

Abstract
Dicer is a multi-domain RNase III-related endonuclease responsible for processing double-stranded RNA (dsRNA) to small interfering RNAs (siRNAs) during a process of RNA interference (RNAi). It also catalyses excision of the regulatory microRNAs from their precursors. In this work, we describe the purification and properties of a recombinant human Dicer. The protein cleaves dsRNAs into ∼22 nucleotide siRNAs. Accumulation of processing intermediates of discrete sizes, and experiments performed with substrates containing modified ends, indicate that Dicer preferentially cleaves dsRNAs at their termini. Binding of the enzyme to the substrate can be uncoupled from the cleavage step by omitting Mg2+ or performing the reaction at 4°C. Activity of the recombinant Dicer, and of the endogenous protein present in mammalian cell extracts, is stimulated by limited proteolysis, and the proteolysed enzyme becomes active at 4°C. Cleavage of dsRNA by purifed Dicer and the endogenous enzyme is ATP independent. Additional experiments suggest that if ATP participates in the Dicer reaction in mammalian cells, it might be involved in product release needed for the multiple turnover of the enzyme.
Keywords: gene silencing/ribonucleases/RNAi/RNase III/siRNA
Introduction
In many eukaryotes, double-stranded RNA (dsRNA) inhibits gene expression in a sequence-specific manner by triggering degradation of mRNA. This effect, referred to as RNA interference (RNAi), has many features in common with post-transcriptional gene silencing (PTGS) in plants and quelling in Neurospora crassa. RNAi/PTGS appears to function as a defence system against viruses and in preventing transposon movement. RNAi also offers a way to inactivate genes of interest and, thus, provides a powerful tool to study gene function (reviewed by Hannon, 2002; Hutvágner and Zamore, 2002a).
Genetic and biochemical studies have revealed that RNAi/ PTGS is a very complex reaction, probably involving dozens of different proteins and intersecting with other cellular processes. Ongoing work is now beginning to unravel some of the mechanistic aspects of RNAi. Experi ments carried out in Drosophila cell or embryo extracts, recapitulating the RNAi reaction in vitro, demonstrated that dsRNA is first processed into ∼21 nucleotide (nt), small interfering RNAs (siRNAs) (Hammond et al., 2000; Zamore et al., 2000; Bernstein et al., 2001; Elbashir et al., 2001b). siRNAs are also implicated in RNAi/PTGS in other eukaryotes (Hamilton and Baulcombe, 1999; reviewed by Hannon, 2002; Hutvágner and Zamore, 2002a) and their role in mediating RNAi is supported by the demonstration that synthetic 21mer duplexes induce an RNAi in various organisms, both in vivo and in vitro (Caplen et al., 2001; Elbashir et al., 2001a,b; Nykänen et al., 2001). siRNAs may affect mRNA degradation by two distinct mechanisms. In Caenorhabditis elegans, they appear to act as primers for the cellular RNA-dependent RNA polymerase (RdRP), converting the target mRNA into dsRNA. Such nascent dsRNA is then fragmented, resulting in elimination of the target mRNA and the generation of a new population of siRNAs (Sijen et al., 2001). Although a similar mechanism may also operate in Drosophila (Lipardi et al., 2001), the major pathway of mRNA degradation in flies appears to be different, involving a multi-component nuclease, called RISC (Hammond et al., 2000; Nykänen et al., 2001). siRNAs associated with RISC target homologous mRNAs for destruction by cleaving them within the region complementary to the siRNA (Hammond et al., 2000; Zamore et al., 2000; Elbashir et al., 2001b; Nykänen et al., 2001).
Irrespective of the mechanism of mRNA degradation, the generation of siRNAs is central to RNAi, and the RNase III-like enzyme (named Dicer) responsible for pro cessing of dsRNA to siRNAs has recently been identified in Drosophila, C.elegans and mammals (Bernstein et al., 2001; Billy et al., 2001; Ketting et al., 2001; Knight and Bass, 2001). Dicer is a large (∼220 kDa) multi-domain protein present in all eukaryotes studied to date, with the exception of budding yeast. Its domains include a putative DExH/DEAH RNA helicase/ATPase domain, a PAZ signature, two neighbouring RNase III-like domains and a dsRNA-binding domain (RBD) (Figure 1A). dsRBD and RNase III domains are most certainly involved in dsRNA binding and cleavage, but the functions of the remaining domains are not known. In Drosophila extracts, RNAi requires ATP (Zamore et al., 2000; Nykänen et al., 2001). Studies performed with Drosophila and C.elegans extracts, and with the immunoprecipitates (IPs) containing Drosophila Dicer, indicated that one of the ATP-dependent steps is the processing of dsRNA to siRNAs (Zamore et al., 2000; Bernstein et al., 2001; Ketting et al., 2001; Nykänen et al., 2001). Consequently, it has been proposed that the helicase/ATPase domain of Dicer may promote translocation of the enzyme along the dsRNA, or a structural rearrangement of the substrate required for the cleavage (Bernstein et al., 2001; Nykänen et al., 2001; Hutvágner and Zamore, 2002a). However, our previous assays with mouse P19 embryonal carcinoma (EC) cell extracts indicated that ATP has no major effect on dsRNA cleavage in the mammalian system (Billy et al., 2001). The PAZ domain (Cerutti et al., 2000) may be responsible for the interaction of Dicer with PPD (PAZ and PIWI Domain) proteins, also involved in RNAi and related reactions (reviewed by Schwarz and Zamore, 2002).

Fig. 1. Schematic structure of Dicer and SDS–PAGE analysis of recombinant Dicer preparations. (A) Domains of human Dicer. Amino acid positions corresponding to domain borders, and a P-loop sequence with the K70A mutation are indicated. (B) Dicer-HisN (1 µg), Dicer-HisC (0.5 µg) and Dicer-HisC P-loop mutant K70A (1 µg) were analysed by 8% SDS–PAGE. Proteins were stained with GelCode Blue Stain Reagent (Pierce). Lanes M, protein size markers in kDa (BenchMark Protein Ladder, Invitrogen).
The biological role of Dicer is not confined to RNAi. In Drosophila and C.elegans, and also in human HeLa cells, Dicer is required in addition for the formation of microRNAs (miRNAs), a novel class of small 21-nt-long RNA regulators of gene expression (Grishok et al., 2001; Hutvágner et al., 2001; Ketting et al., 2001). Most likely, >100 different miRNAs, all excised from 70- to 80-nt-long precursor hairpins, operate in animals. With the exception of let-7 and lin-4, two miRNAs regulating development in C.elegans, their function is unknown (Schwarz and Zamore, 2002). Mutations in Dicer-like proteins in different organisms have developmental phenotypes (Ray et al., 1996; Jacobsen et al., 1999; Ketting et al., 2001; Knight and Bass, 2001), probably resulting from the compromised formation of miRNAs. The demonstration that EC cells (Billy et al., 2001) and embryonic stem cells (our unpublished results) contain much higher levels of Dicer than cultured differentiated cells is consistent with a role for Dicer in early development.
As an initial step towards a better understanding of the biochemistry of Dicer and its role in RNAi, we describe the overexpression and purification of different forms of the recombinant human protein. We characterize their ionic and co-factor requirements, and substrate specificity, and compare some properties of recombinant and endogenous enzymes.
Results
Preparations of recombinant Dicer
To prepare a recombinant human Dicer, we assembled a full-length cDNA encoding the protein. Proteins encoded by this cDNA and cDNAs characterized independently by Provost et al. (2002), and also assembled recently by the National Center for Biotechnology Information (Protein Data Bank entry XP_028308), have an identical sequence. This sequence differs at several positions from that reported by Matsuda et al. (2000). Comparison of human and mouse (Nicholson and Nicholson, 2002) cDNAs indicates that the two encoded proteins are 92% identical.
The cDNA insert was cloned into baculovirus vectors suitable for overexpression of proteins containing a His6 tag at the N- or C-terminus (referred to as Dicer-HisN and Dicer-HisC; for discussion of a probable N-terminus of the protein, see Materials and methods) in insect cells. Dicer-HisN was purified on nickel-nitrilotriacetic (Ni/NTA)–agarose beads, followed by chromatography on a Mono-Q column, and Dicer-HisC on Talon (cobalt) resin, followed by Ni/NTA beads. Purification of Dicer-HisC was performed either in the presence or absence of reducing agents. SDS–PAGE indicated that Dicer-HisC preparations were of higher purity than those of Dicer-HisN (Figure 1B). Recombinant proteins were stored at –20°C in buffers containing 50% glycerol, retaining activity for at least 5 months.
Recombinant proteins process dsRNA to siRNAs
Initial characterization of the nuclease activity of recombinant proteins was performed with the internally 32P-labelled 130 bp dsRNA. Both Dicer-His6 preparations were able to cleave dsRNA into ∼22-nt-long siRNA-like products (Figure 2). No 22 nt RNAs were generated when proteins were incubated with a single-stranded (ss) RNA (data not shown). The extent of cleavage of dsRNA by Dicer-His6 preparations was dependent on the concentration of recombinant proteins in the reaction and on the duration of incubation. Notably, incubation with Dicer resulted in accumulation of processing intermediates having lengths diagnostic of a gradual removal of one or more ∼22 nt siRNA units (Figure 2).

Fig. 2. Recombinant Dicer preparations cleave dsRNA into ∼22-nt-long siRNA-like products. (A) Effect of increasing amounts (indicated at the top) of Dicer-HisC (lanes 2–6) and Dicer-HisN (lanes 7–11) on dsRNA cleavage. Lane M, RNA size markers (90, 70, 45, 40, 27 and 21 nt). Lane 1, reaction with no Dicer added. (B) Kinetics of dsRNA cleavage by Dicer-HisC (25 ng). The size of generated siRNA-like products (21–23 nts) was confirmed by co-electrophoresis with alkaline and RNase T1 ladders.
Proteolysis stimulates activity of the recombinant and endogenous Dicer
While looking for conditions to improve the efficiency of the enzyme, we noted that pre-incubation of either the Dicer-HisN or Dicer-HisC preparation with proteinase K (ProtK) markedly stimulates their activity (Figure 3). The activation could also be brought about by pre-incubation with ProtK–agarose beads, allowing separation of the proteolysis and nucleolytic degradation steps (see below). Since both N- and C-terminally modified proteins were stimulated, it is unlikely that the activation is due to the removal of the interfering His6 tag. This conclusion is supported by the finding that activity of the endogenous Dicer, assayed with IPs of the P19 cell extract prepared with the anti-Dicer antibody (Ab) (Billy et al., 2001), is also stimulated by ProtK (Figure 3C). Since activities of the non-treated IPs and the equivalent amount of cell extract were similar (compare lane 0 with E), activation of the former is unlikely to be due to the protease-mediated release of Dicer from beads, association with which could potentially inhibit enzymatic activity. To further eliminate this possibility, attempts were made to detach Dicer from IP beads with an excess of the peptide used for immunization, but they were unsuccessful. Two other proteases, LysC and GluC, also stimulated activity of the protein by 2- to 3-fold (data not shown).
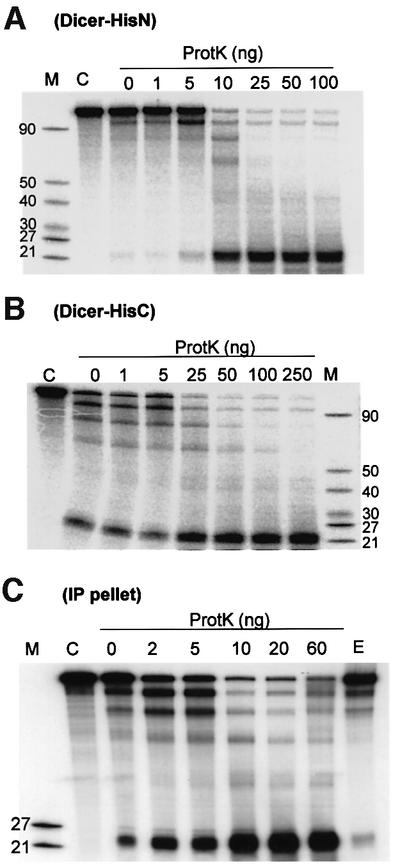
Fig. 3. Proteolysis stimulates activity of Dicer-HisN (A), Dicer- HisC (B) and IPs containing endogenous Dicer (C). Reactions contained 10 ng (A) and 25 ng (B) of recombinant Dicer, or 2 μl of IP beads (C). Amounts of added ProtK are indicated at the top. Lane C in (A) and (B), reactions incubated with no Dicer added. Lane E in (C), reaction containing the amount of P19 cell extract equivalent to the amount of IP beads used in other lanes.
Dicer-HisC used in the experiments shown in Figures 2 and 3 was purified in the absence of reducing agents. Comparison of Dicer-HisC preparations purified in the presence or absence of β-mercaptoethanol and DTT indicated that inclusion of reducing agents lowers the basal activity of the protein by 2- to 3-fold. However, following treatment with ProtK, activities of both preparations were similar (data not shown). Buffers used for the preparation of Dicer-HisN contained DTT and, similarly to Dicer-HisC prepared under reducing conditions, Dicer- HisN had a relatively low basal activity and was strongly activated by proteolysis (Figure 3, compare A with B).
Requirements for Dicer activity
Dicer-HisC prepared under non-reducing conditions was used to optimize cleavage conditions, and used in most of the other experiments described below. Maximal activity was found at pH 6.5–6.9 and at 50–100 mM NaCl concentration; addition of NaCl to 0.2 M inhibited the reaction by >90% (see Supplementary figure A available at The EMBO Journal Online). The reaction required the presence of Mg2+, the optimal concentration being 1–5 mM; addition of EDTA completely inhibited activity. dsRNA cleavage, though less efficient, was also observed when Mg2+ was replaced by Mn2+ and Co2+, but not Ca2+, Ni2+ or Zn2+ (Supplementary figure B). Requirements of the ProtK-activated enzyme were similar (data not shown). However, the response to temperature of native and ProtK-treated Dicer differed substantially. With non-treated Dicer, no cleavage was observed at 4°C, and cleavage at 25°C occurred ∼2× slower than at 37°C. In contrast, the enzyme pre-treated with ProtK beads showed considerable activity even at 4°C (Supplementary figure C).
Estimation of the efficiency of recombinant proteins indicated that under optimal reaction conditions and following 30 min incubation, only ∼0.1 mol of the siRNA product is generated per mole of enzyme. ProtK treatment increased this ratio up to ∼0.5. In the experiments presented above, the concentration of dsRNA was 0.3–0.5 nM. When cleavage of the 130 bp dsRNA substrate was assayed at 30 nM concentration, ∼0.5 mol of siRNA was formed per mole of the native protein. It is unknown whether this low number is due to only a small fraction of the recombinant enzyme being active or if it reflects a low catalytic efficiency of the enzyme (see Discussion).
Processing of dsRNA into siRNAs is ATP independent
Experiments carried out with Drosophila and C.elegans extracts, and IPs containing the Drosophila Dicer, demonstrated that processing of dsRNA into siRNAs is strongly stimulated by addition of ATP (see Introduction). How ever, our previously performed assays with P19 cell extracts indicated that ATP has no major effect on dsRNA cleavage in the mammalian system (Billy et al., 2001). Dicer activity measured in IPs of the mouse P19 and human HeLa cell extracts also did not require addition of ATP, and was not affected by pre-incubation with either calf intestine phosphatase (CIP) or hexokinase (HK) and glucose, treatments performed to eliminate residual ATP (Figure 4A). Likewise, addition of non-hydrolysable ATP analogues, such as AMPPCP or AMPCPP, had no effect on dsRNA cleavage. ATP was also without effect when combined with the ATP regeneration system, creatine phosphate and creatine kinase (data not shown). Parallel experiments carried out with extracts prepared from Drosophila KC167 cells demonstrated a strong stimulatory effect of ATP on dsRNA processing in this system (data not shown), consistent with the results of other investigations (Zamore et al., 2000; Bernstein et al., 2001; Nykänen et al., 2001).

Fig. 4. Processing of dsRNA by human Dicer is ATP independent. (A) Effect of different nucleotides (2 mM; complexed with Mg2+) and ATP depletion conditions on processing of dsRNA by Dicer present in IPs of P19 (left and middle gels) and HeLa (right gel) cell extracts. Lanes S, reactions without addition of IP beads and nucleotide. Lanes ‘Control CIP’ and ‘Control HK’, reactions pre-incubated without CIP, and HK and glucose addition, respectively. Lower panel, quantification of the P19 (black bars, averages of two independent experiments) and HeLa (grey bars, data from the gel shown) IPs. (B) Effect of different nucleotides on processing of dsRNA by Dicer-HisC (25 ng). Lane S, reaction without addition of Dicer and nucleotide. Lower panel, quantification of the data.
We investigated whether activity of the recombinant human Dicer is stimulated by ATP. As shown in Figure 4B, ATP had no appreciable effect on cleavage of dsRNA. Similarly, addition of ATP analogues, or 1 mM GTP, CTP, UTP, dATP, ADP, NAD or NADP, had no influence on Dicer activity (Figure 4B; data not shown). Pre-incubation of the enzyme with either HK and glucose, or CIP, did not affect either the kinetics or the extent of the cleavage, further arguing against the requirement for ATP (Supplementary figure D).
Dicer contains an evolutionarily conserved DExH/DEAH ATPase/RNA helicase domain (Figure 1A). The reported ATP requirement of the dsRNA cleavage in Drosophila extracts is generally attributed to a potential role of this domain in translocation of the enzyme along the dsRNA, or to a structural rearrangement of the substrate required for the cleavage (Bernstein et al., 2001; Ketting et al., 2001; Nykänen et al., 2001; Hutvágner and Zamore, 2002a). We wanted to eliminate the possibility that the observed ATP independence of the recombinant mammalian enzyme is due to ATP remaining associated with the ATPase/helicase domain, even following HK or CIP treatment. To this end, we prepared the Dicer-HisC mutant K70A, bearing the K to A substitution in the P-loop motif of the ATPase/helicase domain (Figure 1). In all investigated ATPases/helicases, and also other P-loop-containing proteins, this mutation strongly inhibits nucleotide binding and enzymatic activity (Gross and Shuman, 1998; Levin and Patel, 1999, and references therein). The DExH motif of Dicer, DECH, contains an evolutionarily conserved cysteine. Hence, the mutant and wild-type (wt) proteins were purified under both reducing and non-reducing conditions, and all preparations were assayed for dsRNA processing and ATPase activities. Nuclease assays have revealed that wt and mutant proteins purified under either reducing (Figure 5) or non-reducing (data not shown) conditions have similar activity, independent of the presence or absence of ATP. However, to date, we have been unable to demonstrate ATPase activity for any Dicer preparation, also when reactions included ssRNA, dsRNA, ssDNA or dsDNA (data not shown). Likewise, assays with the P19 cell extract IPs provided no reliable evidence of ATPase activity associated with Dicer.

Fig. 5. Mutation K70A in the P-loop of the putative ATPase/helicase domain of Dicer has no effect on dsRNA cleavage. Reactions contained 25 ng of Dicer-HisC purified under reducing conditions, and were incubated for the times indicated in the absence or presence of 3 mM ATP.
We have also analysed effects of ATP, its analogues, and different ATP depletion conditions on the activity of wt and K70A mutant proteins at the 30 nM 130 bp dsRNA concentration. The results were similar to those presented above. Likewise, processing of other substrates, a 700 bp dsRNAs or 130 bp dsRNAs strongly differing in the GC content (71 versus 29%), was not dependent on ATP hydrolysis (data not shown). In summary, our findings argue strongly against ATP being required for the cleavage of dsRNA by the human Dicer.
Interactions of Dicer with dsRNA studied on native gels
For more insight into the mechanism of dsRNA cleavage by Dicer, we investigated the formation of complexes between Dicer and dsRNA, using non-denaturing gels. The enzyme was incubated with dsRNA under conditions that do not allow cleavage: either at 4°C in the presence of Mg2+, or at 25°C in the presence of EDTA. As shown in Figure 6, in both conditions, the incubation of 130 bp dsRNA with increasing amounts of Dicer resulted in the formation of complexes whose mobility decreased as a function of increasing protein concentration. The appearance of several distinct bands suggests that more than one Dicer molecule can bind to the 130 bp dsRNA. Taken together with the results shown in Supplementary figures B and C, these findings indicate that binding and cleavage steps of the Dicer-catalysed reaction can be uncoupled by carrying out the incubation at 4°C, or by omitting Mg2+.
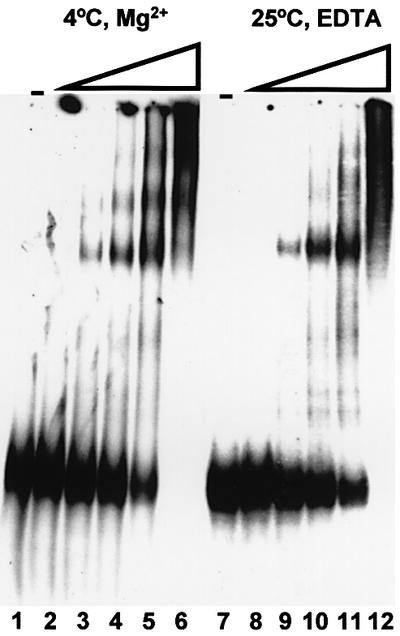
Fig. 6. Complexes between Dicer and dsRNA, visualized on non- denaturing gels. Increasing amounts of Dicer-HisC (0, 2, 6, 20, 60 and 200 ng, respectively, in lanes 1–6 and 7–12) were incubated with the 32P-labelled 130 bp dsRNA either at 4°C in the presence of 2 mM Mg2+ (lanes 1–6) or at 25°C in the presence of 2 mM EDTA (lanes 7–12).
We have also carried out native gel analysis of reactions incubated at 37°C in the presence of Mg2+, under conditions that allow cleavage of dsRNA. In parallel, aliquots of these reactions were analysed on denaturing gels. This comparison, performed with dsRNA substrates of different lengths and either native or ProtK-beads-activated Dicer, revealed that in general less radioactivity migrates at the position corresponding to siRNA on native gels as compared with denaturing gels (Figure 7A and B). This difference was particularly evident for reactions containing higher concentrations of Dicer, both native and ProtK treated (Figure 7; data not shown). To explain this apparent inconsistency, the native gel lanes representing reactions performed with the 70 bp dsRNA and either native or proteolysed Dicer were cut into 1 cm pieces and the recovered RNA was subjected to electrophoresis on a denaturing gel. As shown in Figure 7C, for the ProtK-treated enzyme, the siRNA-like 22 nt RNA was found in association with a complex marked X in Figure 7, while for the native enzyme, ∼50% of ∼22 nt RNA was associated with slow-migrating complexes labelled Y and Z. Additional analyses, allowing better separation of complexes Y and Z, indicated that both contain ∼22 nt RNA (data not shown). Although further work is needed to establish the identity of complexes X, Y and Z, it is possible that complex Z, reproducibly seen with reactions incubated under cleavage conditions but not following incubations at 4°C or in the presence of EDTA (Figure 6; data not shown), represents Dicer associated with the siRNA product, while complex Y is an aggregate thereof. Association of Dicer with its reaction product would provide a plausible explanation for the apparent low catalytic activity of the enzyme (see Discussion).
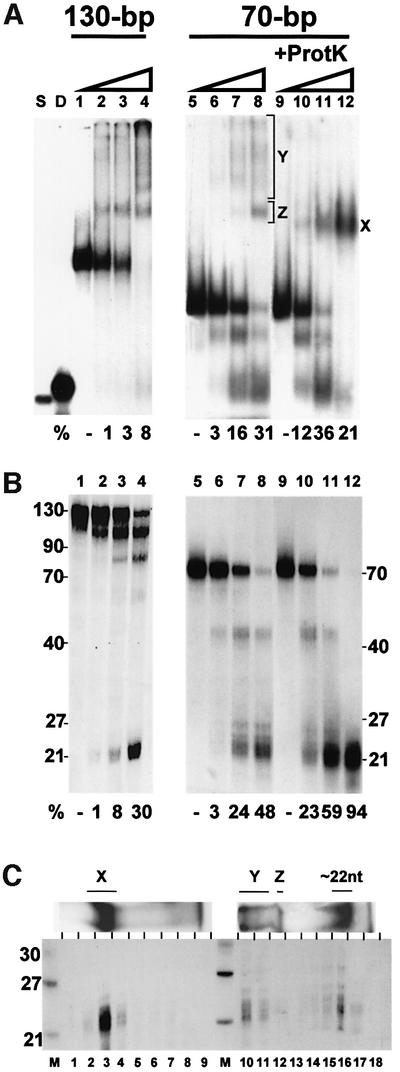
Fig. 7. Analysis of Dicer cleavage reactions on native (A) and denaturing (B) gels, and demonstration that complexes X, Y and Z contain siRNA products (C). (A and B) Increasing amounts of Dicer-HisC (0, 10, 30 and 100 ng), either native (lanes 1–8) or pre-incubated with ProtK beads (lanes 9–12), were incubated with 1 fmol of the 32P-labelled 130 or 70 bp dsRNA at 37°C for 30 min. Halves of each reaction were analysed on two different gel types. Lanes S and D, ss and ds 21 nt siRNA markers. The percentage of radioactivity representing siRNA products is indicated below each lane. (C) Lanes from the non-denaturating gel (shown at the top), representing analysis of reactions containing 70 bp dsRNA and 100 ng of either proteolysed (lanes 1–9) or native (lanes 10–18) Dicer, were cut into 1 cm pieces. RNA was eluted, ethanol precipitated with a tRNA carrier and electrophoresed on an 8 M urea gel. Positions of complexes X, Y and Z are indicated.
Dicer preferentially cleaves dsRNAs at their termini
Previous work has indicated that dsRNAs shorter than ∼100 bp are generally much less effective inducers of RNAi in vivo and in vitro than longer RNAs (Tuschl et al., 1999; Hammond et al., 2000; Parrish et al., 2000; Elbashir et al., 2001b). Likewise, the efficiency of dsRNA processing into siRNAs in Drosophila extracts was found to be length dependent, with 29 bp dsRNA being processed very inefficiently (Bernstein et al., 2001; Elbashir et al., 2001b). To provide a possible explanation for these findings, dsRNAs ranging in length from 30 to 130 bp were compared for their ability to act as substrates for the recombinant Dicer. Processing of dsRNAs of 30, 40, 50, 70 and 130 bp to ∼22 nt RNAs occurred with comparable efficiencies (Figure 8A). For 130, 70 and 50 bp substrates, intermediates of processing with lengths diagnostic of the removal of one or more ∼22 nt siRNA units were readily visible. In addition, processing of 40 (Figure 8A) and 30 bp RNA (data not shown) resulted in the accumulation of short fragments, whose length (∼18 and ∼7–8 nt, respectively) suggests that they represent terminal cut-off products. Accumulation of these terminal fragments, as well as intermediates of discrete sizes, is consistent with a step-wise excision of siRNAs from dsRNA termini.

Fig. 8. Processing of dsRNA of different lengths (A) or terminal structures (B). dsRNAs of indicated length (5 fmol) were incubated either in the absence or presence of 40 ng of Dicer-HisC. Equivalent amounts of counts were applied to the denaturing gel. (B) Terminal GAAA tetra-loops and RNA–DNA duplexes delay cleavage of dsRNA substrates. Upper section, schematic structure of different substrates. Lengths of relevant ds regions are indicated. Lower section, kinetics of processing of different substrates, and effect of 15 min pre-incubation at 4°C on kinetics. The identity of substrates and times of incubation (in min) are indicated at the top. Lanes C, control incubations without Dicer-HisC. Lanes C′, control reactions subjected to only pre-incubation at 4°C. Quantification of the data is shown at the bottom. All kinetic experiments have been reproduced at least twice.
We prepared several derivatives of the 130 bp dsRNA to assess the importance of free RNA termini for the cleavage reaction (Figure 8B). dsRNAtetra bears GAAA tetra-loops at either end. This sequence confers high stability on the loop through an additional base pair between the first guanosine and the third adenosine. dsRNAdna is a 77 bp RNA bearing either 26 or 27 bp RNA–DNA duplexes at the ends. Using a 50 bp RNA–DNA duplex, we verified that Dicer is unable to cleave the RNA–DNA hybrid (results not shown). dsRNArna serves as a control for dsRNAdna and contains 26 and 27 nt terminal RNA oligoribonucleotides in place of oligodeoxynucleotides.
All the RNAs listed above acted as processing substrates (Figure 8B). However, comparison of the kinetics of processing of dsRNAtetra and dsRNAdna with that of dsRNA and dsRNArna revealed that cleavage of the former substrates is initiated with a 5–10 min delay. This could be due to hindrance by the terminal tetra-loops and RNA–DNA duplexes of the interaction with Dicer, necessitating that the enzyme binds, possibly with slower kinetics, to internal regions of the substrate. To address this possibility, dsRNAtetra and dsRNAdna were pre-incubated with the enzyme for 15 min at 4°C, conditions allowing binding but not cleavage of the substrate, prior to the incubation at 37°C. As seen in Figure 8B, the pre-incubation eliminated the delay in siRNA accumulation, indicating that binding of Dicer to the internal sites may indeed be a limiting factor during processing of terminally modified substrates. Consistent with the initiation of cleavages at different internal sites, accumulation of processing intermediates was considerably less pronounced for dsRNAtetra than control dsRNA. However, treatment of dsRNAdna yielded discrete intermediates similar to those seen during cleavage of dsRNA or dsRNArna. Hence, dsRNAdna, despite having non-cleavable RNA–DNA ends, apparently allows a selection of site(s) preferentially used for initiation of processing by the enzyme. Dicer possibly binds to the terminal RNA–DNA duplexes and helps in aligning additional enzyme molecules at the internal RNA–RNA region of the substrate.
dsRNA substrates used in the experiments described above contained blunt termini bearing 5′-triphosphates on both RNA strands. However, siRNA duplexes produced in the Dicer cleavage reaction are expected to contain 2 nt 3′-overhangs bearing 5′-monophosphate groups (Elbashir et al., 2001b), and such terminal structures should be present in processing intermediates acting as Dicer substrates in consecutive rounds of cleavage of long dsRNAs. Using sets of 30 bp dsRNAs with either 5′-monophosphate or 5′-triphosphate ends and either blunt or 2 nt 3′-overhang ends, we found that all these RNAs are processed with similar efficiencies (data not shown).
Discussion
This work represents a first comprehensive characterization of the activity of purified Dicer, a key enzyme in RNAi/PTGS. We have established procedures for purifying the N- and C-terminally His6-tagged recombinant human proteins, Dicer-HisN and Dicer-HisC. Notably, Dicer-HisC was purifed by successive fractionation on Co-containing resin and Ni/NTA beads. Despite a similar principle of affinity separation, only a combination of both matrices reproducibly yielded a high-purity protein; preparations applied to only one type of beads contained numerous impurities. Dicer-HisC, used in most of the experiments described in this work, was prepared under either reducing or non-reducing conditions. The latter preparations were more active then those prepared in the presence of reducing agents. Surprisingly, pre-incubation of recombinant proteins with ProtK and some other proteases markedly stimulated their activity. Activity of the endogenous Dicer present in IPs of P19 cell extracts was also stimulated by pre-incubation with ProtK. A dissection of the mechanisms underlying the effects of proteolysis and reducing agents on enzyme activity requires further experimentation. Provost et al. (2002) have also independently succeeded in preparing an active recombinant human Dicer.
Many reaction requirements and properties of Dicer are similar to those established for the RNase III from E.coli and yeast. All enzymes require divalent cations for cleavage of the substrate. As with the E.coli RNase III (Li et al., 1993), Mg2+ could be substituted by Mn2+ and Co2+. Mg2+ and Mn2+, but not Co2+, are active with the Schizosaccharomyces pombe enzyme (Rotondo and Frendewey, 1996). As previously demonstrated for the E.coli RNase III (Chelladurai et al., 1993), Mg2+ ions are required by Dicer for cleavage of the dsRNA substrate but not for its binding. Likewise, incubation at 4°C allows binding to, but not cleavage of, dsRNA by Dicer (Figure 6). These properties allow separation of the dsRNA binding and cleavage steps of the reaction.
Previous experiments carried out with cell extracts originating from Drosophila and C.elegans, and with IPs containing the Drosophila Dicer, demonstrated that cleavage of dsRNA into siRNAs is strongly stimulated by the addition of ATP (Zamore et al., 2000; Bernstein et al., 2001; Ketting et al., 2001; Nykänen et al., 2001). However, ATP appeared not to have much effect on dsRNA cleavage in the mammalian system (Billy et al., 2001). We have now measured the activity of different preparations of the recombinant Dicer, and also IPs prepared from extracts of mouse P19 and human HeLa cells with anti-Dicer Abs. Cleavage of dsRNA was never stimulated by addition of ATP. Cleavage was also not affected by the addition of an excess of non-hydrolysable ATP analogues or treatments performed to eliminate any residual ATP.
A role for ATP in siRNA formation, demonstrated for Drosophila and C.elegans extracts, has generally been linked to the ATPase/helicase domain of Dicer and its hypothetical function in either moving the enzyme along the dsRNA or a local unwinding of the substrate, possibly required for the cleavage (Bernstein et al., 2001; Ketting et al., 2001; Nykänen et al., 2001; Hutvágner and Zamore, 2002a). We found that the K to A change in the P-loop of the Dicer ATPase/helicase domain, a mutation known to eliminate or at least strongly inhibit nucleotide binding and activity of enzymes containing related domains (Gross and Shuman, 1998; Levin and Patel, 1999), has no effect on dsRNA cleavage by Dicer. Although this result provides an additional argument against the requirement of ATP for the dsRNA cleavage, it has to be interpreted with caution. To date, we have been unable to demonstrate ATPase activity in recombinant Dicer preparations, and hence to directly assess functionality of the K70A mutation. Additional factors may be required to activate the ATPase-like domain of Dicer. Another possibility is that, despite employing different forms of recombinant proteins and purification protocols, none of the Dicer preparations used in this work was fully functional.
Some additional observations may serve as arguments against the dsRNA cleavage per se being ATP dependent. First, processing of various substrates, including perfectly complementary dsRNAs, by RNase III, a ‘prototype’ enzyme of Dicer, does not require ATP (reviewed by Nicholson, 1999). Secondly, a putative homologue of Dicer in Dictyostelium is apparently devoid of the ATPase/helicase domain. In contrast, a domain with homology to the Dicer ATPase/helicase domain is present in RdRP-like proteins of Dictyostelium, suggesting that ATP may be required downstream of the dsRNA cleavage reaction (Martens et al., 2002). Detailed analysis of the ATP requirement for RNAi in Drosophila extracts identified two ATP-dependent steps in the pathway. In addition, ATP was utilized for maintenance of 5′-phosphates on siRNAs (Nykänen et al., 2001).
If ATP is not needed for dsRNA cleavage itself, what other steps leading to the increased siRNA yields, as demonstrated in Drosophila and C.elegans extracts, could be influenced by this factor? ATP hydrolysis, catalysed by either Dicer or another protein interacting with it, could be required for release of the siRNA product from the enzyme and its transfer to the downstream RNAi component, such as a RISC nuclease. Alternatively, and possibly in conjunction with the product release, ATP hydrolysis could help in a structural rearrangement of Dicer required for a next round of substrate binding and catalysis. Our findings that significantly less radioactivity migrates at a position corresponding to siRNA on native as compared with denaturing gels, and that the deficit of the siRNA product on native gels is accompanied by accumulation of complexes probably representing Dicer (or its ProtK-generated fragment) associated with siRNAs (Figure 7), support the first scenario. This possibility is further strengthened by the recent observation of Hutvágner and Zamore (2002b) that pre-let-7 RNA, which must undergo Dicer-catalysed processing in the Drosophila extract, is considerably more effective in targeting degradation of a model RNA than the preformed siRNA-like let-7 duplex. The structural rearrangement explanation is supported by our observations that the activity of different recombinant Dicer preparations, and of the endogenous protein, is stimulated by proteolysis (Figure 3), and that the proteolysed enzyme becomes active at 4°C (Supple mentary figure C). The catalytic site is possibly occluded by another domain of Dicer, and access to it entails a temperature-dependent rearrangement, a requirement at least partially alleviated by proteolysis.
We consider that ATP is unlikely to function in translocation of the enzyme along the dsRNA substrate. In Drosophila extracts, excision of miRNAs from hairpin precursors is ATP dependent, as is the processing of long dsRNAs (Hutvágner et al., 2001). However, miRNA processing involves excision of only a single 21 nt miRNA segment, with no obvious need for Dicer movement along the substrate. In this case also, ATP is more likely to be required for product release and/or a conformational change in the enzyme. Consistent with this possibility, we have found that excision of let-7 RNA from its precursor by the recombinant human Dicer does not require ATP (our unpublished results).
In conclusion, we offer two possible explanations for the apparent discrepancy between the results obtained with mammalian and lower metazoan systems. According to the first, ATP plays a role in increasing the efficiency of siRNA formation in all metazoa by promoting a structural rearrangement of the enzyme and/or product release. With mammalian cell extracts and purified human Dicer, as used in this work, siRNA yields may be ATP independent because these preparations lack some RNAi reaction components, possibly present in extracts of Drosophila and C.elegans. This explanation would be consistent with the apparent low catalytic efficiency of the mammalian enzyme. A second possibility is that mechanisms of dsRNA cleavage by Dicer are fundamentally different in mammals and lower metazoa. For example, Drosophila and C.elegans enzymes might cleave dsRNA by a processive mechanism requiring ATP hydrolysis, while the mammalian Dicer functions distributively and independently of ATP. Comparison of properties and factor requirements of purified Dicer proteins from C.elegans, Drosophila and mammals should help to distinguish between these alternatives.
The accumulation of processing intermediates having lengths diagnostic of the removal of one or more ∼22 nt siRNA units, and experiments performed with substrates containing terminal tetra-loops and RNA–DNA duplexes, indicate that recombinant Dicer preferentially cleaves dsRNAs at their termini. A similar conclusion can be drawn from experiments carried out with P19 and HeLa cell IPs. The ATP-dependent formation of discrete size intermediates, supporting a stepwise excision of siRNAs from dsRNA termini, was also recently observed in C.elegans extracts (Ketting et al., 2001). Previous experiments performed with Drosophila extracts did not reveal any cleavage intermediates, suggesting that Drosophila enzyme cuts dsRNA by a highly processive mechanism (for references, see Zamore, 2001). Nevertheless, it is also very likely that the Drosophila enzyme processes dsRNA progressively from the ends since the mRNA cleavage during RNAi in Drosophila extracts occurs at characteristic ∼22 nt intervals (Zamore et al., 2000).
The demonstrated preference of Dicer to cleave dsRNAs at their termini may be of physiological significance. Such a property might prevent accidental cleavage of extended hairpins located in internal regions of mRNAs and other cellular molecules (Morse et al., 2002). However, as investigated in this work, Dicer is also able to initiate processing, though less effectively, at internal sites of dsRNA substrates. This is consistent with the established function of Dicer in excision of miRNAs from inner regions of hairpin precursors (Grishok et al., 2001; Hutvágner et al., 2001; Ketting et al., 2001).
We have found that dsRNAs as short as 30 bp act as efficient substrates of the purified human Dicer. Previous work with Drosophila extracts indicated that 29 bp dsRNAs are very inefficiently cleaved to siRNAs in embryo extracts (Elbashir et al., 2001b), but more recently Paddison et al. (2002) demonstrated a very effective processing, in extracts originating from suspension cells, of 29 bp and even shorter hairpins. Since 30 bp dsRNAs are cleaved by Dicer only once, they will be useful for a direct assessment of structural features required for RNA processing by the enzyme. Initial experiments performed with a selection of 30 bp substrates, either terminally or internally labelled, demonstrated that neither the recessed nature of the 5′ end, nor its phosphorylation status, is important for the efficient cleavage (our unpublished results). They also indicated that products generated by the recombinant Dicer contain 3′-protruding ends, a feature expected for the reaction catalysed by the RNase III-like enzyme, and also consistent with the findings that siRNA duplexes containing 2 nt 3′-overhangs are optimal effectors of RNAi both in vitro and in vivo (Caplen et al., 2001; Elbashir et al., 2001b,c).
Materials and methods
Dicer cDNAs and expression vectors
A cDNA HH03019 (DDBJ/EMBL/GenBank accession No. AB023145), encoding the human Dicer amino acids 401–1922, cloned in SalI and NotI sites of pBluescript II SK+, was obtained from N.Kusuhara, Kazusa Research Institute, Chiba. A region corresponding to Dicer positions 1–1494 was amplified by RT–PCR, using total HeLa cell RNA and primers ACGCGTCGACATGAAAAGCCCTGCTTTGC (sequence-based DDBJ/EMBL/GenBank accession No. AB028449) and CTGAATTCTGCTTCCATCGTG (SalI and EcoRI sites are underlined), following the Superscript II RNaseH–reverse transcriptase protocol (Life Technologies). The RT–PCR fragment was cut with SalI and EcoRI, and cloned into HH03019 (pre-digested with SalI, and partially with EcoRI) to yield pBS-Dicer. The cDNA insert of pBS-Dicer, and also PCR-generated and otherwise manipulated inserts of plasmids described below, have been sequenced. The ATG selected as an initiation codon for plasmid constructions is an in-frame ATG of the Dicer mRNA 5′ leader. It is preceded by six ATGs, which are either not in-frame or are followed by termination codons. The selected ATG (context TGAATGA) is followed 30 bp downstream by a better context ATG (sequence AGCATGG), which may potentially act as an initiator.
To add a His6 tag to the Dicer C-terminus, the downstream BamHI–NotI fragment in pBS-Dicer was replaced by the equivalent fragment (obtained by PCR, using appropriate primers) containing the sequence CATCACCATCACCATCAC upstream of the stop codon, yielding pBS-Dicer/HisC. Dicer and Dicer-HisC cDNAs were recloned from pBS plasmids into the pENTR1A vector, using the Gateway system (Life Technologies), yielding pENTR-Dicer and pENTR-Dicer-HisC. The non-tagged Dicer cDNA was then switched to the pDEST10 vector by the reaction catalysed by LR Clonase mix (Life Technologies), yielding pDEST10-Dicer, used for expression of the N-terminally His6-tagged Dicer-HisN. Dicer-HisN contains an additional 31 amino acids (DYDIPTTENLYFQGITSLYKKAGFKGTNSVD), encoded by the vector, positioned between the His6 tag and the N-terminus. Dicer-HisC cDNA was switched to the pDEST8 vector for expression of the C-terminally His6-tagged Dicer-HisC. Bacmids for insect cell transformation were generated using the Bac-To-Bac Baculovirus Expression System (Life Technologies). The Dicer P-loop K70A mutant was generated in pBS-Dicer/HisC, using a Quikchange site-directed mutagenesis kit (Stratagene).
Preparation of recombinant Dicers
For protein overexpression, insect Sf9 cells were infected with a virus (virus/cell ratio 1:1) and collected 3 days later. Cells (∼4 × 107) were resuspended in 4 ml of buffer W100 (Tris–HCl pH 7.5, 100 mM NaCl, 1 mM MgCl2, 5 mM β-mercaptoethanol, 10% glycerol and 0.5% Triton X-100) containing 1× protease inhibitor mix without EDTA (Roche) and broken by passing through a 0.45 × 10 gauge needle. Lysates were centrifuged at 17 000 g for 10 min and then at 58 000 r.p.m. (SW60 Beckman rotor) for 1 h. For purification of Dicer-HisC, the final supernatant was mixed with the Talon affinity resin (400 µl; Clontech) pre-washed with buffer W100. After 3 h at 4°C, the resin was washed with buffer W100 (3 × 4 ml), buffer W800 (buffer W100 containing 800 mM NaCl; 3 × 4 ml) and then again with buffer W100 (3 × 4 ml). Protein was eluted with 4 ml of buffer W100 containing 40 mM imidazol. The eluate was directly added to 400 µl of Ni/NTA beads (Qiagen) pre-washed with buffer W100, containing 40 mM imidazol and 1 mM DTT instead of β-mercaptoethanol. Following 3 h at 4°C, the beads were washed similarly to Talon beads and the protein eluted with 2 ml of buffer W100 containing 100 mM imidazol and 1 mM DTT. The eluate was dialysed against buffer D (as buffer W100 but containing 50% glycerol, 0.1% Triton X-100 and 1 mM DTT) (2 × 2 h, 1 l each time) and stored at –20°C. To eliminate any Ni or Co ions potentially released from affinity beads, aliquots of purified Dicer were subjected to additional extensive dialysis against buffer D containing 20 mM EDTA, followed by dialysis against buffer D containing 1 mM EDTA. This treatment had no effect on either cleavage or ATPase activity of Dicer.
For purification of Dicer-HisC under non-reducing conditions, an identical procedure was used, except that all buffers were devoid of DTT or β-mercaptoethanol.
Purification of Dicer-HisN was carried out under reducing conditions and all buffers contained 1 mM DTT. Initial steps were identical as in the purification of Dicer-HisC, except that a high-speed centrifugation and Talon steps were omitted. The 17 000 g supernatant was directly mixed with pre-washed Ni/NTA beads. After 12 h at 4°C, the beads were packed into a column. The column was consecutively washed with buffers W100, W800 and W100, and the protein eluted with buffer W100 containing 100 mM imidazol. Fractions containing Dicer activity (3 × 1 ml) were collected and the buffer was changed back to W100 using the Millipore Biomax 30K concentrator. The sample was applied to a Mini-Q PC 3.2/3 column (Amersham) equilibrated with buffer W100. The column was washed extensively with buffer W100 and protein eluted with a gradient of 10 ml of buffer W100 and 10 ml of buffer W1000 (buffer W100 containing 1 M NaCl). Fractions (2 × 0.4 ml) corresponding to the peak of activity eluting at ∼0.4 M NaCl were collected. They were concentrated, concomitant with a buffer exchange to W100, in a Millipore concentrator. Proteins were stored at –20°C in buffer W100 containing 50% glycerol, 0.1% Triton X-100 and 1 mM DTT. Protein concentration was calculated using Bradford reagent with BSA as a standard.
Cell extracts and IPs
P19 and HeLa cell growth, and preparation of cytoplasmic extracts and IPs with the anti-Dicer Ab D347 coupled to protein A beads, were as described previously (Billy et al., 2001).
dsRNA substrates
Details of their preparation are specified in Supplementary data.
Assays of Dicer activity
dsRNA processing assays (10 µl) contained 30 mM Tris–HCl pH 6.8, 50 mM NaCl, 3 mM MgCl2, 0.1% Triton X-100, 15–25% glycerol and 1 mM DTT when indicated. Amounts of Dicer and other included components and modified conditions are specified in the legends. If not indicated otherwise, reactions contained 3–5 fmol of dsRNA internally labelled with [α-32P]UTP (final sp. act. 30 or 150 Ci/mmol; Amersham), and were incubated for 30 min at 37°C. For ATP depletion, reactions were pre-incubated for 20 min at 37°C with 2 mM (for purified Dicer) or 10 mM (for IPs) glucose and 1 U of hexokinase (HK, Sigma), or with 1 U of CIP (Roche). We have verified, using P19 cell extracts and a bioluminescence ATP assay kit (Sigma), that the procedure with 10 mM glucose decreases ATP to <100 nM. For analysis of activity in IPs, reactions contained 2–5 µl of IP beads. BSA (10 µg) was additionally added to reactions in the experiment shown in Figure 3C. For proteolytic activation, reactions were pre-incubated for 10 min at 37°C, under assay conditions, with the indicated amounts of ProtK (Sigma) prior to dsRNA addition, or 1 µg of Dicer-HisC was pre-incubated with ProtK–agarose beads (Sigma; 3 mg containing 0.8 U of ProtK, for 5 min at 37°C). Reactions were analysed on 10% polyacrylamide/8 M urea gels, which were processed for autoradiography or quantification, using the Storm 860 PhosphorImager (Molecular Dynamics). For quantification, a percentage of radioactivity corresponding to siRNA products was calculated in relation to either the input or a sum of the bands, representing the substrate, intermediates and siRNA products present in individual lanes. For calculation of molar ratios of Dicer to generated siRNA products, the Dicer-HisC purity was assumed to be 75%.
For analysis of complexes on native gels, assays contained Dicer-HisC prepared under non-reducing conditions, 1 fmol of 32P-labelled dsRNA, and either 2 mM MgCl2 or 2 mM EDTA. Samples were electrophoresed (5 V/cm) for 5 h at 4°C on a 4% polyacrylamide gel containing 45 mM Tris–borate pH 8.0 and 0.1% Triton X-100.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank J.Hofsteenge for valuable discussions and P.Provost for exchanging unpublished results. F.A.K. is supported by a long term EMBO fellowship. Friedrich Miescher Institute is a part of Novartis Research Foundation.
References
- Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- Billy E., Brondani,V., Zhang,H., Muller,U. and Filipowicz,W. (2001) Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc. Natl Acad. Sci. USA, 98, 14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplen N.J., Parrish,S., Imani,F., Fire,A. and Morgan,R.A. (2001) Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl Acad. Sci. USA, 98, 9742–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti L., Mian,N. and Bateman,A. (2000) Domains in gene silencing and cell differentiation proteins: the novel PAZ domain and redefinition of the Piwi domain. Trends Biochem. Sci., 25, 481–482. [DOI] [PubMed] [Google Scholar]
- Chelladurai B., Li,H., Zhang,K. and Nicholson,A.W. (1993) Mutational analysis of a ribonuclease III processing signal. Biochemistry, 32, 7549–7558. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001a) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Lendeckel,W. and Tuschl,T. (2001b) RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev., 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Martinez,J., Patkaniowska,A., Lendeckel,W. and Tuschl,T. (2001c) Functional anatomy of siRNAs for mediating efficient RNAi in D.melanogaster embryo lysate. EMBO J., 20, 6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A. et al. (2001) Genes and mechanisms related to RNAi regulate expression of the small temporal RNAs that control C.elegans evelopmental timing. Cell, 106, 23–34. [DOI] [PubMed] [Google Scholar]
- Gross C.H. and Shuman,S. (1998) The nucleoside triphosphatase and helicase activities of vaccinia virus NPH-II are essential for virus replication. J. Virol., 72, 4729–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A.J. and Baulcombe,D.C. (1999) A species of small antisense RNA in posttranscriptional gene silencing in plants. Science, 286, 950–952. [DOI] [PubMed] [Google Scholar]
- Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- Hannon G.J. (2002) RNA interference. Nature, 418, 244–251. [DOI] [PubMed] [Google Scholar]
- Hutvágner G. and Zamore,P.D. (2002a) RNAi: nature abhors a double-strand. Curr. Opin. Genet. Dev., 12, 225–232. [DOI] [PubMed] [Google Scholar]
- Hutvágner G. and Zamore,P.D. (2002b) A microRNA in a multiple-turnover RNAi enzyme complex. Science, 297, 2056–2060. [DOI] [PubMed] [Google Scholar]
- Hutvágner G., McLachlan,J., Pasquinelli,A.E., Balint,E., Tuschl,T. and Zamore,P.D. (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science, 293, 834–838. [DOI] [PubMed] [Google Scholar]
- Jacobsen S.E., Running,M.P. and Meyerowitz,E.M. (1999) Disruption of an RNA helicase/RNAse III gene in Arabidopsis causes unregulated cell division in floral meristems. Development, 126, 5231–5243. [DOI] [PubMed] [Google Scholar]
- Ketting R.F., Fischer,S.E., Bernstein,E., Sijen,T., Hannon,G.J. and Plasterk,R.H. (2001) Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C.elegans. Genes Dev., 15, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight S.W. and Bass,B.L. (2001) A role for the RNAse III enzyme DCR-1 in RNA interference and germ line development in C.elegans. Science, 293, 2269–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M.K. and Patel,S.S. (1999) The helicase from hepatitis C virus is active as an oligomer. J. Biol. Chem., 274, 31839–31846. [DOI] [PubMed] [Google Scholar]
- Li H.L., Chelladurai,B.S., Zhang,K. and Nicholson,A.W. (1993) Ribonuclease III cleavage of a bacteriophage T7 processing signal. Divalent cation specificity, and specific anion effects. Nucleic Acids Res., 21, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipardi C., Wei,Q. and Paterson,B.M. (2001) RNAi as random degradative PCR: siRNA primers convert mRNA into dsRNAs that are degraded to generate new siRNAs. Cell, 107, 297–307. [DOI] [PubMed] [Google Scholar]
- Martens H., Novotny,J., Oberstrass,J., Steck,T.L., Postlethwait,P. and Nellen,W. (2002) RNAi in Dictyostelium: the role of RNA-directed RNA polymerases and double-stranded RNase. Mol. Biol. Cell, 13, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S., Ichigotani,Y., Okuda,T., Irimura,T., Nakatsugawa,S. and Hamaguchi,M. (2000) Molecular cloning and characterization of a novel human gene (HERNA) which encodes a putative RNA-helicase. Biochim. Biophys. Acta, 1490, 163–169. [DOI] [PubMed] [Google Scholar]
- Morse D.P., Aruscavage,P.J. and Bass,B.L. (2002) RNA hairpins in noncoding regions of human brain and C.elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl Acad. Sci. USA, 99, 7906–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson A.W. (1999) Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev., 23, 371–390. [DOI] [PubMed] [Google Scholar]
- Nicholson R.H. and Nicholson,A.W. (2002) Molecular characterization of a mouse cDNA encoding Dicer, a ribonuclease III ortholog involved in RNA interference. Mamm. Genome, 13, 67–73. [DOI] [PubMed] [Google Scholar]
- Nykänen A., Haley,B. and Zamore,P.D. (2001) ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell, 107, 309–321. [DOI] [PubMed] [Google Scholar]
- Paddison P.J., Caudy,A.A., Bernstein,E., Hannon,G.J. and Conklin,D.S. (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev., 16, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S., Fleenor,J., Xu,S., Mello,C. and Fire,A. (2000) Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol. Cell, 6, 1077–1087. [DOI] [PubMed] [Google Scholar]
- Provost P., Dishart,D., Doucet,J., Frendewey,D., Samuelsson,B. and Rådmark,O. (2002) Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J., 21, 5864–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S., Golden,T. and Ray,A. (1996) Maternal effects of the short integument mutation on embryo development in Arabidopsis. Dev. Biol., 180, 365–369. [DOI] [PubMed] [Google Scholar]
- Rotondo G. and Frendewey,D. (1996) Purification and characterization of the Pac1 ribonuclease of Schizosaccharomyces pombe. Nucleic Acids Res., 24, 2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz D.S. and Zamore,P.D. (2002) Why do miRNAs live in the miRNP? Genes Dev., 16, 1025–1031. [DOI] [PubMed] [Google Scholar]
- Sijen T., Fleenor,J., Simmer,F., Thijssen,K.L., Parrish,S., Timmons,L., Plasterk,R.H. and Fire,A. (2001) On the role of RNA amplification in dsRNA-triggered gene silencing. Cell, 107, 465–476. [DOI] [PubMed] [Google Scholar]
- Tuschl T., Zamore,P.D., Lehmann,R., Bartel,D.P. and Sharp,P.A. (1999) Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev., 13, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore P.D. (2001) RNA interference: listening to the sound of silence. Nat. Struct. Biol., 8, 746–750. [DOI] [PubMed] [Google Scholar]
- Zamore P.D., Tuschl,T., Sharp,P.A. and Bartel,D.P. (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell, 101, 25–33. [DOI] [PubMed] [Google Scholar]