

Abstract
L1 elements are ubiquitous human transposons that replicate via an RNA intermediate. We have reconstituted the initial stages of L1 element transposition in vitro. The reaction requires only the L1 ORF2 protein, L1 3′ RNA, a target DNA and appropriate buffer components. We detect branched molecules consisting of junctions between transposon 3′ end cDNA and the target DNA, resulting from priming at a nick in the target DNA. 5′ junctions of transposon cDNA and target DNA are also observed. The nicking and reverse transcription steps in the reaction can be uncoupled, as priming at pre-existing nicks and even double-strand breaks can occur. We find evidence for specific positioning of the L1 RNA with the ORF2 protein, probably mediated in part by the polyadenosine portion of L1 RNA. Polyguanosine, similar to a conserved region of the L1 3′ UTR, potently inhibits L1 endonuclease (L1 EN) activity. L1 EN activity is also repressed in the context of the full-length ORF2 protein, but it and a second cryptic nuclease activity are released by ORF2p proteolysis. Additionally, heterologous RNA species such as Alu element RNA and L1 transcripts with 3′ extensions are substrates for the reaction.
Keywords: genome evolution/human genome/retroelement/retrotransposon
Introduction
The completion of the human genome sequence has revealed the sheer abundance, diversity and importance of our transposons (Lander, 2001). Transposition is an ongoing process, actively changing the genome, occasionally for the worse (Kazazian and Moran, 1998; Ostertag and Kazazian, 2001a; Gilbert et al., 2002; Symer et al., 2002). L1 transposition has recently been suggested as a mechanism for exon shuffling (Moran et al., 1999). More passively, transposon sequences have been co-opted by the cell for a wide variety of functions including use as gene regulatory sequences and centromeric heterochromatin (Howard et al., 1995; Boeke and Stoye, 1997; Laurent et al., 1997). Due to their high copy number, these sequences are often substrates for homologous recombination and rearrangement (Meuth, 1989). Transposons are therefore a source of plasticity for the genome.
The element responsible for the vast majority of transposition in humans is the L1 retrotransposon (Figure 1A). The majority of L1s in the genome are 5′ truncated (Boissinot et al., 2001; Szak et al., 2002). As most truncated (and non-truncated) L1s are flanked by variable-length target site duplications, this process is typically thought to be due to a premature termination of reverse transcription rather than recombinational 5′ deletion. Elements that are full length contain both 5′ and 3′ UTRs and two non-overlapping open reading frames (ORFs). ORF1 has been shown to code for an RNA-binding protein specifically associated with L1 RNA (Hohjoh and Singer, 1997) and to form ribonucleoprotein particles with L1 RNA in vivo (Hohjoh and Singer, 1996). Recently, the murine L1 ORF1 protein was shown to have nucleic acid chaperone activity: ORF1 encouraged annealing of complementary sequences and promoted the formation of the most stable nucleic acid hybrid possible (Martin and Bushman, 2001). ORF2 encodes an endonuclease (L1 EN) that is required for retrotransposition (Feng et al., 1996; Moran et al., 1996). The nicking specificity of L1 EN mirrors the sequence at the sites of L1 insertion in vivo, and the biochemical requirements of its nucleic acid recognition have been investigated (Feng et al., 1996; Cost and Boeke, 1998). Briefly, the L1 EN is specific for DNA within a range of structural and sequence parameters, with minor groove width being of particular importance. The DNA sequence that best correlates with these requirements is TnAn, with nicking occurring mainly at the TpA and flanking phosphodiesters. A hotspot for L1 EN nicking occurs between the bla gene and the origin of replication on pBluescript, as this region contains many TnAn sequences (Feng et al., 1996). Nicking at such sequences is generally inhibited by chromatinization; interestingly however, cleavage of some non-consensus sites is enhanced (Cost et al., 2001). The ORF2-encoded L1 reverse transcriptase (RT) contains seven conserved domains and is significantly similar to the telomerase RT (Xiong and Eickbush, 1990; Eickbush, 1997). Following the RT domain is a cysteine-rich domain of unknown function. Interestingly, the proteins encoded by L1 elements work preferentially in cis, that is, preferentially on the RNA from which they were translated (Boeke, 1997; Esnault et al., 2000; Wei et al., 2001). Despite this preference, members of the Alu class of retroelements are believed to misappropriate L1 proteins in order to proliferate (Smit, 1996; Boeke, 1997; Esnault et al., 2000).

Fig. 1. (A) The human L1 retrotransposon. EN, endonuclease domain; RT, reverse transcriptase domain; ZN cysteine-rich domain; vTSD, variable target site duplication. The 5′ UTR contains an internal promoter (arrow); the 3′ UTR, a polyG and polyA sequence. (B) Protein purification. L1 ORF2p purification was analyzed by electrophoresis and western blotting and silver staining (right panel). T, total lysate; S, supernatant; P, pellet; F, column flow-through; W, wash; 1–9, 0.5 ml GSH elution fractions. (C) Reaction and detection scheme. Incubation of the reaction components results in formation of branched TPRT products. Branched molecules are detected by PCR with primers JB1179 and 1180, followed by Southern blotting with the JB2296 probe. (D) TPRT by L1 ORF2p. Lane 1, full reaction; lanes 2–7, full reaction less the indicated omission; lane 8, to ensure that the products observed in lane 1 were not the result of PCR-mediated target DNA–cDNA recombination, reactions 3 (containing cDNA but no target DNA) and 4 (containing target DNA but no cDNA) were mixed before PCR; lane 9, a full reaction, but with a large excess of AMV RT substituted for L1 ORF2p. The sizing standard used here and throughout is a MspI digest of pBR322, consisting of fragments of the following number of base pairs: 622, 527, 404, 307, 242, 238, 217, 201, 190, 180, 160, 147 and smaller fragments.
A model of the first steps of retrotransposition of the R2Bm element has been derived from the biochemical work of Luan and Eickbush (Luan et al., 1993). In the R2Bm model, called target-primed reverse transcription (TPRT), an element-encoded endonuclease nicks the target DNA, generating an exposed 3′ hydroxyl that serves as a primer for reverse transcription of the element’s RNA. The mechanism of second-strand synthesis and nick repair is unknown. The R2Bm and L1 elements are both non-LTR polyA transposons, but otherwise share little structural similarity (Malik et al., 1999). In contrast to the semi-specific apurinic/apyrimidinic (AP)-endonuclease-related L1 EN, the R2Bm endonuclease is a type IIs restriction-like enzyme with a CCHC motif, and is specifically targeted to a sequence in the insect rDNA (Yang et al., 1999). The R2Bm enzyme is C-terminal, located after the RT; the L1 EN is at the N-terminus, before the RT. In place of the endonuclease at the N-terminus of R2Bm is a consensus zinc-finger motif proposed to be involved in DNA binding (Yang et al., 1999). Additionally, the R2Bm element completely lacks an ORF1 protein. Given these substantial differences, our results were surprising: we found that the basic mechanism of transposition initiation is conserved, as the L1 ORF2 protein (ORF2p) can carry out a TPRT reaction. The reaction faithfully recapitulates many aspects of in vivo L1 transposition, and displays several mechanistically and evolutionarily interesting behaviors.
Results
An in vitro TPRT assay
To investigate the mechanism of L1 retrotransposition, we have reconstituted several steps of this reaction in vitro. The ORF2 protein of the highly active L1.3 retrotransposon was purified by affinity chromatography (Figure 1B), and assayed according to the scheme depicted in Figure 1C for its ability to initiate the transposition reaction. Definition of the TPRT model coupled with the discovery of an EN domain at the N-terminus of L1 ORF2p (Feng et al., 1996) suggested that an early step in the process of L1 transposition might be the synthesis of L1 cDNA utilizing a nick in the target plasmid to prime reverse transcription. When L1 ORF2 protein was incubated with L1.3 3′ end RNA and a suitable DNA target, L1 ORF2p produced a distribution of branched molecules as detected by PCR amplification (Figure 1D). Success of this reaction depended upon the presence of the L1 RNA and the target DNA (Figure 1D), as well as free deoxynucleotides and Mg2+ (data not shown). The size distribution of the branched molecules formed was dependent upon two variables: the position of the site of nicking on the target DNA and the length of the polyA tail on the transposed RNA (see Materials and methods). The minimum product length expected from the reaction was 174 bp, corresponding to an insertion in the target DNA exactly at the 3′ end of the JB1180 PCR primer. While the large majority of products exceeded this length, a small amount of shorter products were formed due to internal initiation of L1 ORF2p RT or by utilization of truncated RNAs. Most amplified TPRT products were 275–400 bp long, corresponding to cDNA insertion into the hotspot region of the plasmid. Although L1 ORF2p can produce a minimal amount of cDNA in the absence of a DNA target (data not shown), the material seen in lane 1 was not the result of artifactual cDNA/target DNA recombination during PCR, as it was not produced when cDNA and target DNA were mixed after the TPRT reaction but before PCR (Figure 1D, lane 8). Additionally, AMV RT was unable to substitute for L1 ORF2p in the TPRT reaction, even at high concentrations (Figure 1D, lane 9).
TPRT products resemble in vivo L1 insertions
Targeting of L1 transposition in vivo is not random. While multiple factors, including the accessibility of chromatin, may influence transposon insertion on a global scale, targeting of insertion at the nucleotide level is dictated by the specificity of the ORF2p EN domain (Feng et al., 1996; Jurka, 1997; Cost and Boeke, 1998; Cost et al., 2001). If the branched molecules detected in Figure 1 were authentic intermediates in L1 transposition, then the distribution of in vitro insertion sites should reflect the specificity of the L1 EN domain. PCR products generated in Figure 1D were cloned and sequenced. The sites of L1 cDNA insertion into the target DNA are mapped in Figure 2A, along with the major and minor sites of L1 EN nicking on this DNA sequence. In vitro TPRT exhibited extremely non-random targeting, with a large majority of the recovered cDNAs correctly targeted (Figure 2A and D).
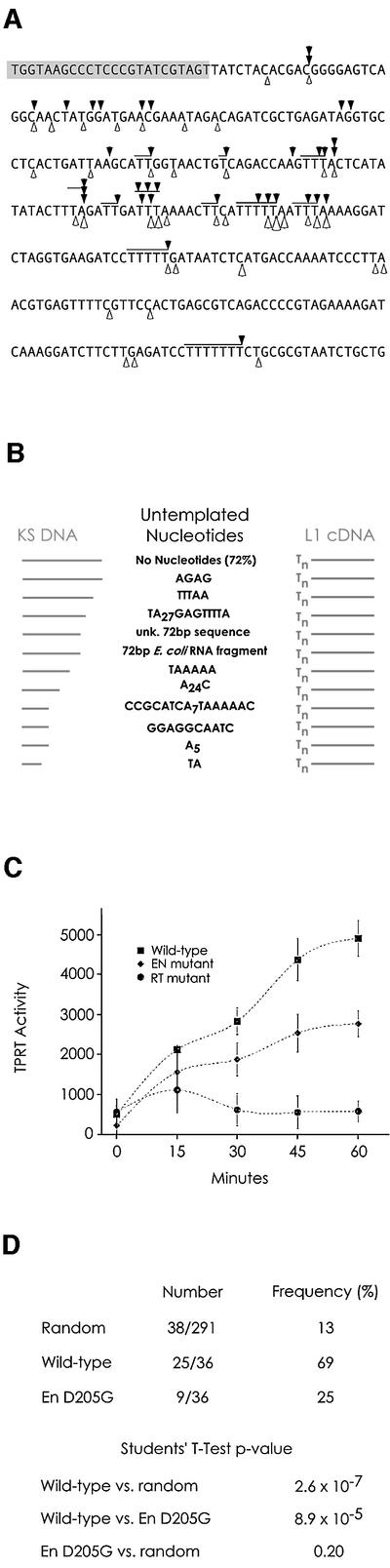
Fig. 2. (A) In vitro transposition insertions. L1 EN nicking sites, white arrowheads; observed L1 insertion sites, black arrowheads; JB1180 PCR primer, shaded nucleotides; ambiguity in the exact site of insertion due to microhomology between the polyT of the L1 cDNA and the target DNA, horizontal lines. (B) Untemplated nucleotides are sometimes found at transposon insertion sites. (C) Transposition activity of wild-type and mutant ORF2 proteins. Diamonds, wild-type ORF2p; squares, EN mutant ORF2p; triangles, RT mutant ORF2p. (D) Targeting of transposition. (A) contains 291 nt, 38 of which are defined as L1 target sites (see Materials and methods). Random insertion into this sequence would therefore yield an apparent targeting frequency of 13%. When wild-type L1 ORF2p was used, 25/36 L1 insertions were targeted, whereas only 9/36 insertions with EN mutant protein were. The complete set of sequenced L1 insertions exists as Supplementary information for this paper and is available from J.D.B. at http://www.bs.jhmi.edu/MBG/boekelab/boeke_lab_homepage.
In addition to retaining insertion site specificity, our TPRT assay also partially recapitulated another aspect of L1 biology. In vivo and in vitro, most L1 insertions contain L1 cDNA with a variable length polyT tail directly joined to the target DNA. However, short stretches of nucleotides, often simple repeats of high A–T content, were found between genomic L1 polyT tails and the target site duplication at a frequency of 13% (2092/15921) overall and 12% (56/479) for TA subset L1s (Szak et al., 2002). We found similar (presumably untemplated) nucleotides at the junction of the L1 cDNA and the target DNA in 28% (11/38) of in vitro insertions (Figure 2B). The extra nucleotides observed in vivo were mainly of the structure [TAAA(A)n]n; several of the extra-nucleotide sequences seen in our assay are similar to this type, but most are of apparently random sequence. While it is possible that some of these nucleotides came from aberrant extension of the RNA by T7 polymerase, we also saw such nucleotides in cDNAs from non-polyadenylated transcripts (data not shown). Additionally, extra-nucleotide addition by L1 RT has been observed with Ty1/L1 hybrid elements (Dombroski et al., 1994; Teng et al., 1996), with the R2Bm element (Luan and Eickbush, 1995) and with the Mauriceville plasmid RT (Chiang et al., 1994).
TPRT can occur at pre-formed nicks and breaks
L1 transposition in vivo is dependent upon the activities of both the EN and RT domains (Feng et al., 1996; Moran et al., 1996). When in vitro TPRT was attempted with protein containing an active site mutation in the RT domain sufficient to abolish all detectable RT activity, TPRT activity was undetectable (Figure 2C). Surprisingly, a similarly deleterious mutation of the EN domain resulted in the apparent retention of appreciable TPRT activity. However, rather than the targeted insertion seen with wild-type ORF2 protein, branched molecules recovered from reactions using EN mutant protein were much more randomly scattered across the target DNA (Figure 2D). This observation suggested that such molecules resulted from usage of spurious nicks generated by a low-level nicking activity found to be present in the reaction. Indeed, the use of pre-existing cellular nicks has been postulated to account for the existence of genomic L1 elements found in a sequence context inconsistent with L1 EN activity (Hutchison et al., 1989). We directly tested this hypothesis by assaying TPRT activity on DNA molecules pre-nicked at specific locations. When pGC89 plasmid previously nicked by various restriction enzymes was used as the target DNA (Figure 3A), TPRT activity was observed at the site of the nicks, both in the hotspot region (data not shown) and in a normally transposition-incompetent region (Figure 3B).

Fig. 3. L1 TPRT can utilize pre-existing 3′ hydroxyls for transposition. (A) Pre-nicking reaction scheme. pGC89 DNA was pre-nicked with various restriction enzymes outside of the nicking and transposition hotspot region of the plasmid, then used in the TPRT reaction. (B) TPRT products are produced at the pre-nicked sites. Predicted product sizes are 252, 268 and 285, for DraI (D1 on the figure), HindIII (H3) and HincII (H2), respectively. HindIII ‘star’ nicking activity results in a band at ∼250 nt. As the pGC89 substrate used in this experiment contains four DraI sites, only 1/4th of the nicks in the plasmid are at the assayed DraI site, reducing the intensity of the DraI band four-fold relative to the other enzymes; the other sites are unique. (C) Pre-digestion of target pBluescript KS–DNA into linear fragments. (D) Transposition products are produced via utilization of either a blunt-end DSB (DraI, D1, TTT/AAA), or a four base overhang (5′ overhang, BspHI, B1, T/CATGA, all BspHI sites are also NlaIII sites; 3′ overhang, NlaIII, N3, CATG/). The region of the plasmid assayed in this experiment is within the L1 ENp nicking and L1 ORF2p TPRT hotspot region of the plasmid. Predicted molecular weights of TPRT-derived PCR products: DraI, 303 bp; NlaIII, 361 bp; BspHI, 357 bp. A band from primer–L1 cDNA fusion is seen near the bottom of all lanes in this panel. The products of the normal TPRT reaction appear lighter in this gel only because the efficiency of transposition using DSBs necessitated a shorter exposure.
When the standard plasmid target DNA was digested with restriction enzymes (Figure 3C) and then used in the TPRT assay, a substantial amount of the total TPRT activity was directed to the ends of the DNA fragments (Figure 3D). Blunt-ended fragments were much better substrates than either 5′ or 3′ overhang fragments. Utilization of blunt-ended fragments as DNA targets allowed for TPRT in the absence of EN activity. As the ORF2 protein is capable of using 3′ hydroxyls found at nicks and double-strand breaks (DSBs) generated in trans, we conclude that the nicking and reverse transcription phases of the TPRT reaction can be uncoupled.
RNA requirements for reverse transcription
We observed that L1 ORF2p had precocious template switching tendencies in a conventional primer–template RT assay (Mathias et al., 1991), as there was a substantial size difference between the template RNA and the cDNA produced (Figure 4A). Such activity was also detected with reverse transcription of an A20 ribo-oligonucleotide template, with which the products of reverse transcription reached lengths of several hundred nucleotides (data not shown). Evidence of template switching was also obtained from the sequences of in vitro L1 transposon insertions (Figure 2B, lanes 5 and 6), in which the joining of at least two heterologous cDNAs to the L1 cDNA was observed.

Fig. 4. (A) In a homopolymer RT assay (polyA RNA, oligo-dT primer), L1 ORF2p produces RT products far in excess of the molecular weight of the template, indicative of template switching activity; AMV RT does not. (B) TPRT of various RNA species. The end-point of the L1 and Alu RNAs are indicated with the dotted line (not to scale). RNA number 4 has 38 nt of vector RNA after the polyA tail. URA3, fragment of S.cerevisiae URA3 RNA with and without a polyA tail. (C) Distribution of cDNA initiation points. Shown below each histogram is a full-length cDNA (from 3′ to 5′), with the positions of the reverse-transcribed polyG and polyA tracts indicated by ‘Cn’ and ‘TTTTTT’, respectively. Position zero marks the end of L1 sequence and the beginning of the polyA tail. The actual positions of the cDNA initiation are plotted in the histogram grouped into 10 nt bins. For the first cDNA (generated from RNA number 1 in B), many cDNAs end beyond the designed position at nucleotide 14 due to the addition of extra A residues to the RNA by T7 polymerase (see Materials and methods). Transposition of RNA number 4 (the second cDNA from the top) yields two distinct populations of cDNA initiation points. Mutation of either the polyG or the polyA sequence (histograms three and four, respectively) removes the bias towards internal initiation of cDNA synthesis, although only the polyA mutation results in a statistically significant difference. Student’s two-tailed t-test comparison of wild type versus mutant, bins –20–30 with bins 30–60; polyG p-value = 0.17; polyA p-value = 0.05. For all three cDNAs from 3′ extended transcripts, all initiation points in the 50–60 bin occurred at nucleotide 53, the end of the transcript. The following number of highly truncated (endpoint <–30) cDNAs were excluded from the statistical analysis: wt 3′ extended RNA, 3; polyG mutant, 4; polyA mutant, 3.
We carried out a limited deletion analysis of the 366 nt L1 3′ RNA in an effort to define any cis requirements for efficient TPRT (Figure 4B). Simple deletion of the polyA tail of the RNA (lane 2) had a modest effect on the efficiency of TPRT, whereas a more substantial 3′ truncation (lane 1) reduced the amount of TPRT products. Interestingly, a hybrid RNA containing L1 3′ RNA joined to 38 nt of vector sequence could be used as a substrate for TPRT at low levels (lane 4). We detected efficient TPRT of the Alu element RNA (lanes 5 and 6), a transposon long thought to utilize L1-encoded proteins for its mobilization. TPRT products from an irrelevant RNA were also formed at reasonable efficiency, however (lanes 7 and 8), suggesting that the molecular basis for the RNA selectivity of L1 is not likely to reside in the ORF2 protein (Esnault et al., 2000; Wei et al., 2001). Generally, a 3′ terminal polyA tail modestly stimulated TPRT activity of the transcripts.
In addition to assaying bulk TPRT activity with the different RNAs, analysis of discrete cDNAs produced by TPRT proved informative as well. Sequencing of the insertion events from the chimeric RNA number 4 (which has a 3′ extension of 38 nt after the polyA tail) revealed a bimodal distribution of L1 cDNA initiation points (Figure 4C). Excluding the three highly truncated insertions, about half (13/24) of the inserted cDNAs ended at or near the 3′ end of the RNA and half (11/24) in the vicinity of the polyT region. While L1 ORF2p RT can reverse transcribe the full length of 3′ extended transcripts, we infer from these results that reverse transcription of extended RNAs is often guided to begin internally within a window set by a cis-acting RNA sequence.
One possible candidate for this sequence is the evolutionarily conserved polyguanosine stretch (25/34 nt are Gs) in the L1 3′ UTR (Furano, 2000). We found a polyG RNA sequence to be a potent inhibitor of L1 EN activity (rG20 IC50 ≈ 50 nM), in contrast to a polyA sequence (rA20 IC50 ≈ 50 µM; Figure 5B). Under physiological conditions, the polyG stretch in the L1 3′ UTR can adopt a ‘G-quartet’ secondary structure (Howell and Usdin, 1997). Complete disruption of a similar structure in a polyI homopolymer by conversion from the sodium to the lithium salt (Figure 5C, see Materials and methods) had no effect on the inhibitory activity of this sequence (Figure 5D), suggesting that a quartet structure is not required for L1 EN inhibition.

Fig. 5. L1 EN is inhibited by RNA. (A) L1 EN nicking activity was assayed by following conversion of the quickly migrating supercoiled KS–plasmid to the slowly migrating open-circular form. (B) L1 EN nicking of a supercoiled plasmid was challenged with 10-fold dilutions (100 µM–100 nM; 100 µM–10 nM for G20) of the indicated RNA oligo. Unlike the related CCR4 nuclease (Chen et al., 2002), L1 EN has neither polyA-specific RNA exonucleolytic activity, nor any detectable nucleolytic activity on RNA (data not shown). (C) Quartet structures (as assayed by differential light absorbance and thermal melting) are disrupted when polyI is converted to a lithium salt. (D) Quartet structures are not required for inhibition of L1 EN nicking. Ten-fold dilutions (100 ng/µl–10 pg/ml); no difference was seen even at two-fold dilutions (data not shown). sc., supercoiled; oc., open circular.
Furthermore, we investigated whether the polyG or polyA tract was required to observe the bimodal cDNA endpoint distribution observed in Figure 4C. Despite its ability to markedly inhibit L1 EN activity, mutation of the endogenous polyG sequence within the L1 3′ UTR RNA only partially affected where cDNA was begun on 3′ extended transcripts (Figure 4C). In contrast, substitution of non-polyA sequence for the L1 polyA stretch in a 3′ extended RNA significantly altered the position of cDNA initiation such that the large majority (24/30) began near the 3′ end of the RNA (Figure 4C). We conclude that L1 ORF2p RT can recognize and initiate reverse transcription at internal polyA RNA sequences.
Second-strand L1 DNA synthesis
Completion of the transposition reaction requires a second round of TPRT. Molecules resulting from complete L1 insertions will therefore have a junction of the 5′ end of second-strand DNA to the target DNA at the site of the second nick (Figure 6A). A PCR assay (analogous to the one used to detect transposon 3′ end insertion in Figure 1C and D) revealed the existence of a population of such molecules in our in vitro reaction (Figure 6B). Like 3′ TPRT, the formation of second-strand L1 DNA was dependent upon the activity of the L1 RT and (to a lesser extent) EN domains (Figure 6B). Cloning and sequencing of these PCR products allowed for the site of 5′ end insertion to be determined for six independent 5′ end insertions (Figure 6C). Of the six, three contained a single extra thymidine, one was missing the 5′-most nucleotide of the L1 cDNA, one missing the first seven cDNA nucleotides, and one 5′ junction was neither missing nor had gained nucleotides. As L1 retrotransposition does not result in fixed spacing between the 5′ and 3′ TPRT reactions, the exact position of the 3′ end TPRT for these insertions is unknown.

Fig. 6. Creation of second-strand L1 cDNA. (A) The full L1 transposition reaction requires the utilization of two nicks in the target DNA. Arrows indicate the position of the primers (JB1180 and 2NP) used for PCR. (B) Five prime end insertion. Lane 1, wild-type ORF2 protein; lane 2, EN mutant ORF2p; lane 3, RT mutant ORF2p. (C) Black arrowheads indicate the position of target DNA–L1 5′ cDNA junctions.
L1 EN activity is repressed in the full-length ORF2 protein
In contrast to the robust RT activity present in full-length ORF2p, initial assays for L1 EN nicking revealed little EN activity (Figure 7B, lane 1; data not shown). The observation (in Figure 3) that pre-nicking or pre-breaking of the DNA target could stimulate TPRT with wild-type and EN mutant protein implied that nicking activity was in fact rate-limiting in the reaction. One possibility is that the conformation of the EN domain in the context of the full-length ORF2 protein renders it unable to efficiently nick DNA. We tested this hypothesis by examining the EN activity of proteolytic fragments of L1 ORF2p. Treatment of ORF2p with Factor Xa protease [a procedure originally intended to specifically remove the glutathione S-transferase (GST) affinity tag] resulted in the unexpected scission of the EN domain from the ORF2p, without appreciable release of the GST domain (Figure 7A). When ORF2 protein treated with the highest concentration of Factor Xa used in Figure 7A (Figure 7B, lanes 2, 5 and 8), or an excess of Factor Xa (complete EN proteolytic release, lanes 3, 6, 9 and 12) was assayed for nicking ability, activity was detected in a proteolysis-dependent manner. Similar results were obtained with a wide variety of less specific proteases (data not shown). The nicking activity released by EN cleavage had a DNA sequence specificity identical to that of the purified EN domain, as it cleaved at and near the TpA bond of a TnAn oligonucleotide (Cost and Boeke, 1998). Bona fide L1 EN activity is stimulated by the addition of DMSO to the reaction (Cost and Boeke, 1998; Figure 7C); the nicking activity released by Factor Xa proteolysis reacted similarly (lanes 11 and 12). When EN mutant ORF2 protein was proteolyzed, no EN activity was released. In contrast, proteolysis of RT mutant ORF2p resulted in activation of EN nicking. We conclude that the nicking activity released upon proteolysis of ORF2p was L1 EN activity, and that L1 EN is negatively regulated in the context of the full-length ORF2 protein.

Fig. 7. L1 EN activity is masked in full-length ORF2 protein. (A) Treatment of L1 ORF2p with Factor Xa results in removal of the EN domain, as monitored by immunoblot with a L1 EN antibody. The amounts of Factor Xa used are as follows: lane 1, zero; lane 2, 0.0625 µg; lane 3, 0.25 µg; lane 4, 1 µg. (B) Proteolyzed ORF2p exhibits L1 EN activity on a double-stranded, 5′ end-labelled DNA target. The position of the TpA bond is indicated. The final concentration of DMSO added in lanes 11 and 12 was 25%. Factor Xa: lanes 1, 4, 7 and 11, zero; lanes 2, 5 and 8, 1 µg; lanes 3, 6, 9 and 12, 3 µg. (C) L1 EN activity is increased and full-length L1 ORF2p RT activity is decreased by DMSO addition. The graph for L1 EN is a quantitation and plot of the gel in Cost and Boeke (1998), figure 6a.
Surprisingly, a second cryptic nuclease activity was also released from the ORF2 protein upon proteolysis, resulting in a reduction to 5′ terminal mono- and dinucleotides of a substantial fraction of the input DNA (Figure 7B, lanes 1–3). Consistent with this nuclease activity residing in the RT portion of ORF2p, it was absent from RT mutant proteolyzed ORF2p (lanes 4–6), and like RT activity, was repressed by the addition of DMSO (Figure 7B, lanes 11 and 12, and Figure 7C). Interestingly, however, this nuclease activity required a wild-type EN domain (Figure 7B, lanes 7–9).
Discussion
Given the extensive similarities between the products of our in vitro reaction and the products of L1 retrotransposition in vivo, we infer that the double-ended TPRT reaction reported here is in fact L1 retrotransposition. As such, this system provides the first direct evidence that the human L1 element uses a TPRT mechanism for retrotransposition (Figure 8). By extension, it is likely that this mechanism applies to the numerous retroelements found in diverse lineages bearing an AP-like endonuclease.

Fig. 8. The L1 TPRT model. (A) The polyA sequence positions the L1 RNA for reverse transcription; the polyG sequence could inhibit L1 EN activity. Competitive inhibition is shown for simplicity, but non-competitive inhibition could also be possible. (B) The polyG RNA may be removed from the L1 EN domain, perhaps by DNA binding or action of L1 ORF1 (Martin et al., 2000; Martin and Bushman, 2001), and L1 EN nicks the chromosome at its consensus site. (C) The nicked DNA moves to the RT active site and the newly generated 3′ hydroxyl primes reverse transcription.
The overall inefficiency of the TPRT reaction suggests that other factors may be necessary for robust transposition in vivo. Histones or other chromosome-associated proteins may well serve as cofactors for the reaction. Addition of L1 ORF1 protein to the reaction mix might also increase the efficiency of TPRT. Interestingly, the mouse L1 ORF1 protein has recently been shown to have an RNA chaperone activity, postulated to be involved in formation of a L1 polyA:chromosomal polyT RNA:DNA hybrid at the transposon insertion site (Martin and Bushman, 2001). A clear requirement of this model is complementarity between the transposon RNA and the DNA target. While we find no such requirement, ORF1p-mediated RNA:DNA hybrid formation could be required for high-efficiency TPRT. Alternately, ORF1p chaperone activity may be involved in mediating a dynamic ORF2p–RNA interaction such as proposed in our model (Figure 8A and B), or could be required for attainment of a specific requisite RNA secondary structure. Attempts were made to functionally complement the latter activity by thermal denaturation of L1 RNA followed by slow cooling. Such manipulation had no effect on TPRT activity (data not shown). Also possible is that cotranslational RNA:protein positioning (or similar post-translational heat shock protein-promoted associations) is required for highly efficient transposition; such a requirement has been found for human telomerase (Holt et al., 1999).
Remarkably, we found that the ORF2 protein can utilize the 3′ hydroxyls of pre-existing nicks to initiate reverse transcription. This activity could explain the origin of those L1 elements inserted into local sequence environments that lack features associated with good L1 EN cleavage sites (e.g. GC rich regions). Consistent with this in vitro result, an EN-mutant L1 element was shown to be capable of transposition in non-homologous end-joining-deficient cell lines (containing high levels of steady-state DNA damage due to mutations in either XRCC4 or DNA-PKcs) (Morrish et al., 2002). The in vitro usage of DSBs by the EN mutant ORF2p was somewhat weaker than the wild-type protein. We note that Morrish et al. (2002) report a similar result, using a slightly different mutation than ours (D205A versus our D205G), in that the EN-mutant L1 achieved 76% of wild-type activity in V3 cells (DNA-PKcs mutant) and 89% in XR-1 cells (XRCC4 mutant). Using a second EN domain mutation (H230A), the values were 28 and 52%, respectively. Perhaps even in the absence of EN catalysis, the EN domain is required for substrate DNA recognition; the mutant proteins may be partially defective in such recognition.
The 3′ hydroxyls at DSBs are quite readily used for the priming of reverse transcription. In particular, we observed that the blunt-ended DSB was able to be used more efficiently than either the 5′ or 3′ overhang-containing ends. While it is possible that this difference is simply a function of the sequence present at the site of the break, given the apparent absence of a local sequence bias in in vivo EN-independent insertions (Morrish et al., 2002) and the fact that the 3′ terminal nucleotide is the same in both the blunt-end and 5′ overhang DSB (the best and least well used, respectively), we suspect that this bias genuinely reflects the substrate preferences of the transposition reaction.
LTR retrotransposon Pol proteins require proteolytic processing for activity. While there are multiple mechanisms capable of explaining our observation that EN activity is released upon ORF2p proteolysis, the possibility that L1 ORF2p is normally proteolyzed in vivo must be considered. The proteolysis experiment revealed an unexpected second nuclease activity that requires extensive further investigation. This activity may be an endonucleolytic activity of the RT domain, similar to that described for telomerase and RNA polymerase (Collins and Greider, 1993; Kassavetis and Geiduschek, 1993). Oddly, however, an inactivating mutation in the EN domain abolishes this nuclease activity (without affecting the RT activity of the EN mutant protein as assayed by the homopolymer assay). While the mechanistic basis of this observation remains unclear, this nuclease activity must require some form of EN–RT domain cooperation.
L1 RT’s propensity to fall off of or switch its template may play a role in generating the many internally rearranged and deleted L1 transposons found in the genome as well as some processed pseudogenes (Ostertag and Kazazian, 2001b). Similar chimeric products have been observed during L1 pseudogene formation (Wei et al., 2001), EN-independent retrotransposition (Morrish et al., 2002), TPRT of the R2Bm element (Bibillo and Eickbush, 2002) and L1 RT-mediated DSB repair in yeast (Teng et al., 1996). RT disassociation followed by a shift in the register between the L1 RNA and the cDNA, followed by resumption of reverse transcription (an intramolecular template switch) could lead to a telomerase-like expansion of the polyA tail during transposition. Untemplated nucleotide addition (Figure 2B) followed by intramolecular template switching may be responsible for the generation of the short simple T(A)3–4 repeats occasionally found at the 3′ end of L1s, as the analogous repeats of the I-factor and unagi transposons undergo retrotranspositionally dependent expansion and contraction (Chaboissier et al., 2000; M.Kajikawa and N.Okada, personal communication). In contrast to the extra nucleotides observed at the 3′ junction (Figure 2B), the 5′ terminal modifications are substantially shorter and simpler, perhaps reflecting an RT more securely tethered to the DNA, and/or one unable to template switch due to the absence of a homopolymer template.
The L1 ORF2 protein is able to transpose L1 RNA with a 3′ extension of vector RNA after the polyA tail, an observation that supports the demonstration of L1- mediated 3′ transduction in cultured cells (Moran et al., 1999). In more than half of recovered in vitro transpositions with a 3′ extended RNA, reverse transcription initiated near the end of the L1 sequence rather than at the end of the chimeric RNA, suggesting that L1 may be able to compensate for inherently inefficient transcriptional termination (Eickbush, 1999) by internal initiation of reverse transcription. The L1 3′ transduction in vivo may be limited by such internal initiation events.
Our data suggest that this internal initiation is the result of L1 RT’s recognition of polyA RNA. While an enticing model, given hypotheses of L1/Alu transposition (Boeke, 1997), two considerations suggest that this specificity determinant is not sufficient in vivo to avoid high frequency 3′ transduction. In vivo 3′ extended transcripts are likely to be terminally polyadenylated, creating a competition for L1 ORF2p polyA binding between the internal and 3′ terminal sequence. Secondly, Wei et al. observed that polyadenylated retrotransposition-defective L1 RNA is retrotransposed in trans more efficiently than polyadenylated non-L1 RNAs, suggestive of a specific L1 RNA–protein interaction (Wei et al., 2001). The unagi non-LTR retrotransposon RT specifically recognizes a stem–loop structure in the unagi 3′ RNA (M.Kajikawa and N.Okada, personal communication); the R2Bm retrotransposon protein recognizes a structure in the R2Bm 3′ UTR (Luan and Eickbush, 1995; Mathews et al., 1997). Both L1 RNAs 1 and 2 in Figure 4B contain deletions that remove thermodynamically plausible stem–loops from the L1 3′ UTR (data not shown). Further mutagenesis will be required to dissect this sequence.
The polyG specificity of RNA-mediated L1 EN inhibition is remarkable. L1 RNA has two prominent G-rich regions: the 3′ UTR polyG sequence and the first 19 nt of the 5′ UTR (11 of which are G). Because L1 RNA with a large 3′ UTR deletion that removes the polyG sequence can undergo transposition (Moran et al., 1996), this sequence is not absolutely required for the TPRT reaction. A caveat to this conclusion is that the SV40 promoter inserted into the L1 3′ UTR in this mutant has many G-rich stretches that could potentially functionally substitute for the native polyG stretch. Even in the absence of a required role in transposition, the 3′ UTR polyG might aid in regulation of L1 EN activity or transposition in vivo, as shown in Figure 8A. Alternatively, if the L1 integration complex contains a dimer of L1 ORF2 protein, the 5′ polyG sequence could inhibit the EN activity of the second ORF2 molecule, perhaps delaying the creation of the second chromosomal nick until RT reaches the 5′ end of the transcript, displacing the 5′ polyG RNA. Such a sequential nicking mechanism might be advantageous, as it would postpone potentially dangerous DSB creation until after the completion of reverse transcription.
The 11% of the genome that consists of Alu element DNA has long been thought to result from transposition by L1 machinery; our observation of significant TPRT of Alu templates is consistent with this hypothesis. However, given ORF2p’s ability to transpose an irrelevant RNA, the step at which Alu RNA competes with L1 RNA for access to the transposition machinery must occur before reverse transcription. Template switching from L1 to Alu RNA during reverse transcription of the polyA tail could provide an additional opportunity for Alu to subvert L1 replication. Competitive template switching as a mechanism for Alu transposition would be molecularly ‘silent’, as the polyT cDNA derived from the L1 RNA would be indistinguishable from Alu RNA derived sequence.
Materials and methods
Protein purification
GST was fused to the C-terminus of the ORF2 protein from the L1.3 (Sassaman et al., 1997) retrotransposon, incorporated into the pro-baculoviral genome and expressed in Hi-Five Trichoplusia ni cells using the Bac-to-Bac expression system (Life Technologies). 108 cells were infected at a m.o.i. of 0.1–1.0 for 3 days, and then pelleted and resuspended in 10 ml ice cold buffer A [150 mM KCl, 50 mM Tris–HCl pH 7.5, 10% glycerol, 5 mM dithiothreitol (DTT), 1% Triton X-100 and 1 mM PMSF]. Cells were lysed by sonication and the debris was pelleted at 2500 g for 10 min. The lysate was bound to 0.5 ml GSH–Sepharose for 16 h at 4°C. The resin was applied to a column and washed with 20 vols of buffer A, 20 vols of buffer A plus 250 mM KCl, and eluted with 5 vols of buffer A plus 250 mM KCl plus 10 mM GSH. The eluate was dialyzed and concentrated against buffer A. This procedure yielded highly pure protein (>95%, excluding free GST). Mutant versions of L1.3 ORF2p (EN-, D205G; RT-, D702Y) were prepared in parallel. Immunoblotting was performed using a mouse antibody directed against the L1 EN domain.
TPRT reactions
Components of the TPRT reaction included: ∼1 ng (6 fmol) L1.3 ORF2–GST protein, 40 ng (20 fmol) supercoiled pBluescript KS–, 60 ng (460 fmol) L1.3 3′ RNA, 10 U RNAsin (Promega), 100 µM each dNTP, 50 mM HEPES pH 8.0, 50 mM KGlu, 5 mM Mg(OAc)2 and 10 mM DTT. Nick priming reactions (20 µl) were incubated at 37°C for 2 h then heat inactivated at 68°C for 20 min. One microlitre of reaction mix was amplified by PCR for 30 cycles of 94, 65 and 72°C (30 s each), with an initial denaturation at 94°C for 4 min and a final extension at 72°C for 7 min. PCR reactions contained 200 µM each dNTP, 1.5 mM MgCl2, 50 mM KCl, 10 mM Tris pH 8.3, 2.5 U of Amplitaq polymerase (Perkin Elmer) and 0.5 µM of each oligo.
Oligonucleotides
JB1179 (5′-GGGGAGGGATAGCATTGGGAGATA-3′, identical to nucleotides 5887–5910 of the L1.3 sequence) and JB1180 (5′-TGG TAAGCCCTCCCGTATCGTAGT-3′, the complement of nucleotides 2061–2084 of the pBluescript KS– sequence) were used to PCR amplify TPRT products in Figures 1D, 2, 3D and 4B, lanes 1–4. PCR was performed with JB1180 in combination with JB3800 (5′-ATAGCA TTGGGAGATATACCTAAT-3′, identical to nucleotides 5895–5918 of the L1.3 sequence) for Figure 4C (as JB1179 overlaps the polyG region). Oligo 2NP (5′-CTGAGAATGATGGTTTCCAATTTC-3′, the complement of nucleotides 5712–5732 of the L1.3 sequence), was used in combination with JB1180 to amplify the 5′ junctions between the target DNA–L1 cDNA in Figure 6B. The T7 primer (5′-GTAATACGA CTCACTATAGGG-3′, identical to nucleotides 625–645 of the pGC89 sequence) was used in combination with JB1179 to amplify TPRT products at pre-nicked sites in pGC89 (Figure 3B). The PCR reactions were separated using 5% polyacrylamide gels in 1× TTE, then electrotransferred to Genescreen membrane (DuPont) in 0.5× TBE, and probed with 5′ [32P]end-labeled JB2296 (5′-AGCATGGCACAT GTATAC-3′, identical to nucleotides 5951–5968 of the L1.3 sequence) for all blots except Figures 4B and 6B; JB2296′ (5′-CACCAGC ATGGCACATGT-3′, identical to nucleotides 5947–5964 of the L1.3 sequence) for Figure 4B (as the last four nucleotides of the JB2296 are deleted in RNA number 1), and 2NPR (5′-ATCCATGTCCCTACAAAG GAT-3′) for Figure 6B. Oligonucleotides JB2297 and 3918 were used to amplify Alu and URA3 TPRT products respectively. Quantitation of TPRT products was performed in quintuplicate by dot–blot hybridization of the PCR products followed by phosphoimager analysis.
RNAs
L1 3′ RNA was produced by in vitro T7 polymerase-driven transcription of nucleotides 5672–6036 of L1.3 from pQF325 linearized with BsaI. T7 transcription was performed for 1.5 h in 50 µl of buffer containing 2 µg DNA template, 40 mM Tris pH 7.5, 20 mM MgCl2, 50 µg/ml bovine serum albumin, 500 µM each NTP, 10 mM DTT and 100 U T7 polymerase (Life Technologies) and 40 U of RNAsin (Promega). Although designed to produce a transcript with a 14 nt polyadenylate tail, >50% of the transcripts contained polyA tails of 14–75 nt, as seen previously with transcripts ending in A (Milligan and Uhlenbeck, 1989; Luan et al., 1993). RNAs corresponding to the L1 3′ end without a terminal polyA tail (pQF325 cut with AccI or RsaI), L1 3′ RNA with additional 3′ nucleotides (pQF325 cut with MseI), the tPA-25 Alu (Ludwig et al., 1992) (pQF155 cut with NsiI) and tPA-25 Alu without a polyA tail (pQF155 cut with BstNI) were generated by analogous means. The Saccharomyces cerevisiae URA3 RNA fragments were generated by T7 transcription of PCR amplicons of the URA3 gene generated with an identical 5′ primer (5′-ACTGTAATACGACTCACTATAGGGCAC ACGGTGTGGTGGGCCC-3′) and either 5′-CTCAAATATCGTTC CCAG-3′ or 5′-TTTTTTTTTTTTTTCTCAAATATGCTTCCCAG-3′. In the transcription construct pGC114, the 34 nt polyG region (5861–5894) was replaced with 5′-ATCTCATTCATCCTATGC ATCCTCAACAGCTCCA-3′. In pGC115, the 18 nt polyA sequence (6020–6037, including the last T at position 6022) was replaced with 5′-TCGTTATGCATTCGTCTT-3′. All pQF325 derived RNAs contain 47 nt of vector-derived RNA at the 5′ end.
EN nicking
Sites of EN nicking on the hotspot region of Bluescript (Figure 2A, white arrowheads) were mapped by comparison of an EN nicked end-labeled PCR product generated with JB1180 and JB3462 (5′-AGTGGA ACGAAAACTCACGT-3′) and a DMSO/piperidine ladder. EN nicking of supercoiled plasmids was performed as described previously (Cost and Boeke, 1998), with the addition of the appropriate concentration of RNA oligo, N20 (Dharmacon). Quartet dependence experiments were performed in potassium containing buffer with polyI (Sigma; average length, 190 nt) instead of polyG, as polyG quartets melt at >100°C and are indifferent as to the coordinated metal ion (Howard and Miles, 1982). PolyI is equally inhibitory as polyG (data not shown). Metal ion exchange was performed by extensive dialysis against LiCl then water, and assayed by measurement of light absorbance at 247 nm as a function of temperature (Howard and Miles, 1982).
Plasmid construction and site-specific nicking
pGC89 was made by ligation of JB1457 (5′-GCCCGGTTTTTTA AAAAAGGCCCG-3′) and its annealed complement JB1458 into the EcoRV site of pBluescript KS–. Site-specifically nicked pGC89 was produced by preparative electrophoretic separation of nicked plasmids generated by reaction of 200 ng supercoiled pGC89, 1 U of either DraI, HindIII or HincII, and ethidium bromide at final concentrations of 100, 50 or 12.5 µg/ml, respectively. Some nicking outside of the restriction enzyme consensus site was observed with all three enzymes (data not shown). We chose to examine TRPT using pre-existing nicks in this region of the plasmid because there is normally very little TPRT activity in this region, probably due to its high GC content.
RT assay
RT activity was assayed by incubation of 10 µg/ml polyA RNA (average length = ∼200 nt; Pharmacia), 0.7 µg/ml dT12–18, 10 mM MgCl2, 0.2 µM dTTP, 50 mM Tris pH 8.0, 275 mM β-mercaptoethanol and a trace amount of [32P]α-dTTP. After 1.5 h, reactions were separated using denaturing polyacrylamide gels.
Proteolysis of ORF2p
Proteolysis of ∼5 ng of ORF2p was carried out for 2 h at 37°C in 60 mM NaCl, 50 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM CaCl2, with Factor Xa (Boehringer Mannheim) as indicated in the legend to Figure 6. Nicking was assayed as in Cost and Boeke (1998).
Acknowledgments
Acknowledgements
We thank all members of the Boeke lab for insightful and productive discussions, John Moran and Norihiro Okada for communicating data prior to publication, Nick Cozzarelli for partial financial support of G.C., and Haig Kazazian and Elena Kouvabina for encouragement. We are grateful to Yolanda Eby and Laurance Decourty for excellent technical assistance, and to Paul Miller and Tomoko Hamma for use of their Varian Cary 3E spectrophotometer. This work was supported by NIH grant CA16519.
References
- Bibillo A. and Eickbush,T.H. (2002) The reverse transcriptase of the R2 non-LTR retrotransposon: continuous synthesis of cDNA on non-continuous RNA templates. J. Mol. Biol., 316, 459–473. [DOI] [PubMed] [Google Scholar]
- Boeke J.D. (1997) LINEs and Alus: the polyA connection. Nat. Genet., 16, 6–7. [DOI] [PubMed] [Google Scholar]
- Boeke J.D. and Stoye,J.P. (1997) Retrotransposons, endogenous retro viruses and the evolution of retroelements. In Coffin,J.M., Hughes,S.H. and Varmus,H.E. (eds), Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 343–435. [PubMed]
- Boissinot S., Entezam,A. and Furano,A.V. (2001) Selection against deleterious line-1-containing loci in the human lineage. Mol. Biol. Evol., 18, 926–935. [DOI] [PubMed] [Google Scholar]
- Chaboissier M.C., Finnegan,D. and Bucheton,A. (2000) Retrotrans position of the I factor, a non-long terminal repeat retrotransposon of Drosophila, generates tandem repeats at the 3′ end. Nucleic Acids Res., 28, 2467–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Chiang,Y.C. and Denis,C. (2002) CCR4, a 3′–5′ poly(A) RNA and ssDNA exonuclease, is the catalytic component of the cytoplasmic deadenylase. EMBO J., 21, 1414–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C.C., Kennell,J.C., Wanner,L.A. and Lambowitz,A.M. (1994) A mitochondrial retroplasmid integrates into mitochondrial DNA by a novel mechanism involving the synthesis of a hybrid cDNA and homologous recombination. Mol. Cell. Biol., 14, 6419–6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K. and Greider,C.W. (1993) Tetrahymena telomerase catalyzes nucleolytic cleavage and nonprocessive elongation. Genes Dev., 7, 1364–1376. [DOI] [PubMed] [Google Scholar]
- Cost G.J. and Boeke,J.D. (1998) Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry, 37, 18081–18093. [DOI] [PubMed] [Google Scholar]
- Cost G.J., Golding,A., Schlissel,M.S. and Boeke,J.D. (2001) Target DNA chromatinization modulates nicking by L1 endonuclease. Nucleic Acids Res., 29, 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombroski B.A., Feng,Q., Mathias,S.L., Sassaman,D.M., Scott,A.F., Kazazian,H.H. and Boeke,J.D. (1994) An in vivo assay for the reverse transcriptase of human retrotransposon L1 in Saccharomyces cerevisiae. Mol. Cell. Biol., 14, 4485–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickbush T.H. (1997) Telomerase and retrotransposons: which came first? Science, 277, 911–912. [DOI] [PubMed] [Google Scholar]
- Eickbush T. (1999) Exon shuffling in retrospect. Science, 283, 1465–1467. [DOI] [PubMed] [Google Scholar]
- Esnault C., Maestre,J. and Heidmann,T. (2000) Human LINE retrotransposons generate processed pseudogenes. Nat. Genet., 24, 363–367. [DOI] [PubMed] [Google Scholar]
- Feng Q., Moran,J., Kazazian,H. and Boeke,J.D. (1996) Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell, 87, 905–916. [DOI] [PubMed] [Google Scholar]
- Furano A.V. (2000) The biological properties and evolutionary dynamics of mammalian LINE-1 retrotransposons. Prog. Nucleic Acid Res. Mol. Biol., 64, 255–294. [DOI] [PubMed] [Google Scholar]
- Gilbert N., Prigge,S.L. and Moran,J.V. (2002) Genomic deletions created upon LINE-1 retrotransposition. Cell, 110, 315–325. [DOI] [PubMed] [Google Scholar]
- Hohjoh H. and Singer,M.F. (1996) Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J., 15, 630–639. [PMC free article] [PubMed] [Google Scholar]
- Hohjoh H. and Singer,M.F. (1997) Sequence-specific single strand RNA binding protein encoded by the human LINE-1 retrotransposon. EMBO J., 16, 6034–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt S.E. et al. (1999) Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev., 13, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard F.B. and Miles,H.T. (1982) Poly(inosinic acid) helices: essential chelation of alkali metal ions in the axial channel. Biochemistry, 21, 6736–6745. [DOI] [PubMed] [Google Scholar]
- Howard B.H., Russanova,V.R. and Englander,E.W. (1995) Alu silencing mechanisms: implications for the modulation of local chromatin structure. In Maria,R.J. (ed.), The Impact of Short INterspersed Elements (SINEs) on the Host Genome. R.G.Landes, Austin, TX, pp. 133–141.
- Howell R. and Usdin,K. (1997) The ability to form intrastrand tetraplexes is an evolutionarily conserved feature of the 3′ end of L1 retrotransposons. Mol. Biol. Evol., 14, 144–155. [DOI] [PubMed] [Google Scholar]
- Hutchison C.A. III, Hardies,S.C., Loeb,D.D., Shehee,W.R. and Edgell,M.H. (1989) LINEs and related retroposons: long interspersed repeated sequences in the eucaryotic genome. In Berg,D.E. and Howe,M.M. (eds), Mobile DNA. American Society of Microbiology, Washington, DC, pp. 593–617.
- Jurka J. (1997) Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl Acad. Sci. USA, 94, 1872–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassavetis G.A. and Geiduschek,E.P. (1993) RNA polymerase marching backward. Science, 259, 944–945. [DOI] [PubMed] [Google Scholar]
- Kazazian H.H. Jr and Moran,J.V. (1998) The impact of L1 retrotransposons on the human genome. Nat. Genet., 19, 19–24. [DOI] [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Laurent A.M., Puechberty,J., Prades,C., Gimenez,S. and Roizes,G. (1997) Site-specific retrotransposition of L1 elements within human alphoid satellite sequences. Genomics, 46, 127–132. [DOI] [PubMed] [Google Scholar]
- Luan D.D. and Eickbush,T.H. (1995) RNA template requirements for target DNA-primed reverse transcription by the R2 retrotransposable element. Mol. Cell. Biol., 15, 3882–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan D.D., Korman,M.H., Jakubczak,J.L. and Eickbush,T.H. (1993) Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell, 72, 595–605. [DOI] [PubMed] [Google Scholar]
- Ludwig M., Wohn,K.D., Schleuning,W.D. and Olek,K. (1992) Allelic dimorphism in the human tissue-type plasminogen activator (TPA) gene as a result of an Alu insertion/deletion event. Hum. Genet., 88, 388–392. [DOI] [PubMed] [Google Scholar]
- Malik H.S., Burke,W.D. and Eickbush,T.H. (1999) The age and evolution of non-LTR retrotransposable elements. Mol. Biol. Evol., 16, 793–805. [DOI] [PubMed] [Google Scholar]
- Martin S.L. and Bushman,F.D. (2001) Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol. Cell. Biol., 21, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.L., Li,J. and Weisz,J.A. (2000) Deletion analysis defines distinct functional domains for protein–protein and nucleic acid interactions in the ORF1 protein of mouse LINE-1. J. Mol. Biol., 304, 11–20. [DOI] [PubMed] [Google Scholar]
- Mathews D.H., Banerjee,A.R., Luan,D.D., Eickbush,T.H. and Turner,D.H. (1997) Secondary structure model of the RNA recognized by the reverse transcriptase from the R2 retrotransposable element. RNA, 3, 1–16. [PMC free article] [PubMed] [Google Scholar]
- Mathias S.L., Scott,A.F., Kazazian,H.H.,Jr, Boeke,J.D. and Gabriel,A. (1991) Reverse transcriptase encoded by a human retrotransposon. Science, 254, 1808–1810. [DOI] [PubMed] [Google Scholar]
- Meuth M. (1989) Illegitimate recombination in mammalian cells. In Berg,D.E. and Howe,M.M. (eds), Mobile DNA. American Society of Microbiology, Washington, DC, pp. 833–860.
- Milligan J.F. and Uhlenbeck,O.C. (1989) Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol., 180, 51–62. [DOI] [PubMed] [Google Scholar]
- Moran J.V., Holmes,S.E., Naas,T.P., DeBerardinis,R.J., Boeke,J.D. and Kazazian,H.H.,Jr (1996) High frequency retrotransposition in cultured mammalian cells. Cell, 87, 917–927. [DOI] [PubMed] [Google Scholar]
- Moran J.V., DeBerardinis,R.J. and Kazazian,H.H.,Jr (1999) Exon shuffling by L1 retrotransposition. Science, 283, 1530–1534. [DOI] [PubMed] [Google Scholar]
- Morrish T.A., Gilbert,N., Myers,J.S., Vincent,B.J., Stamato,T.D., Taccioli,G.E., Batzer,M.A. and Moran,J.V. (2002) DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet., 31, 159–165. [DOI] [PubMed] [Google Scholar]
- Ostertag E.M. and Kazazian,H.H.,Jr (2001a) Biology of mammalian retrotransposons. Ann. Rev. Genet., 35, 501–538. [DOI] [PubMed] [Google Scholar]
- Ostertag E.M. and Kazazian,H.H.,Jr (2001b) Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res., 11, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassaman D.M., Dombroski,B.A., Moran,J.V., Kimberland,M.L., Naas,T.P., DeBerardinis,R.J., Gabriel,A., Swergold,G.D. and Kazazian,H.H.,Jr (1997) Many human L1 elements are capable of retrotransposition. Nat. Genet., 16, 37–43. [DOI] [PubMed] [Google Scholar]
- Smit A.F.A. (1996) The origin of interspersed repeats in the human genome. Curr. Opin. Genet. Dev., 6, 743–748. [DOI] [PubMed] [Google Scholar]
- Symer D.E., Connelly,C., Szak,S.T., Caputo,E.M., Cost,G.J., Parmigiani,G. and Boeke,J.D. (2002) Human L1 retrotransposition is associated with genetic instability in vivo. Cell, 110, 327–338. [DOI] [PubMed] [Google Scholar]
- Szak S.T., Pickeral,O.K., Makalowski,W., Boguski,M.S., Landsman,D. and Boeke,J.D. (2002) Molecular archeology of L1 insertions in the human genome. Genome Biol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng S.C., Kim,B. and Gabriel,A. (1996) Retrotransposon reverse-transcriptase-mediated repair of chromosomal breaks. Nature, 383, 641–644. [DOI] [PubMed] [Google Scholar]
- Wei W., Gilbert,N., Ooi,S.L., Lawler,J.F., Ostertag,E.M., Kazazian,H.H., Boeke,J.D. and Moran,J.V. (2001) Human L1 retrotransposition: cis preference versus trans complementation. Mol. Cell. Biol., 21, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y. and Eickbush,T.H. (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J., 9, 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Malik,H.S. and Eickbush,T.H. (1999) Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc. Natl Acad. Sci. USA, 96, 7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]