

Abstract
In Escherichia coli, the major cytoplasmic domain (C6) of the polytopic membrane protein lactose permease (LacY) is exposed to the opposite side of the membrane from a neighboring periplasmic domain (P7). However, these domains are both exposed on the periplasmic side of the membrane in a mutant of E.coli lacking phosphatidylethanolamine (PE) wherein LacY only mediates facilitated transport. When purified LacY was reconstituted into liposomes lacking PE or phosphatidylcholine (PC), C6 and P7 were on the same side of the bilayer. In liposomes containing PE or PC, C6 and P7 were on opposite sides of the bilayer. Only the presence of PE in the liposomes restored active transport function of LacY as opposed to restoration of only facilitated transport function in the absence of PE. These results were the same for LacY purified from PE-containing or PE-lacking cells, and are consistent with the topology and function of LacY assembled in vivo. Therefore, irrespective of the mechanism of membrane insertion, the subdomain topological orientation and function of LacY are determined primarily by membrane phospholipid composition.
Keywords: lactose permease/membrane protein assembly/phosphatidylethanolamine/phospholipid/topology
Introduction
The assembly of polytopic membrane proteins requires the targeting of a nascent polypeptide to the membrane, recognition, integration and orientation of transmembrane (TM) domains, and proper formation of tertiary and quaternary structure. How a membrane protein attains its final form in vivo is not known. Whether there are constraints imposed on the folding process by the translocase/insertase assembly machinery in addition to those dictated by the principles underlying protein–lipid interactions is also not clear (Chin et al., 2002). Does the membrane lipid composition impose constraints on orientation of TM domains or simply provide a hydrophobic environment for protein folding? Many proteins follow the positive inside rule whereby the TM domains are organized so that those positively charged hydrophilic domains connecting TM domains face the cytoplasm in bacteria (von Heijne, 1992). The molecular basis for this organization is due partly to the negative inward membrane potential (Andersson and von Heijne, 1994) and partly to the interaction of positively charged protein domains with headgroups of the anionic phospholipids phosphatidylglycerol (PG) and cardiolipin (CL) (van Klompenburg et al., 1997).
The most compelling evidence for a specific role for phospholipids in membrane protein organization is the requirement for phosphatidylethanolamine (PE) for the proper folding, topological organization and function of lactose permease (LacY) of Escherichia coli (Bogdanov et al., 1996, 1999; 2002; Bogdanov and Dowhan, 1998). LacY is organized in the inner membrane with 12 TM domains connected by hydrophilic domains exposed alternatively to the cytoplasm and the periplasm (Figure 1). LacY couples downhill movement of a proton with the uphill movement of substrate to drive active transport, but can also facilitate the equilibration of substrate across the membrane in the absence of a proton electrochemical gradient. LacY assembled in a mutant of E.coli lacking PE cannot accumulate substrate against a concentration gradient but can still facilitate substrate transport. When LacY is assembled in cells lacking PE, TM domains I–VI and the hydrophilic domains from the N-terminus through the major cytoplasmic domain (C6) are topologically inverted with respect to the membrane bilayer. Periplasmic domain P7 to the C-terminus retains native topology. The conformation of domain P7 is also altered, as determined by lack of recognition by a conformation-specific monoclonal antibody. Normal topology of at least the misoriented C6 domain can be restored post-assembly and in vivo by biosynthetically introducing PE into the membrane of PE-lacking cells. LacY active transport function is also restored. Therefore, the topological organization of this polytopic membrane protein is determined during assembly primarily by membrane phospholipid composition. In addition, the loss of native conformation of domain P7 due to assembly in PE-lacking cells can be restored in vitro after partial denaturation and renaturation in the presence of PE but not PG, CL or phosphatidylcholine (PC; foreign to E.coli). Most remarkable is that topological organization, once established, is not static but is dynamic in response to changes in lipid environment. What is not known is whether the principles governing protein–lipid interactions are primarily responsible for the organization of LacY in the membrane or whether intervention is required by the protein assembly machinery of the cell.

Fig. 1. Secondary structure model of LacY. Topology organization and TM domains of LacY are based on biochemical determination (Kaback et al., 2001) and cysteine accessibility in whole cells and inside-out membrane vesicles (Bogdanov et al., 2002). Rectangles indicate putative helical TM domains numbered from the N-terminus (NH2) to the C-terminus (COOH) to which His6 was added. The hydrophilic domains connecting the TM domains are numbered sequentially and their putative topological disposition in PE-containing cells is indicated by the prefix of ‘C’ for cytoplasmic (IN) or ‘P’ for periplasmic (OUT). An ‘X’ and the single-letter amino acid code of the replaced amino acid followed by the residue number indicate the position of single cysteine replacements in a cysteine-less derivative of LacY.
In this report, we investigated whether the primary determinant of LacY topology is the interaction between the protein and the membrane phospholipids. Since LacY was not rapidly degraded when assembled in PE-lacking cells, we postulated that it resides in the membrane as a stable folded protein at an energy minimum (Bogdanov et al., 2002). We further postulated that addition of PE to the membrane destabilizes the protein and stimulates the topological reorganization to the native state. If protein sequence is written specifically for a given phospholipid environment, then the topology and function of purified LacY assembled in vitro into liposomes should be dependent on the lipid composition of the liposomes and not the lipid composition of the cells from which LacY is derived. Previous reports demonstrated that PE was required for active transport in reconstituted proteoliposomes containing LacY (Chen and Wilson, 1984). In this report, we demonstrate that the topological organization and transport function of LacY reconstituted into protein-free liposomes are determined solely by the phospholipid composition independent of the cellular protein assembly machinery or the lipid composition of the cells from which LacY was derived.
Results
Purification of LacY
Cysteine-less, C-terminal hexa-histidine (His6)-tagged LacY with a single cysteine either in C6 at position H205 or in P7 at position F250 (see Figure 1) was expressed in either PE-containing strain AL95/pDD72GM or PE-lacking strain AL95. The phospholipid composition of all strains was verified before and after growth (Bogdanov and Dowhan, 1995). LacY was purified from the above four strains by nickel affinity chromatography without addition of phospholipids during the purification. Protein purification was monitored by SDS–PAGE followed by either staining with Coomassie Blue or detection by western blot analysis (Figure 2). A broad band centered at an apparent molecular mass of ∼36 kDa was detected by both methods in the last step of the procedure and after reconstitution into proteoliposomes. As noted previously, LacY displays apparent multiple species because it is not completely denatured by SDS (Bogdanov and Dowhan, 1998). Few or no higher molecular weight aggregates of LacY were detected in any of the samples. About 1 mg of LacY was obtained from 10 g wet weight of cells. Similar results were obtained for each of the four strains.

Fig. 2. SDS–PAGE analysis of LacY. Proteins were resolved by SDS–PAGE (12% gels) and visualized by Coomassie Blue staining (lanes 1–5) or western blot analysis with pAb raised against LacY (lanes 6–9). The results shown are for C6(+) LacY. Lane 1, protein molecular weight standards with the indicated molecular weights; lanes 2 and 6, membranes (10 µg); lanes 3 and 7, DM extract (10 µg); lanes 4 (0.3 µg) and 8 (0.5 µg), LacY eluted from a Ni-NTA column with 200 mM imidazole; lanes 5 (0.3 µg) and 9 (0.5 µg), LacY reconstituted into PE liposomes.
Characterization of proteoliposomes
Each of the four preparations of LacY was reconstituted into three different liposomes prepared with the following phospholipids: acetone/ether-washed E.coli polar lipids (as supplied by Avanti Polar Lipids) designated as PE liposomes and containing PE, PG and CL; the anionic component of E.coli polar lipids isolated by chromatography designated as PG–CL liposomes; and the PG–CL liposomes supplemented with PC to 70% by weight and designated as PC liposomes. Proteoliposome composition is designated by the LacY derivative present (a single cysteine replacement in cysteine-less LacY in either C6 or P7); the cell source of LacY, either PE-containing (+) or PE-lacking (–); and the dominant lipid component, either PE, PG–CL or PC. Therefore, proteoliposomes made from liposomes containing PC, PG and CL and cysteine-less LacY derivative H205C purified from PE-lacking cells are designated C6(–)/PC.
The ability to generate and maintain a membrane potential for each proteoliposome preparation was taken as an indication that the vesicles were sealed and capable of providing energy to support active transport of substrate by LacY. Proteoliposomes with internal 50 mM potassium phosphate buffer pH 7.5 were diluted 100-fold into 50 mM sodium phosphate buffer pH 7.5 containing valinomycin. A positive outward membrane potential was generated by outward diffusion of potassium ions. The magnitude of the membrane potential was monitored after formation by the addition of 3,3′-dipropylthiadicarbocyamine iodine (DiSC) (Figure 3). The initial rise in fluorescence is due to dilution of the dye followed by a rapid decline as the dye is concentrated and self-quenched in the vesicles in response to the membrane potential. Addition of the ionophore nigericin allows the inward diffusion of potassium in exchange for a proton, dissipation of the membrane potential (high buffering capacity prevents ΔpH formation) and exit of the dye with an associated increase in fluorescence. The difference in DiSC fluorescence between samples with and without nigericin [added either before (Figure 3A) or after (Figure 3B–D) DiSC additon] was proportional to the membrane potential and corrected for any changes in fluorescence due to interaction of the dye with the proteoliposome. A nearly identical membrane potential was generated for the same amount of proteoliposome energized independently of the lipid composition. In all cases, the membrane potential was maintained for a minimum of 10 min, as indicated by the stable low fluorescence after DiSC addition. The results shown in Figure 3 are representative of all proteoliposomes used in the experiments to follow.

Fig. 3. Detection of membrane potential generated in proteoliposomes. Proteoliposomes (10 µl) of the indicated lipid composition containing 50 mM potassium phosphate pH 7.5 and C6(–) LacY were diluted 100-fold into 50 mM sodium phosphate pH 7.5 containing valinomycin at time zero. DiSC or nigericin (Nig) was added as indicated, and fluorescence was recorded as described in Materials and methods.
The internal volume of the proteoliposomes with different phospholipid compositions was measured as described in Materials and methods. Proteoliposomes (1 µl) containing 0.1 µg of LacY and 50 µg of phospholipid had an internal volume of 0.061 ± 0.0004 µl for PE proteoliposomes, 0.051 ± 0.0002 µl for PG–CL proteoliposomes and 0.054 ± 0.0009 µl for PC proteoliposomes. These values are in good agreement with previously published internal volumes of proteoliposomes containing LacY (Garcia et al., 1983; Chen and Wilson, 1984).
Active transport and facilitated transport of LacY in different proteoliposomes
The active transport function of LacY reconstituted into proteoliposomes of different phospholipid make up was measured by [14C]lactose accumulation after generation of a membrane potential across the bilayer. Proteoliposomes with internal buffer containing 50 mM potassium phosphate pH 7.5 were treated with valinomycin and then diluted into 50 mM sodium phosphate pH 7.5. A positive outward membrane potential was imposed across the proteoliposome membrane by creating an outward-directed K+ diffusion gradient. [14C]Lactose was added and samples were taken at the indicted times, filtered, washed and dried. LacY is functionally symmetrical, and the direction of lactose movement is independent of orientation (Teather et al., 1977). The direction of lactose transport is dependent on either the downhill movement of substrate until it is equilibrated across the membrane (facilitated transport) or the uphill movement of substrate coupled to the inward movement of protons in response to the positive outward membrane potential (active transport) (Vitanen et al., 1986). The radioactivity taken up by the proteoliposomes was converted to lactose concentration and normalized to the amount of LacY in the sample. Figure 4 shows the transport function of LacY in different proteoliposomes. The results for C6 (Figure 4A) and P7 (Figure 4B) proteoliposomes are essentially the same. Although the activity of LacY isolated from PE-lacking cells was consistently lower than LacY isolated from PE-containing cells (addressed later), LacY reconstituted into PE liposomes showed high levels of active transport no matter what the cell source. Addition of carbonyl cyanine p-(trifluoromethoxyl) phenylhydrazone (FCCP) (Figure 4A) to equilibrate the positive inward movement of protons associated with active uptake of lactose abolished the accumulation of lactose to the level observed with liposomes lacking PE, confirming that active transport was being measured. The level of active transport normalized to the amount of LacY present agreed well with previously published results for reconstituted LacY (Consler et al., 1993; Newman et al., 1981).

Fig. 4. Measurement of active and facilitated transport by proteoliposomes. Lactose uptake (nmol/mg protein) was measured in energized (active transport) (A and B) or de-energized (facilitated) (C) proteoliposomes of the following composition as described in Materials and methods. In (A) and (C), all LacY was C6, and in (B) all LacY was P7. The cell source of LacY is indicated by (+) for PE-containing cells or (–) for PE-lacking cells, followed by the designation for the lipid make up of the proteoliposomes as follows: (+)/PE (filled squares), (–)/PE (filled circles), (–)/PE with FCCP added at 90 s (open circles), (+)/PG–CL (open upright triangles), (–)/PG–CL (open inverted triangles) and (–)/PC (filled diamonds). The results are the average of at least three independent experiments, and the bars represent the standard deviation from the mean that was greater than the size of the symbols.
LacY reconstituted into PG–CL or PC proteoliposomes showed little or no active transport. The very low level of accumulation is probably due to facilitated transport (see below), as indicated by the similarity to the uptake in PE proteoliposomes in the presence of FCCP. Since all proteoliposomes generate a membrane potential (Figure 3) and have similar internal volumes, the results demonstrate that LacY, isolated from either PE-containing or PE-lacking cells, displays active transport only when reconstituted into proteoliposomes containing PE. PC cannot substitute for PE in supporting active transport. When PE was added back to PG–CL to reconstitute PE liposomes, followed by the addition of LacY, full active transport was observed (not shown), ruling out the presence of an inhibitor in the PG–CL preparation and confirming the requirement for PE to support active transport.
To determine whether proteoliposomes carried out facilitated transport of lactose, 100 µl of proteoliposomes containing 50 mM potassium phosphate pH 7.5 were diluted 10-fold into 50 mM potassium phosphate pH 7.5 containing valinomycin, FCCP and [14C]lactose as described in Materials and methods. Higher proteoliposome and lactose concentrations were required to detect uptake and to account for the higher apparent Km for facilitated uptake of lactose (Vitanen et al., 1986), respectively. The presence of FCCP prevented the generation of a positive inward H+ gradient by coupled lactose–H+ symport. Liposomes prepared in the same way, but lacking LacY, did not take up lactose, consistent with the equilibration process in proteoliposomes being LacY mediated (not shown). The low background values recorded fo liposomes lacking LacY were subtracted in the experiments. Figure 4C shows the facilitated transport of LacY in proteoliposomes containing different lipid compositions. The steady-state level of lactose accumulation in all proteoliposomes approximated (120 nmol/mg) that for PG–CL and PC proteoliposomes measured under energized conditions (Figure 4A and B), and there was no difference dependent on phospholipid composition. There fore, LacY carried out facilitated transport of substrate independently of liposome composition.
Orientation of LacY in proteoliposomes
The topology of hydrophilic domains C6 and P7 in proteoliposomes was established by determining the accessibility of single cysteine residues in these domains to sulfhydryl reagents. 3-(N-maleimidylpropionyl) biocytin (MPB) is a relatively membrane-impermeable, biotinylated maleimide and will preferentially label a cysteine exposed on the outside of proteoliposomes. The MPB-labeled proteins were separated by SDS–PAGE, transferred to nitrocellulose and detected by avidin– horseradish peroxidase (HRP). To label the cysteine on the interior, proteoliposomes were solubilized with β-d-octylglucoside (OG) before MPB labeling. 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), another fully membrane-impermeable non-biotinylated maleimide, was used to block exposed cysteine prior to MPB treatment, thus preventing biotinylation. Figure 5A shows the reactivity of the cysteine in domain C6 of C6(+)/PE and C6(+)/PG–CL proteoliposomes. Domain C6 was biotinylated in PE but not in PG–CL proteoliposomes (Figure 5A, lanes 1 and 5), suggesting that domain C6 is on the outside of PE proteoliposomes, but on the inside of PG–CL proteoliposomes. This was confirmed further by control experiments. When the C6(+)/PE proteoliposomes were solubilized with OG before MPB labeling, the signal did not increase significantly (Figure 5A, lane 2). Pre-treatment with and removal of AMS followed by MPB treatment (Figure 5A, lane 3) or followed by OG and then MPB treatment (lane 4) resulted in no biotinylation, supporting the accessibility to all cysteines from outside the proteoliposomes and insignificant cysteine sequestered on the inside of the liposomes. However, in C6(+)/PG–CL proteoliposomes, cysteine can only be labeled after permeabilization with OG (Figure 5A, lane 6), and is not blocked by treatment with and removal of AMS prior to OG and MPB treatment (lane 8), supporting complete sequestering of the cysteine inside the proteoliposome. Identical results (Figure 5B) were obtained for C6(–)/PE (lanes 1–4) and C6(–)/PG–CL (lanes 5–8) proteoliposomes as were obtained above for C6(+)/PE and C6(+)/PG-CL, respectively, except that in the former PE proteoliposomes 5–10% of the cysteine was sequestered (Figure 5B, lane 4), indicating the possibility of some mixed topology.
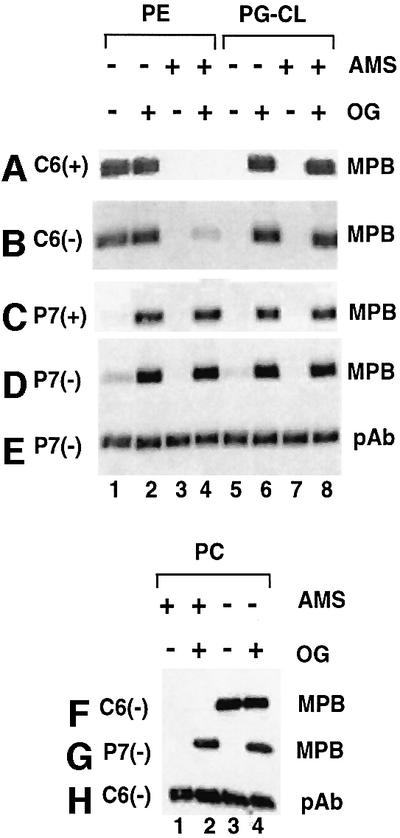
Fig. 5. Determination of the orientation of domains C6 and P7 of LacY in proteoliposomes. Proteoliposomes containing the indicated lipid content (PE, PG–CL or PC), LacY with a single cysteine in the C6 or P7 domain, and prepared from either PE-containing (+) or PE-lacking (–) cells were treated with MPB directly or after pre-treatment with and removal of AMS (+), or after solubilization with OG (+). Samples were dissolved in 1% SDS and subjected to SDS–PAGE. Biotinylation with MPB or the total amount of LacY was detected after transfer to a solid support using avidin–HRP (A–D, F and G) or pAb (E and H).
The accessibility of the cysteine in domain P7 for P7(+)/PE and P7(+)/PG–CL proteoliposomes is shown in Figure 5C. Cysteine in these samples could only be labeled when the sample was permeabilized with OG prior to MPB treatment (Figure 5C, lanes 2, 4, 6 and 8), consistent with sequestration of the P7 domain inside the proteoliposomes. Treatment with and removal of AMS prior to OG treat ment did not protect against biotinylation (Figure 5C, lanes 4 and 8). The same results (Figure 5D) were obtained with P7(–)/PE (lanes 1–4) and P7(–)/PG–CL (lanes 5–8) proteoliposomes as those above for the P7(+)/PE and P7(+)/PG–CL, respectively, except that ∼5% of the former cysteine was accessible from the outside in both PE (lane 1) and PG–CL proteoliposomes (lane 5), again suggesting some mixed topology. The presence of an equal amount of protein was demonstrated in each sample by western blot analysis of an equal volume of each reaction mixture (Figure 5E) and is representative of all samples presented in Figure 5.
Therefore, in PE proteoliposomes, LacY was oriented with the C6 domain on the outside and the P7 domain on the inside. In PG–CL proteoliposomes, both the C6 and P7 domains were on the inside. There was no difference in the orientation for LacY isolated from PE-containing or PE-lacking cells, except that LacY isolated from PE-lacking cells showed ∼5–10% of the protein in an orientation opposite from that of the majority of the protein. This may explain the slightly lower active transport activity observed for both C6(–)/PE and P7(–)/PE proteoliposomes (Figure 4A and B).
Given the lack of active transport function of LacY in PC proteoliposomes, the results of topological determination of LacY in PC proteoliposomes were surprising (Figure 5F and G). Domain C6 was blocked from biotinylation by treatment with AMS (Figure 5F, lanes 1 and 2) and was fully accessible from the outside of proteoliposomes (lane 3) with no increase in accessibility after OG treatment (lane 4). In contrast, domain P7 was only accessible after OG treatment (Figure 5G, lanes 2 and 4). No biotinylation occurred without OG treatment (lanes 1 and 3). Therefore, in both PC and PE proteoliposomes, LacY orients with the C6 domain on the outside and the P7 domain on the inside, indicating that a zwitterionic phospholipid governs topological orientation but is not sufficient to support active transport, which was only observed when PE was present. The presence of equal amounts of protein was demonstrated by western blot analysis (Figure 5H).
Discussion
Membrane proteins generally are thought to be inserted into the lipid bilayer in only one stable orientation whether they possess a single TM span or multiple TM spans. We previously showed that, in vivo, LacY adopts at least two different membrane topological organizations depending on the presence or absence of the zwitterionic phospholipid PE (Bogdanov et al., 2002). The topological organization of LacY is also dynamic and changes in response to membrane phospholipid composition. These findings raise an interesting question as to whether protein topogenesis is determined primarily by protein–lipid interactions or depends on the lipid requirements of the components of the assembly machinery. Phospholipids might exert their effect on membrane protein topology either directly by interacting with the topogenic signals of newly inserted proteins or indirectly by influencing the assembly machinery. The former possibility is reasonable based on simple equilibrium and lowest energy considerations within a given lipid environment, but ignores the complexity of the assembly process. The latter possibility represents a reasonable alternative, since many of the components of the assembly machinery are known to be lipid dependent (Van Voorst and De Kruijff, 2000). Previous conclusions that anionic lipids play a direct role as topological determinants did not rule out the possibility of the influence of lipids on known and unknown components of the assembly machinery (van Klompenburg et al., 1997). The establishment of the primary determinant of topology is important in defining the pathway and factors that determine the final organization of a protein in the membrane. In order to establish the role phospholipids play in the topological organization of membrane proteins, we turned to a system of reconstitution of purified membrane protein into liposomes of defined composition and free of other protein components.
Cysteine-less LacY containing a single cysteine replacement in either domain C6 or P7 was expressed in PE (+) and PE (–) cells and purified to near homogeneity by established procedures. Similar yields and activity of LacY from both cell types further support the conclusion that LacY assembly in membranes containing only PG and CL is stable and structured. Since a variation of the methods used previously was employed to prepare proteoliposomes, and LacY from PE (–) cells had never been reconstituted, proteoliposomes were first characterized with respect to the ability to generate a membrane potential, define an internal volume and transport lactose in either the active or facilitated mode. All of these parameters are clear indicators that the vesicles independent of phospholipid composition were sealed to low molecular weight solutes. Since the internal volumes of all the vesicle preparations were approximately the same and in agreement with previous reports, then the nearly identical change in fluorescence after energization of the vesicles indicated a very similar positive outward membrane potential. In all cases, an internal volume accessible by lactose was only observed when LacY was present, consistent with general impermeability to hydrophilic solutes but specific facilitated transport of lactose. Finally, only in proteoliposomes containing PE was active accumulation of lactose observed dependent on a membrane potential. Liposomes lacking PE displayed non-energy-dependent facilitated equilibration of lactose without accumulation, even though a membrane potential was established. The specificity for PE, the lack of substitution by PC and the magnitude of active uptake of lactose are in good agreement with previously reported results (Garcia et al., 1983; Chen and Wilson, 1984). These results clearly mimic the in vivo requirement specifically for PE to support active transport of lactose and lack of lipid specificity to support facilitated transport.
The major question to be addressed was the topological organization of LacY as a function of liposome phospholipid composition. The reconstitution method employed resulted in a 95% (limit of detection) or better uniform topological orientation of LacY molecules from PE (+) cells, but in two different orientations dependent on the liposome phospholipid composition (see Figure 6). On reconstitution of LacY from PE (–) cells, ∼5–10% of the molecules were not oriented uniformly with respect to the bilayer, which may explain the consistently lower active transport function for this source of LacY. The impermeability of the vesicles and the opposite orientation of subdomains C6 and P7 in PE (+) liposomes were confirmed further by the consistent inaccessibility of subdomain P7 unless the vesicles were disrupted. In the case of PE and PC liposomes, there was a clear difference between the complete accessibility of C6 to and the complete protection of P7 from biotinylation. The fact that the central region of LacY, and presumably the whole molecule, was in the opposite orientation (i.e. P7 facing the lumen) to that found in the cell was not surprising since LacY was inserted from the outside of partially detergent-disrupted liposomes whereas in the cell LacY is inserted into the membrane from the lumen side of the cell. The overriding and most significant conclusion is that in PE or PC liposomes, LacY assembles with C6 and P7 on opposite sides of the bilayer, as it does in PE (+) cells. In PG–CL liposomes, C6 and P7 were on the same side of the bilayer, as they are in PE (–) cells. The final topology of LacY after reconstitution was also not dependent on the different topologies attained during assembly in vivo in the PE-containing or PE-lacking host cells from which LacY was isolated. Therefore, we conclude that membrane protein topology is determined primarily by the interaction of a given protein with its native phospholipid environment independent of the cellular protein assembly machinery. The latter certainly has a catalytic role in facilitating TM domains in attaining their most favorable orientation within a given environment as determined by phospholipid composition.

Fig. 6. Topology adopted by LacY in cells (Bogdanov et al., 2002) or liposomes (this work) with different phospholipid composition. The curved parallel lines indicate the lipid bilayer, with the top of each diagram being the exterior of the cell or liposome. Domain P7 was exposed to the exterior of cells and sequestered to the interior of liposomes irrespective of phospholipid composition. Domain C6 orientation was dependent on phospholipid composition, being on the interior of cells containing PE or liposomes lacking PE, and facing the exterior of cells lacking PE or liposomes containing PE or PC. For simplicity, the possibility that TM VII is not inserted into the membrane as a U-shaped miniloop but is completely outside the bilayer in PG–CL vesicles (Bogdanov et al., 2002) is not depicted.
Since PC does not support the regain of the native conformation of domain P7 during in vitro renaturation of LacY (Bogdanov et al., 1999) and does not support active transport function, it was surprising that C6 domain topology in PC liposomes was the same as in PE liposomes and therefore opposite to that in PG–CL vesicles. These results indicate that the zwitterionic nature of PE and PC is sufficient for determining topology but that some other specific property of PE is required to support active transport. The net positive charge of both PE and PC versus the anionic character of PG–CL may affect other properties of the bilayer such as net charge density or the thickness of the bilayer. Since the length and the hydrophobicity of the TM domains of LacY are not homogeneous, the greater diversity in properties of a liposome containing both anionic and zwitterionic phospholipids may be required for proper topological organization of LacY. In particular, the lower hydrophobicity of TM VII, which contains two negatively charged amino acids (Kaback et al., 2001), may either allow only partial insertion into an anionic bilayer as depicted in Figure 6 or result in its complete exclusion (Bogdanov et al., 2002) due to charge repulsion. PE also differs from PC in several important aspects. The former can form hydrogen bonds, exchange protons and, dependent on fatty acid composition and temperature, is a non-bilayer-forming lipid. The latter cannot form hydrogen bonds or exchange protons, and is a bilayer-forming lipid. The polymorphic properties of PE appear not to be important because in previous studies investigating lipid properties that supported the proper conformation of domain P7 of LacY, bilayer-forming PE species were required (Bogdanov et al., 1999). However, the hydrogen bonding and proton exchange properties of PE are plausible candidates to support the required symport of a proton along with lactose in order to couple substrate uptake with the proton electrochemical potential across the membrane. Alternatively, PC may disrupt a proton wire, may adversely affect the pKa of critical residues in a proton wire or may disrupt the proximity of critical residues by not supporting the proper conformation of domain P7 of LacY.
The evidence that membrane protein sequence is written for a specific lipid environment is most convincing for LacY, but there are several examples that support this concept. The P-glycoprotein is localized to mammalian cytoplasmic membranes and is an ATP-binding cassette transporter responsible for multidrug resistance. Like LacY (Prive and Kaback, 1996; Venkatesan et al., 2000a,b,c), it is a highly flexible protein that undergoes large conformational changes during its catalytic cycle (Rosenberg et al., 2001) or biogenesis (Moss et al., 1998). In its native host, the protein exhibits 12 TM domains with both the N- and C-terminus exposed to the cytoplasm. When expressed in E.coli, the N-terminal half of the protein assumes the same topology as in the native host. However, TM domain VII no longer spans the membrane, TM domains VIII–XII assume an inverted orientation, and the C-terminus, which includes the nucleotide-binding domain, is exposed to the periplasmic side of the membrane (Linton and Higgins, 2002). Similarly, a citrate carrier of Klebsiella pneumoniae displays 11 TM domains when inserted into dog pancreas endoplasmic reticulum (ER) membranes but only nine TM domains when expressed in E.coli (van Geest et al., 1999). An outer membrane protein (OEP7) of the chloroplast assumes its native topological organization when reconstituted into outer membrane lipids but an opposite topology when reconstituted into total chloroplast lipids (Schleiff et al., 2001). Several proteins exhibit both different topologies and functions depending on their natural subcellular membrane location. The L envelope protein of hepatitis B virus exists in two topological forms dependent on subcellular localization (Lambert and Prange, 2001). Ductin exists as a subunit of the vacuolar H+-ATPase and in an opposite orientation in ER membranes as a component of the connexon channel of gap junctions (Dunlop et al., 1995). An ER epoxide hydroxylase is found with a different topology in the sinusoidal plasma membrane where it mediates bile acid transport (Zhu et al., 1999). Do these differences originate during membrane insertion followed by differences in subcellular distribution, or are they induced by changes in lipid composition as proteins move through different organelles to their final destination?
The above observations may also explain the difficulties encountered in expressing functional membrane proteins in heterologous organisms. The common practice of using PC in place of PE to study E.coli membrane proteins is certainly questionable in the absence of functional and structural studies to ensure that the native organization is retained. A similar argument can be made against using a single or simple mixture of phospholipids in studying eukaryotic membrane proteins. Host organisms should be designed to express foreign lipids in order to accommodate expression of foreign membrane proteins (Dowhan, 1997).
Certainly the translocase/insertase assembly machinery plays a major catalytic role in ensuring both delivery to and proper assembly of nascent chains of complex membrane proteins, although some proteins of the thylakoid membrane appear to be inserted spontaneously (Schleiff and Klosgen, 2001). Despite the lack of a complete understanding of the fine details of membrane protein assembly, our studies emphasize the importance of lipid–protein interactions as strong and direct determinants of protein topology and further emphasize the importance of studying membrane protein assembly as well as structure and function in the context of a native lipid environment.
Materials and methods
Materials
Escherichia coli polar lipid extract (with and without acetone/ether extraction) and dioleoyl-PC were purchased from Avanti Polar Lipids. Diethylaminoethyl cellulose (DE52) was purchased from Whatman. Nitrocellulose sheets (pore size 0.45 mm) for immunoblotting were purchased from Schleicher and Schuell. The ECL kit, HRP-labeled secondary antibody and [d-glucose-1-14C]lactose were obtained from Amersham Pharmacia Biotech. Ni-NTA Superflow was purchased from Qiagen. GFTS filters (0.22 µm) were purchased from Millipore. Modified Lowry protein reagent assay, Folin and Ciocalteu phenol reagent, and avidin–HRP were purchased from Pierce. Valinomycin, nigericin, lactose, DNase and FCCP were purchased from Sigma. β-d-Dodecylmaltoside (DM) was purchased from Anatrace. Pefabloc was purchased from Roche Molecular Biochemicals. Polyclonal antibody (pAb) against the C-terminal dodecapeptide of LacY and plasmids pT7-5/C-less lacY/H205C and pT7-5/C-less lacY/F250C (Frillingos et al., 1998) were provided by Dr H.R.Kaback (University of California, Los Angeles). AMS, MPB and DiSC were purchased from Molecular Probes. Vivaspin concentrators (30 000 molecular weight cut off) were purchased from Sartorius. Bio-spin 6 columns were purchased from Bio-Rad.
Bacterial strains, plasmids and growth conditions
Strain AL95 (pss93::kanR lacY::Tn9) was used as the host strain (Bogdanov et al., 2002). This strain cannot make PE and is not viable without either plasmid pDD72GM (pssA+ genR and pSC101 temperature-sensitive replicon) (Bogdanov et al., 2002) or growth media containing 50 mM MgCl2 (DeChavigny et al., 1991). Strain AL95 (grown at 37°C) lacks PE, and strain AL95/pDD72GM (grown at 30°C) contains the normal E.coli complement of phospholipids including PE. LacY was expressed from plasmids (ampR, ColE1 replicon) encoding a single cysteine replacement in cysteine-less LacY at either H205 (pT7-5/C-less lacY/H205C) in the C6 cytoplasmic domain connecting TM VI and VII or F250 (pT7-5/C-less lacY/F250C) in the periplasmic domain P7 connecting TM VII and VIII (see Figure 1). LacY was engineered with a His6 tag at the C-terminus to facilitate purification and was expressed under control of OPlac by growth of cells in the presence of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were grown in LB-rich medium containing ampicillin (100 µg/ml) as required and 50 mM MgCl2.
Preparation of PE-lacking E.coli lipids
Pre-swollen anion exchanger DE52 (100 g) was washed in water, suspended in 1 M acetic acid and degassed for 30 min. The resin was equilibrated in 2.4 M ammonium acetate pH 7.4 and washed once with water, twice with 80% methanol/0.2 M ammonium acetate pH 7.4 and once with chloroform/methanol/0.4 M ammonium acetate (2/3/1, by vol.) pH 7.4. The resin was suspended in the last wash buffer, poured into a chromatography column and washed with two column volumes of chloroform/methanol/water (2/3/1, by vol.).
The E.coli total lipid extract (400 mg) that had not been extracted with acetone/ether was dissolved in 20 ml chloroform/methanol/water (2/3/1, by vol.) and applied to the column at room temperature. The column was washed with chloroform/methanol/water (2/3/1, by vol.) to remove all the PE and neutral lipids, followed by chloroform/methanol/0.2 M ammonium acetate pH 7.4 (2/3/1, by vol.) to elute PG and CL free of PE. Solvent was removed by rotary evaporation under vacuum.
LacY purification
Escherichia coli cells were grown to an OD600 of 0.8, induced by addition of IPTG and grown until cell arrest occurred. Purification of LacY was carried out at 4°C or on ice by a modification of a published procedure (le Coutre et al., 1997) as follows. The major modification was to eliminate the addition of exogenous phospholipids during the purification and to change the pH of some buffers. Cells were harvested by centrifugation, and the cell pellet (10 g wet weight) was suspended in 50 ml of 50 mM potassium phosphate pH 7.5, 5 mM MgSO4, 10 mM β-mercaptoethanol (BME), 0.5 mM Pefabloc and 30 µg/ml DNase. Cells were disrupted with a French press and unbroken cells removed by centrifugation at 13 000 gav for 20 min. Membranes were collected by centrifugation at 100 000 gav for 4 h and washed once by suspension in and centrifugation from 20 ml of 50 mM potassium phosphate pH 7.5, 10 mM BME, 0.5 mM Pefabloc and 5 M urea. Membranes were solubilized in 20 ml of DM at a final concentration of 2% (w/v) in 50 mM potassium phosphate pH 7.5, 0.2 M NaCl, 10 mM BME, 0.5 mM Pefabloc (extract-wash buffer). The DM extract was isolated by centrifugation at 100 000 gav for 30 min and bound to 3.5 ml of Ni-NTA Superflow in batch by shaking for 2 h. The nickel resin was washed with extract-wash buffer until the A280 baseline was reached. This was followed by three washes with 50 mM potassium phosphate, 10 mM BME, 10% glycerol and 0.008% DM, first at pH 7.5, secondly at pH 6.0, and thirdly at pH 7.5 with 20 mM imidazole until the A280 baseline was reached. LacY was eluted with the pH 7.5 buffer containing 200 mM imidazole. Purified samples were analyzed by SDS–PAGE and visualized by Coomassie Blue staining and western blot analysis. Fractions containing LacY were pooled and concentrated to ∼1 mg/ml by centrifugation using a Vivaspin concentrator.
Preparation of proteoliposomes containing LacY
Escherichia coli lipid extracts lacking PE (prepared by DE52 chromatography) with or without addition of PC or containing PE (acetone/ether washed) were dissolved in chloroform/methanol (9/1, v/v) and processed by a modification of a published procedure (Vitanen et al., 1986). The solvent was removed first under a stream of oxygen-free nitrogen and then under a vacuum to obtain a thin layer of dry lipids. The lipids were suspended in 50 mM potassium phosphate pH 7.5, 2 mM BME, 1.5% OG to yield a lipid concentration of 50 mg/ml. OG was removed by dialysis against 50 mM potassium phosphate pH 7.5, and the resulting liposomes were frozen in liquid nitrogen and stored at –70°C. For reconstitution with LacY, liposomes were thawed, OG was added to 1.5%, mixed with purified LacY in a 500:1 ratio (lipid to protein, w/w) and incubated at room temperature with gentle agitation for 10 min. To remove OG, the mixture was diluted 30-fold into 50 mM potassium phosphate pH 7.5, 2 mM BME, and centrifugated at 100 000 gav for 4 h. Stock preparations of proteoliposomes (final concentration of 50 mg/ml phospholipid and 100 µg/ml LacY) were suspended in 50 mM potassium phosphate pH 7.5, 2 mM BME and stored at –70°C. Prior to use, proteoliposomes were subjected to three cycles of freeze/thaw/sonication (every cycle 4 s, total 12 s) in a bath sonicator.
Determination of membrane potential
The ability of proteoliposomes to generate and maintain a membrane potential was determined by reduction in fluorescence of a positively charged dye due to its internalization and concentration in response to a positive outward membrane potential (Fendler et al., 1996; Toyomizu et al., 2002). Proteoliposomes (10 µl) prepared in 50 mM potassium phosphate pH 7.5 were diluted into 990 µl of 50 mM sodium phosphate pH 7.5, 0.4 µM valinomycin and 0.25 µM fluorophore DiSC. Addition of 5 µl of 1 mM nigericin was used to dissipate the membrane potential, and the increase in fluorescence was taken as a measure of the membrane potential. The excitation and emission wavelengths used were 650 and 675 nm, respectively. All incubations were at 30°C.
Determination of internal volume of proteoliposomes
Proteoliposomes (80 µl) were brought to a final volume of 100 µl in 50 mM potassium phosphate pH 7.5 containing 20 µM valinomycin, 20 µM FCCP and 2.7 mM [14C]lactose (3.7 Ci/mol) to equilibrate the internal volume with radiolabeled lactose. Aliquots (15 µl) at various times were diluted rapidly into 3 ml of ice-cold 50 mM potassium phosphate at pH 7.5, and filter washed as described for the transport assays (see below) to determine when radiolabel had fully equilibrated with the internal volume (time zero sample). Once equilibrium was reached, the remaining proteoliposomes were diluted 200-fold into 50 mM potassium phosphate at pH 7.5 and monitored until there was no further decline in retained radiolabel (diluted sample) as measured by filtration. The diluted samples relative to the time zero samples retained <1% of the label. Liposomes lacking LacY did not accumulate radiolabel in the time zero samples. The differences between the time zero and diluted sample radiolabel and the specific radioactivity of lactose were used to calculate the internal volume of the proteoliposomes (Garcia et al., 1983). All incubations were at 30°C.
Sugar transport assays
To measure membrane potential-driven active transport, 10 µl of proteoliposomes containing 50 mM potassium phosphate pH 7.5 were diluted into 990 µl of 50 mM sodium phosphate pH 7.5 containing 0.22 mM [14C]lactose (2.7 Ci/mol) and 5 µM valinomycin (Vitanen et al., 1986). For measurements of facilitated diffusion, 100 µl of proteoliposomes were diluted into 900 µl of 50 mM potassium phosphate pH 7.5 containing 10 µM valinomycin, 20 µM FCCP and 0.54 mM [14C]lactose (3.7 Ci/mol) (Vitanen et al., 1986). Aliquots of 100–150 µl were removed at various times, quenched with 3 ml of ice-cold 50 mM sodium phosphate pH 7.5, 100 mM LiCl, immediately filtered through GFTS filters and washed using 5 ml of the same buffer. Filters were dried and counted by liquid scintillation. A background value, determined by assaying liposomes prepared in the same way, but lacking LacY, was subtracted routinely. All incubations were at 30°C.
Site-directed labeling of LacY
Stock solutions of proteoliposomes were supplemented to the indicated concentrations of reagents by 10-fold dilutions from stock solutions prepared in water. The following procedure is a modification of methods used on whole cells (Bogdanov et al., 2002) and proteoliposomes (Jung et al., 1998). Proteoliposomes were incubated in 200 µM MPB at room temperature for 20 min to biotinylate LacY, or in 200 µM AMS for 10 min to block water-accessible cysteine. The excess maleimides were inactivated by addition of BME to 1 mM. OG (at 1.5%) was used to disrupt proteoliposomes where indicated. Centrifuging the reaction mixture through a Bio-spin column was used to change the solution surrounding proteoliposomes or to remove AMS/BME. Samples were subjected to SDS–PAGE followed by western blot analysis using the ECL detection method as previously described (Bogdanov et al., 1996) after incubation with avidin–HRP to assess biotinylation or pAb and HRP-conjugated secondary antibody for total LacY. A Bio-Rad Fluor-S Max multilmager was used to record the results.
Determination of protein concentration
Protein content during purification was determined by the Bradford method (Bradford, 1976), and the concentration of LacY in proteoliposomes was determined by a modified Lowry method (Peterson, 1977). The former method is sensitive to the high concentration of lipids in proteoliposomes, and the latter method is sensitive to the high concentration of imidazole in the elution buffer. For measurements of protein content of proteoliposomes, SDS was added in 3-fold excess by weight over protein.
Acknowledgments
Acknowledgements
We thank Dr H.R.Kaback for generously providing the collection of LacY derivatives and LacY antibodies, without which this work would not have been possible, and Gill Verner of Dr Kaback’s laboratory for help with the LacY purification protocol. This work was supported by grant GM-20478 from the National Institutes of Health awarded to W.D.
References
- Andersson H. and von Heijne,G. (1994) Membrane protein topology: effects of ΔµH+ on the translocation of charged residues explain the ‘positive inside’ rule. EMBO J., 13, 2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M. and Dowhan,W. (1995) Phosphatidylethanolamine is required for in vivo function of the membrane-associated lactose permease of Escherichia coli. J. Biol. Chem., 270, 732–739. [DOI] [PubMed] [Google Scholar]
- Bogdanov M. and Dowhan,W. (1998) Phospholipid-assisted protein folding: phosphatidylethanolamine is required at a late step of the conformational maturation of the polytopic membrane protein lactose permease. EMBO J., 17, 5255–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M., Sun,J., Kaback,H.R. and Dowhan,W. (1996) A phospholipid acts as a chaperone in assembly of a membrane transport protein. J. Biol. Chem., 271, 11615–11618. [DOI] [PubMed] [Google Scholar]
- Bogdanov M., Umeda,M. and Dowhan,W. (1999) Phospholipid-assisted refolding of an integral membrane protein. Minimum structural features for phosphatidylethanolamine to act as a molecular chaperone. J. Biol. Chem., 274, 12339–12345. [DOI] [PubMed] [Google Scholar]
- Bogdanov M., Heacock,P.N. and Dowhan,W. (2002) A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J., 21, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chen C.-C. and Wilson,T.H. (1984) The phospholipid requirement for activity of the lactose carrier of Escherichia coli. J. Biol. Chem., 259, 10150–10158. [PubMed] [Google Scholar]
- Chin C.-N., von Heijne,G. and de Gier,J.-W.L. (2002) Membrane proteins: shaping up. Trends Biochem. Sci., 27, 217–271. [DOI] [PubMed] [Google Scholar]
- Consler T.G., Persson,B.L., Jung,H., Zen,K.H., Jung,K., Prive,G.G., Verner,G. and Kaback,H.R. (1993) Properties and purification of an active biotinylated lactose permease from Escherichia coli. Proc. Natl Acad. Sci. USA, 90, 6934–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChavigny A., Heacock,P.N. and Dowhan,W. (1991) Sequence and inactivation of the pss gene of Escherichia coli: phosphatidyl ethanolamine may not be essential for cell viability. J. Biol. Chem., 266, 5323–5332. [PubMed] [Google Scholar]
- Dowhan W. (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem., 66, 199–232. [DOI] [PubMed] [Google Scholar]
- Dunlop J., Jones,P.C. and Finbow,M.E. (1995) Membrane insertion and assembly of ductin: a polytopic channel with dual orientations. EMBO J., 14, 3609–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendler K., Drose,S., Altendorf,K. and Bamberg,E. (1996) Electrogenic K+ transport by the Kdp-ATPase of Escherichia coli. Biochemistry, 35, 8009–8017. [DOI] [PubMed] [Google Scholar]
- Frillingos S., Sahin-Toth,M., Wu,J. and Kaback,H.R. (1998) Cys-scanning mutagenesis: a novel approach to structure function relationships in polytopic membrane proteins. FASEB J., 12, 1281–1299. [DOI] [PubMed] [Google Scholar]
- Garcia M.L., Viitanen,P., Foster,D.L. and Kaback,H.R. (1983) Mechanism of lactose translocation in proteoliposomes reconstituted with lac carrier protein purified from Escherichia coli. 1. Effect of pH and imposed membrane potential on efflux, exchange and counterflow. Biochemistry, 22, 2524–2531. [DOI] [PubMed] [Google Scholar]
- Jung H., Tebbe,S., Schmid,R. and Jung,K. (1998) Unidirectional reconstitution and characterization of purified Na+/proline transporter of Escherichia coli. Biochemistry, 37, 11083–11088. [DOI] [PubMed] [Google Scholar]
- Kaback H.R., Sahin-Toth,M. and Weinglass,A.B. (2001) The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol., 2, 610–620. [DOI] [PubMed] [Google Scholar]
- Lambert C. and Prange,R. (2001) Dual topology of the hepatitis B virus large envelope protein: determinants influencing posttranslational translocation. J. Biol. Chem., 276, 22265–22272. [DOI] [PubMed] [Google Scholar]
- le Coutre J., Narasimhan,L.R., Patel,C.K. and Kaback,H.R. (1997) The lipid bilayer determines helical tilt angle and function in lactose permease of Escherichia coli. Proc. Natl Acad. Sci. USA, 94, 10167–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton K.J. and Higgins,C.F. (2002) P-glycoprotein misfolds in Escherichia coli: evidence against alternating-topology models of the transport cycle. Mol. Membr. Biol., 19, 51–58. [DOI] [PubMed] [Google Scholar]
- Moss K., Helm,A., Lu,Y., Bragin,A. and Skach,W.R. (1998) Coupled translocation events generate topological heterogeneity at the endoplasmic reticulum membrane. Mol. Biol. Cell, 9, 2681–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M.J., Foster,D.L., Wilson,T.H. and Kaback,H.R. (1981) Purification and reconstitution of functional lactose carrier from Escherichia coli. J. Biol. Chem., 256, 11804–11808. [PubMed] [Google Scholar]
- Peterson G.L. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem., 83, 346–356. [DOI] [PubMed] [Google Scholar]
- Prive G.G. and Kaback,H.R. (1996) Engineering the lac permease for purification and crystallization. J. Bioenerg. Biomembr., 28, 29–34. [PubMed] [Google Scholar]
- Rosenberg M.F. et al. (2001) Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J., 20, 5615–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiff E. and Klosgen,R.B. (2001) Without a little help from ‘my’ friends: direct insertion of proteins into chloroplast membranes? Biochim. Biophys. Acta, 1541, 22–33. [DOI] [PubMed] [Google Scholar]
- Schleiff E., Tien,R., Salomon,M. and Soll,J. (2001) Lipid composition of outer leaflet of chloroplasts outer envelope determines topology of OEP7. Mol. Biol. Cell, 12, 4090–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teather R.M., Hamelin,O., Schwarz,H. and Overath,P. (1977) Functional symmetry of the β-galactoside carrier in Escherichia coli. Biochim. Biophys. Acta, 467, 386–395. [DOI] [PubMed] [Google Scholar]
- Toyomizu M., Okamoto,K., Akiba,Y., Nakatsu,T. and Konishi,T. (2002) Anacardic acid-mediated changes in membrane potential and pH gradient across liposomal membranes. Biochim. Biophys. Acta, 1558, 54–62. [DOI] [PubMed] [Google Scholar]
- van Geest M., Nilsson,I., von Heijne,G. and Lolkema,J.S. (1999) Insertion of a bacterial secondary transport protein in the endoplasmic reticulum membrane. J. Biol. Chem., 274, 2816–2823. [DOI] [PubMed] [Google Scholar]
- van Klompenburg W., Nilsson,I., von Heijne,G. and de Kruijff,B. (1997) Anionic phospholipids are determinants of membrane protein topology. EMBO J., 16, 4261–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorst F. and De Kruijff,B. (2000) Role of lipids in the translocation of proteins across membranes. Biochem. J., 347, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan P., Hu,Y. and Kaback,H.R. (2000a) Site-directed sulfhydryl labeling of the lactose permease of Escherichia coli: helix X. Biochemistry, 39, 10656–10661. [DOI] [PubMed] [Google Scholar]
- Venkatesan P., Kwaw,I., Hu,Y. and Kaback,H.R. (2000b) Site-directed sulfhydryl labeling of lactose permease of Escherichia coli: helix VII. Biochemistry, 39, 10641–10648. [DOI] [PubMed] [Google Scholar]
- Venkatesan P., Liu,Z., Hu,Y. and Kaback,H.R. (2000c) Site-directed sulfhydryl labeling of the lactose permease of Escherichia coli: N-ethylmaleimide-sensitive face of helix II. Biochemistry, 39, 10649–10655. [DOI] [PubMed] [Google Scholar]
- Vitanen P., Newman,M.J., Foster,D.L., Wilson,T.H. and Kaback,H.R. (1986) Purification, reconstitution and characterization of the lac permease of E.coli. Methods Enzymol., 125, 429–452. [DOI] [PubMed] [Google Scholar]
- von Heijne G. (1992) Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol., 225, 487–494. [DOI] [PubMed] [Google Scholar]
- Zhu Q.-S., von Dippe,P., Xing,W. and Levy,D. (1999) Membrane topology and cell surface targeting of microsomal epoxide hydrolase. Evidence for multiple topological orientations. J. Biol. Chem., 274, 27898–27904. [DOI] [PubMed] [Google Scholar]