., Faggart, M., et al. 2002. The structure of haplotype blocks in the human genome. Science 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R. 1990. Gene genealogies and the coalescent process. In Oxford surveys in evolutionary biology (eds. D.J. Futuyma and J. Antonovics), pp. 1–44. Oxford University Press, Oxford, UK.
- Hudson, R.R. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Jeffreys, A.J., Kauppi, L., and Neumann, R. 2001. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 29: 217–222. [DOI] [PubMed] [Google Scholar]
- Kauppi, L., Sajantila, A., and Jeffreys, A.J. 2003. Recombination hotspots rather than population history dominate linkage disequilibrium in the MHC class II region. Hum. Mol. Genet. 12: 33–40. [DOI] [PubMed] [Google Scholar]
- Kong, A., Gudbjartsson, D.F., Sainz, J., Jonsdottir, G.M., Gudjonsson, S.A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G., et al. 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31: 241–247. [DOI] [PubMed] [Google Scholar]
- Marth, G.T., Czabarka, E., Murvai, J., and Sherry, S.T. 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166: 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, C.A., Shone, A.C., Kalaydjieva, L., Sajantila, A., and Jeffreys, A.J. 2002. Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat. Genet. 31: 272–275. [DOI] [PubMed] [Google Scholar]
- McVean, G.A., Myers, S.R., Hunt, S., Deloukas, P., Bentley, D.R., and Donnelly, P. 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Phillips, M.S., Lawrence, R., Sachidanandam, R., Morris, A.P., Balding, D.J., Donaldson, M.A., Studebaker, J.F., Ankener, W.M., Alfisi, S.V., Kuo, F.S., et al. 2003. Chromosome-wide distribution of haplotype blocks and the role of recombination hot spots. Nat. Genet. 33: 382–387. [DOI] [PubMed] [Google Scholar]
- Pritchard, J.K. and Przeworski, M. 2001. Linkage disequilibrium in humans: Models and data. Am. J. Hum. Genet. 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich, D.E., Cargill, M., Bolk, S., Ireland, J., Sabeti, P.C., Richter, D.J., Lavery, T., Kouyoumjian, R., Farhadian, S.F., Ward, R., et al. 2001. Linkage disequilibrium in the human genome. Nature 411: 199–204. [DOI] [PubMed] [Google Scholar]
- Reich, D.E., Schaffner, S.F., Daly, M.J., McVean, G., Mullikin, J.C., Higgins, J.M., Richter, D.J., Lander, E.S., and Altshuler, D. 2002. Human genome sequence variation and the influence of gene history, mutation and recombination. Nat. Genet. 32: 135–142. [DOI] [PubMed] [Google Scholar]
- Sabeti, P.C., Reich, D.E., Higgins, J.M., Levine, H.Z., Richter, D.J., Schaffner, S.F., Gabriel, S.B., Platko, J.V., Patterson, N.J., McDonald, G.J., et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R., Weissman, D., Schmidt, S.C., Kakol, J.M., Stein, L.D., Marth, G., Sherry, S., Mullikin, J.C., Mortimore, B.J., Willey, D.L., et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933. [DOI] [PubMed] [Google Scholar]
- Tenaillon, M.I., U'Ren, J., Tenaillon, O., and Gaut, B.S. 2004. Selection versus demography: A multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 21: 1214–1225. [DOI] [PubMed] [Google Scholar]
- Wakeley, J. and Lessard, S. 2003. Theory of the effects of population structure and sampling on patterns of linkage disequilibrium applied to genomic data from humans. Genetics 164: 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., Nielsen, R., Liu-Cordero, S.N., and Ardlie, K. 2001. The discovery of single-nucleotide polymorphisms—and inferences about human demographic history. Am. J. Hum. Genet. 69: 1332–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J.D. and Pritchard, J.K. 2003a. Assessing the performance of the haplotype block model of linkage disequilibrium. Am. J. Hum. Genet. 73: 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J.D. and Pritchard, J.K. 2003b. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 4: 587–597. [DOI] [PubMed] [Google Scholar]
- Wall, J.D., Andolfatto, P., and Przeworski, M. 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, E.C., Sabeti, P., Hutcheson, H., Fry, B., Schaffner, S.F., de Bakker, P.I.W., Varilly, P., Roy, J., Cooper, R., Zeng, Y., et al. 2005. Large-scale survey of variation and signals of selection in 168 immunological genes. Human Genetics (in press). [DOI] [PubMed]
- Wiuf, C. and Posada, D. 2003. A coalescent model of recombination hotspots. Genetics 164: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web site references
- http://www.broad.mit.edu/∼sfs/cosi; authors' Web site.