

Abstract
We analyzed 8.55 million LongSAGE tags generated from 72 libraries. Each LongSAGE library was prepared from a different mouse tissue. Analysis of the data revealed extensive overlap with existing gene data sets and evidence for the existence of ≈24,000 previously undescribed genomic loci. The visual cortex, pancreas, mammary gland, preimplantation embryo, and placenta contain the largest number of differentially expressed transcripts, 25% of which are previously undescribed loci.
Keywords: alternative transcripts, development, serial analysis of gene expression
The laboratory mouse has emerged as a premiere model system for studies of mammalian development and disease. A major obstacle to realizing the full potential of the mouse in these studies is the lack of detailed information on the function of the majority of mouse genes. Gleaning such information will occupy biologists for years to come, but significant acceleration of such efforts can be achieved through systematically identifying the genes expressed in precisely defined cells and tissues at numerous developmental stages. To be of broad general use, these efforts should initially emphasize wild-type animals, be available to the scientific community in a format that is easily analyzed and readily distributed, remain applicable as the mouse genome sequence and its annotation are updated, and have the potential to contribute to the annotation of the genome sequence. To meet these needs, we are using serial analysis of gene expression (SAGE) [LongSAGE (1); SAGE (2)] to develop spatially and temporally specific digital gene-expression profiles throughout development in a total of 200 mouse cells and tissues. The data are made publicly available as they are generated to fuel mouse functional genomics and bioinformatic analyses.
This article provides an analysis of 8.55 million 21-bp tags derived from 72 LongSAGE libraries (see Table 3, which is published as supporting information on the PNAS web site).
Libraries have been sampled to an average depth of >118,000 tags. This sampling depth yields gene-detection sensitivity approximately equivalent to that of fluorescence-based microarray approaches (3) and, thus, is sufficient for detection of abundant and moderately abundant transcripts but likely insufficient for reliable detection of rare transcripts. For deeper sampling, we have retained frozen aliquots of libraries.
Although others have profiled gene-expression levels in the mouse (4-6), the scale of this project and its strong emphasis on development are distinguishing features. Unique achievements of the project include: high-throughput production of SAGE libraries, creation of protocols for the precise microdissection of tissues from numerous stages of development, the refinement of technologies for construction of libraries from nanogram quantities of total RNA, the rapid public release of the data, the creation of protocols for computational analysis of the data, and the construction and distribution of software tools, at our Genome Centre and elsewhere, to facilitate its analysis. For example, the data reported here have been used to construct mouse sagegenie, a software tool available from the Cancer Genome Anatomy Project for analysis of mouse LongSAGE tags (http://cgap.nci.nih.gov/SAGE/#mouse). We present here an overview of the data, focusing on data quality, representation of known genes, and identification of previously undescribed transcripts.
Materials and Methods
Maintenance of Mice and Tissue Collection. C57BL/6J mice were provided with Purina mouse food and autoclaved water ad libitum and maintained at 20°C ± 2°C under a light/dark cycle (light, 5 a.m. to7 p.m. and dark, 7 p.m. to 5 a.m. at the British Columbia Cancer Agency and light, 7 a.m. to 7 p.m. and dark, 7 p.m. to 7 a.m. at the Centre for Molecular Medicine and Therapeutics). Stud males were mated overnight with up to three females; females were inspected for copulation plugs before 10:00 the following morning. Plugged mice were considered to be 0.5 days postcoitum. Mice were assigned to the appropriate Theiler stage at the time of tissue collection to ensure uniformity in the classification of developmental stages.
SAGE Protocol. Mouse tissue samples were collected in either RNAlater (Ambion) or TRIzol reagent (Invitrogen), or they were snap-frozen by using liquid nitrogen. LongSAGE (1) libraries were constructed with at least 5 μg of DNase I-(Invitrogen) or DNA-free-(Ambion) treated total RNA by using the Invitrogen I-SAGE Long kit and protocol. Sequencing reaction products were purified by ethanol precipitation and analyzed on model 3700 and 3730xl capillary DNA sequencers (Applied Biosystems). These template-preparation and sequencing protocols were described by Yang et al. in ref. 7.
Sequence data were collected automatically by using a custom DNA-sequencing laboratory information-management system and processed by trimming reads for sequence quality and removal of nonrecombinant clones and linker-derived tags. Sufficient clones were sequenced to yield ≈100,000 LongSAGE tags per library. On average, 34 LongSAGE tags resulted from each sequencing read. Samples with limiting (submicrogram) amounts of total RNA were subject to an amplification step similar to the SAGELite method (8).
Further methodological details are provided in Supporting Methods, which is published as supporting information on the PNAS web site.
LongSAGE Processing Pipeline. After sequencing, flanking vector sequences were removed and the tags extracted from each sequence read. The SAGE protocols generated concatemers in which the tags were present in pairs (ditags). A sequence quality factor (QF) was derived for each tag by using the following formula:
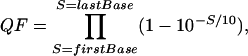 |
where S is the phred score (9) for a particular base, and the value is calculated over all bases in the tag. The quality factor was used in the calculation of tag-sequence-probability values. Further details on the processing of tags and the calculation of tag-sequence-probability values are provided in Supporting Methods.
Tag-Sequence Mapping. Tag sequences were mapped to the genome sequence, Mammalian Gene Collection (MGC) genes (ftp://ftp.ncbi.nih.gov/repository/MGC/MGC.sequences), RefSeq genes (ftp://ftp.ncbi.nih.gov/refseq/daily), and Ensembl genes (Ensembl v20). All mappings were transformed to genomic coordinates (chromosome, position, and strand) on the mouse sequence (assembly 32) (10), with the aid of the Ensembl application programming interface perl api (11). The mapping of RefSeq genes to genome contigs used data from Ensembl. The mapping of MGC genes to genome contigs used data from the University of California, Santa Cruz genome browser site (12).
We counted gene identifiers to calculate the number of gene loci represented by the data. To avoid double-counting different identifiers used to name the same gene in different databases, identifiers found at the same genomic location were assumed to represent the same gene.
“Known” Ensembl genes are those confirmed by full-length sequences deposited in public sequence databases. “Novel” Ensembl genes are those predicted by computational methods and confirmed by ESTs.
Tag-Sequence Classification. The Ensembl database and api (ver. 20) were used to determine the genomic location of ESTs, UniGene clusters, human genome sequence blast (13) hits, and rat genome sequence blast hits. Tag sequences that mapped to MGC, known RefSeq, or known Ensembl genes were classified as annotating high-quality resources. Tag sequences were classified as annotated exon or UTR by using the Ensembl definitions of these regions. The classification was inferred from the annotation of the Ensembl transcript whose 3′ end was closest to the tag's position. Tag sequences that did not map to a transcript and hit introns of Ensembl genes were classified as intron tags.
We noted that many genes in Ensembl were either missing UTRs or had very short UTRs. We assumed that such genes were not fully annotated and, hence, extended the 5′ and 3′ ends of the gene (from the coding start and stop) to compensate. We determined that 90% of known Ensembl genes had a 5′ UTR of ≤456 bases and a 3′ UTR of ≤2,039 bases. We extended genes with shorter UTRs to be equal to these lengths.
Tag sequences that did not correspond to exons, introns, or UTRs of known or novel genes were classified as intergenic.
RT-PCR Validation. An RT-PCR method was used to confirm the presence of transcripts corresponding to singleton longSAGE tags that hit an unannotated genomic sequence. The singleton tags were filtered by removing those that matched against RefSeq sequences (standard, X, and GS), MGC sequences, UniGene sequences, Ensembl EST genes, and Ensembl mappings of ESTs onto the genome. PCR primers were designed by using genomic sequence, Primer3 (14), and custom scripts to generate amplicons with an average length of 120 bp. Primers were designed that flank the tag sequence such that the tag would be included in the amplicon. The amplicons were each amplified from RNA representing the developmental stage and tissue in which the singleton tag was observed. Further details are provided in Supporting Methods.
Control experiments (data not shown) demonstrated that amplicons were RNA-dependent: RNase-A-treated RNA samples failed to produce amplicons, indicating that amplicons were derived from RNA and not from genomic DNA potentially contaminating the RNA.
Representation of Gene Families. Genes in each category were identified by their Gene Ontology (GO) classification (15). In addition, transcription factors reported in Messina et al. (16) were taken directly from that paper. The GO classification of human genes was used and the mouse orthologue determined by using the Ensembl database.
Results and Discussion
Data Overview and Quality Filtering. The 8.55 million LongSAGE tags analyzed for this report represent 924,392 different tag sequences, each of which is derived from a transcript. Preliminary inspection of this tag set suggested that experimental artifacts (sequencing errors, reverse-transcriptase artifacts, etc.) had inflated the number of different tag sequences. This inflation was particularly apparent in the singleton class, where only 29% mapped to a sequence resource (the genome, MGC, RefSeq, or Ensembl). We implemented bioinformatics approaches to recognize erroneous variants of more common tags, reducing the numbers of tag sequences to 769,608. We also developed an approach to assign a “confidence” (i.e., probability) value to each tag sequence, distinguishing high-quality tags from low-quality tags. Employing a quality cutoff of P < 0.2 to the 8.55-million-tag metalibrary yielded 8.24 million tags representing 465,178 different tag sequences, of which the majority [60% of singletons and 88% of nonsingletons (see Fig. 2, which is published as supporting information on the PNAS web site)] could be mapped to at least one mouse-sequence resource. Of these tags, 261,134 of the LongSAGE tag sequences (3.7 million tags comprised of 154,173 singletons and 106,961 nonsingletons) mapped uniquely to the mouse genome. These tags were analyzed further.
Representation of Known Genes. We assessed the representation of known genes in our data by comparing the LongSAGE tag sequences with existing mouse-transcript resources, including MGC mouse (17-19), RefSeq mouse (NM, NR, and XM (20, 21), mouse UniGene (22), Riken Fantom (23), and the Ensembl (24-26) gene sets (Table 1). The tag sequences identified many of the sequences in the high-quality transcript data sets (e.g., 85% and 96% of sequences in RefSeqNM and MGC, respectively). Although these genes are classified as known, our project provides an association between these transcripts and precisely defined developing tissues.
Table 1. Coverage of existing sequence resources by the Mouse Atlas data set.
| Sequence resource | Resource subset | Sequences in resource* | No. of sequences hit (unique)† | Percentage of sequences hit (unique) | Number of sequences hit (all)‡ | Percentage of sequences hit (all) |
|---|---|---|---|---|---|---|
| RefSeq§ | All | 26,520 | 15,399 | 58 | 19,897 | 75 |
| NM | 17,057 | 12,445 | 73 | 14,523 | 85 | |
| NR | 23 | 6 | 26 | 9 | 39 | |
| XM | 9,440 | 2,948 | 31 | 5,365 | 57 | |
| MGC | All | 13,174 | 10,240 | 78 | 12,631 | 96 |
| Ensembl transcripts | All | 32,281 | 18,382 | 57 | 23,241 | 72 |
| Known | 26,004 | 16,909 | 65 | 19,734 | 76 | |
| Novel | 6,277 | 1,473 | 23 | 3,507 | 56 | |
| UniGene clusters¶ | All | 29,111 | 16,752 | 58 | 18,970 | 65 |
| Riken | 3′ | 691,524 | 24,301 | 4 | 291,410 | 42 |
| 5′ | 431,560 | 16,223 | 4 | 226,242 | 52 |
This is the number of sequences present in the sequence resource. For example, the version of RefSeq used contained 9,440 XM sequences
This column provides the number of sequences to which a tag sequence maps, with the requirement that the tag sequence map to only one sequence in the resource. The Riken sequence databases contain many redundant sequences, and, therefore, the number of tag sequences that map uniquely to a single Riken sequence is small
This column provides the number of sequences to which a tag sequence maps. Unlike†, the same tag sequence can be used to confirm multiple resource sequences
Refseq NM sequences represent mature RNA (mRNA) protein-coding transcripts. RefSeq XM sequences are model mRNAs defined during genome annotation. RefSeq NR sequences are noncoding transcripts, including structural RNAs and transcribed pseudogenes
The UniGene clusters utilized for this article were taken from the Ensembl database
The SAGE data also provide experimental evidence for the existence of a significant fraction of computationally predicted genes. For example, 57% of sequences in RefSeq XM and 56% of predicted Ensembl transcripts matched tag sequences.
Representation of Gene Families of Interest. We assessed the representation of classes of genes likely to be of particular interest and for which there were Ensembl-assigned human-mouse orthologues (16, 27), including kinases, phosphatases, G protein-coupled receptors (GPCRs) and transcription factors (see Table 4, which is published as supporting information on the PNAS web site). Most of the genes within each of these classes were found in our data, with the exception of GPCR genes. Of these, only 28% (173 of 615) were “hit” in an annotated exon or UTR by at least one high-quality tag. In contrast, >76% of all kinase (359 of 454) and phosphatase (89 of 117) genes were detected. Seventy-seven percent (966 of 1,247) of mouse genes orthologous to a recently published set of candidate human transcription factors (16) were likewise detected. Expression of GPCR genes is known to be, in general, at low levels and constrained to particular tissues, and this known result appears to be reflected in the SAGE data (Table 3).
Number of Genes Identified. We derived an estimate of the total number of genes represented in the LongSAGE metalibrary for the set of 261,134 uniquely mapping LongSAGE tag sequences identified above. We found that 106,847 LongSAGE tag sequences mapped to 17,890 high-quality annotated genes (from RefSeq NM, MGC, and known Ensembl gene definitions), and an additional 13,939 LongSAGE tag sequences mapped to 4,073 lower-quality predicted genes (from RefSeq XM and predicted Ensembl gene definitions). The total number of observed genes was reduced to 19,865 by the removal of loci redundant between these two sets. This number agreed with previous analyses of the mouse and human genome sequences that yielded estimates of 20,000-30,000 mammalian genes (10, 28). However, there remained 140,348 uniquely mapping tag sequences unaccounted for. Of these, 23,516 tag sequences mapped to a nonredundant set of 12,244 loci predicted from ESTs (UniGene and Ensembl EST genes), leaving 116,622 tag sequences unaccounted for. Some fraction of these may be artifacts in the data, but we believe that many of these tag sequences represent novel transcripts because they map to the genome. We note that 52,255 (36%) of the unaccounted tag sequences map antisense to annotated genes and may have some function related to the regulation of the gene on the opposite strand (29-31). Our interpretation is that the unaccounted tag sequences observed support the existence of many novel, transcribed loci in the C57BLJ/6 genome.
Location of Tag Hits on Genes. We explored the utility of the LongSAGE data for the identification of transcribed features, including the identification of novel transcripts of known genes by using the set of 261,134 of the LongSAGE tag sequences identified above. We assessed whether the tag sequences mapped to exons, to introns, to candidate (putative) UTR regions, or to regions we classified as “intergenic” (Table 2; Materials and Methods). We observed that 21.3% (55,962) of the tag sequences matched annotated exons and UTRs [MGC, RefSeq (NM, NR, and XM), or Ensembl (known and novel genes)], and 22.2% (58,029) mapped to annotated introns or to regions we identified as candidate UTRs, suggesting that they were derived from unannotated exons or UTRs for these genes. The proportion of tag sequences that mapped to either annotated exons or UTRs was higher for more abundant transcripts (increasing from 21.3% for all transcripts to 94.8% for the most abundant transcripts; Table 2), possibly reflecting better annotation accuracy for more abundantly expressed genes.
Table 2. Distribution of uniquely mapping tag sequences to gene features.
| Location | Gene evidence* | All transcripts (A > 0)† | All transcripts expressed at A > 1† | All transcripts expressed at A > 10† | All transcripts expressed at A > 60† | All transcripts expressed at A > 1000† |
|---|---|---|---|---|---|---|
| No of unique locations | – | 261,134 | 106,961 | 25,829 | 8,855 | 424 |
| Annotated exon,‡ % | Known | 12.1 | 17.9 | 23.8 | 28.3 | 34.7 |
| Novel | 0.9 | 1.2 | 1.2 | 1.1 | 0.7 | |
| Annotated UTR,‡ % | Known | 8.0 | 14.6 | 30.9 | 46.0 | 58.0 |
| Novel | 0.3 | 0.5 | 1.0 | 1.2 | 1.4 | |
| Annotated exon or UTR, % | Known or Novel | 21.3 | 34.2 | 56.9 | 61.4 | 94.8 |
| Intron,% | Known | 20.0 | 14.3 | 4.4 | 1.8 | 1.2 |
| Novel | 1.5 | 1.1 | 0.4 | 0.2 | 0 | |
| Putative UTR,‡ % | Known | 0.5 | 0.7 | 0.8 | 0.5 | 0.5 |
| Novel | 0.2 | 0.2 | 0.2 | 0.2 | 0 | |
| Intergenic,‡ % | – | 56.3 | 49.5 | 37.4 | 20.8 | 3.5 |
All percentages are specified to 1 decimal place and, hence, may not add up to 100%.
The known gene category encompasses MGC, RefSeq (NM, NR) and Ensembl “known” genes. The novel gene category encompasses RefSeq (XM) and Ensembl “novel” genes
The abundance (A) is the number of times the tag sequence is observed in the metalibrary. The columns to the right limit the data to the most highly expressed transcripts
Annotated exons and UTRs represent regions of genes annotated as part of the transcript in sequence resources. Ensembl's definitions of the coding regions were used to delineate the exon/UTR boundaries. Transcripts with short or absent UTRs were extended, giving rise to the putative UTR category. Tag sequences falling outside of the boundaries of genes were classified as intergenic
Alternative Transcripts. During our analyses, we found that many annotated genes were identified by several tag sequences. We examined in detail 13,068 known Ensembl genes hit by at least one uniquely mapping tag sequence and found that 64% (8,338) were hit by multiple tag sequences. We inferred that each of these multiple tag sequences was derived from a different transcript from the same locus, produced by alternative splicing (32) or alternative polyadenylation (33). The percentage of genes for which these transcript variants were detected in the metalibrary was 64% for all genes, increasing to 88% for the most abundantly expressed genes (see Table 5, which is published as supporting information on the PNAS web site). These values compare favorably with the 35-60% range reported by others for the percentage of alternatively spliced genes (32, 34). Consistent with our expectation, and in agreement with Zavolan et al. (34), our analysis supports the observation that more highly expressed genes have more detected variants. Over all, we detected an average of 3.3 variants per locus for the 8,338 loci studied. This value increased to 5.0 variants per locus for the most highly expressed loci (Table 5). These numbers are likely an underestimate, because our analysis is restricted to uniquely mapping tags, and our method of detection is able to detect only variants that result in a change of the most distal NlaIII site in the transcripts. Many of the variants appeared to be spatially or temporally regulated; of the 8,338 loci, 4,781 were identified by at least one tag sequence that exhibited a significant change in expression between at least one pair of libraries (with a significance level of P < 0.01 and a change in expression level of at least 2-fold, P value not corrected for multiple tests) (35). Overall, 5,220 (63%) of the 8,338 loci were hit by tag sequences that mapped to different portions of the protein-coding region, leading us to believe that the transcripts may encode different proteins. For 827 of the 5,220 loci, at least one of the tag sequences demonstrated a significant change in expression between at least one pair of libraries, whereas at least one other tag sequence mapping to the same locus did not demonstrate a significant change in expression between the same pair of libraries. For each of an additional 222 loci, at least one pair of tag sequences exhibited significant changes in expression levels in opposite directions between at least one pair of libraries. For 18% of identified loci in the first category and 12% in the second (152 of 827 loci and 27 of 222 loci, respectively), the tag sequences identifying the transcripts mapped to different locations within the coding region of the gene. These results are consistent with the existence of multiple transcripts produced from each of many loci and consistent with the existence of multiple, independently regulated transcripts derived from a single locus, possibly encoding protein isoforms.
Number of Novel Genes. Many tag sequences were intergenic with respect to annotated genes (Table 2). The proportion of tag sequences mapped to intergenic regions decreased with increasing transcript abundance. Some fraction of these “intergenic tag sequences” may represent novel, low-abundance transcripts. Of the 147,143 intergenic tag sequences (56.3% of the 261,134 uniquely mapping tag sequences) in Table 2, 40% (58,762) mapped to regions of the genome containing EST and UniGene matches. Another 40% (58,573) mapped to regions of the mouse genome sequence that were unremarkable, except that they exhibited sequence similarity to either the human or rat genome sequence (highly conserved regions, as specified by Ensembl ComparaDB; parameters described at www.ensembl.org/Multi/helpview?se=1&kw=multicontigview#WholeGenomeSimilarityMatches), providing evidence that these evolutionarily conserved regions are transcribed. Twenty percent (29,808) of the tag sequences mapped to genome regions that, in addition to lacking annotation, also lacked a strong similarity to either the human or rat genome sequence. This latter category may represent transcripts specific to mouse. Approximately 78% of the 88,381 transcripts in the latter two categories were identified by only a high-quality singleton and are likely to be infrequently expressed.
We sought to estimate the number of transcribed loci represented by the 147,143 tag sequences mapping to intergenic regions. This result was achieved by grouping sequences into clusters by using tag proximity in the genome to define group members. To derive clustering parameters, we first considered 16,937 relatively well annotated Ensembl genes for which tags were detected. We used these genes to explore the effect of varying the size of the genomic region used to produce clusters. We specifically asked, for increasing size of the genomic interval, whether tags belonging to single genes were contained within a single cluster (desirable) or split across clusters (undesirable, indicating insufficiently large intervals) or whether a cluster contained more than a single gene (also undesirable, indicating intervals that are too large). Selection of an interval size that was too large or too small would have the effect of under- or overestimating, respectively, the number of potential new loci detected by the intergenic tag sequences.
We plotted the relationship between increasing genomic interval size and the proportion of known genes split across intervals. We also plotted the relationship between genomic interval size and the proportion of intervals that contained multiple genes (see Fig. 3, which is published as supporting information on the PNAS web site). At the intersection of these curves, at an interval size of ≈21 kb, ≈20% of the intervals contain multiple known genes, and 20% of the known genes are split across an interval. Conversely, by using this interval size, ≈80% of the intervals contain only a single gene, and 80% of the known genes are within a single interval. Hence, use of a 21-kb interval size seems to produce a reasonable compromise between the possibilities of over- or underestimating the number of possible new loci.
Use of the 21-kb interval size in clustering yielded an estimate of 34,409 clusters containing only novel intergenic tag sequences. This number represents an estimate of the number of previously undescribed loci detected by the intergenic tag sequences, and 13,888 of these clusters contain two or more tag sequences. The remaining clusters contain only a single tag sequence. Of these remaining clusters, 10,442 are identified by lower-quality singletons (P > 0.05). Excluding these clusters reduces the number of previously untranscribed loci to 23,967. This relatively large number of unannotated previously undescribed loci points to an ongoing need to apply whole-genome unbiased approaches to gene discovery. Our results show that this approach is clearly needed in even apparently well characterized genomes. Such data will be required to generate more comprehensively annotated genome sequences, which, in turn, will be essential to approach an understanding of genome function.
Validation of Singleton Tags. Using RT-PCR, we sought to validate a set of the potentially novel transcripts that were identified by high-quality singletons that mapped to a set of unannotated, unique genomic coordinates. A total of 173 tag sequences were selected for validation. Of these, 96 tag sequences were observed only once in an individual library yet multiple times in other libraries (“library singletons”), and 77 tag sequences were observed only once in the entire metalibrary of 8.55 million tags (“metalibrary singletons”). All of the selected tags were at least 30-kb 3′ and 12-kb 5′ of the nearest annotated Ensembl gene. Some of the singleton tags matched sequences in dbEST. Metalibrary singletons were limited to those assigned probability values <0.05, indicating that they were of the highest quality possible. Library singletons were limited to those with a library probability value <0.05 and a metalibrary probability value <0.00001. Based on these criteria and the availability of RNA remaining after construction of LongSAGE libraries, eight libraries were selected, each providing both metalibrary and library singletons. The genome sequences surrounding the tags were used to design oligonucleotide primers, which were used in RT-PCR assays using individual RNA preparations as the amplification template (see Fig. 4 and Table 6, which are published as supporting information on the PNAS web site). Analysis of the PCR products on agarose gels, followed by comparison of the sizes obtained with the sizes expected based on the genome sequence, revealed that 122 (85%) of the PCR products were within 15% of their expected size. These experiments support the expression of 77% of the metalibrary singletons and 86% of the library singletons. We concluded from this finding that most high-quality singletons, even those mapping only to the genome, appear to represent bona fide transcripts.
Noncoding RNAs. We explored the overlap between the SAGE data and databases of noncoding RNAs, including the portion of the Fantom database containing a reported 15,814 noncoding sequence entries (23). We found SAGE tags in our data for 9,810 (62%) of these sequences. A different data set of ≈4,000 sense-antisense transcripts compiled by Kiyasawa et al. (36), with estimated coding and noncoding status, was also analyzed. Seventy-eight percent (2,111 of 2,717) of the coding members of the data set were matched by our mouse SAGE tags. Sixty-nine percent (808 of 1,174) of the noncoding members of the data set were matched by mouse SAGE tags. The reduced level of overlap between our SAGE data and the coding and noncoding subsets of the Kiyosawa et al. data may be due to the higher incidence of noncoding transcripts which lack a poly(A) tail compared with coding transcripts. Noncoding transcripts lacking a polyA tail are expected to be underrepresented in the SAGE data, because tags are derived from oligo(dT)-primed cDNA.
Summary
The multitude of developmental time points analyzed and the precise dissection of tissues allowed us to construct a detailed view of changes in gene expression levels (Table 3; Fig. 1; and see Fig. 5, which is published as supporting information on the PNAS web site). We have shown that the LongSAGE data provide good coverage of important gene families (Table 4) and insight into novel transcribed loci associated with specific developmental stages and tissues. These characteristics of the data will be exploited to gain insight into how expression changes trigger gross changes in the morphology and function of differentiating tissues.
Fig. 1.

Differential expression of tag sequences among tissue groups. Shown is a graph of tag sequences at least 5-fold (red and yellow bars) and at least 10-fold (green and black bars) differentially expressed (P < 0.05 and P < 0.01, respectively) among tissue groups and all other libraries in all other tissue groups. The yellow and black bars depict the number of differentially expressed tag sequences that hit only unannotated regions of the genome sequence. The blue line indicates the number of libraries in each tissue group. From this data it is evident that the visual cortex, pancreas, mammary gland, preimplantation embryo, and placenta tissue groups contain the largest number of differentially expressed tag sequences, many of which hit unannotated regions of the genome. In contrast, virtually all of the differentially expressed tag sequences in stomach, spleen, and kidney hit annotated regions of the genome. SVZ/VZ, subventricular zones/ventricular zones; Post-I, post implantation; Pre-I, pre implantation; UGS, urogenital sinus.
The Mouse Atlas LongSAGE data are a rich source of novel transcripts and represent the majority of previously identified genes. The data were generated from RNAs purified from tissue samples harvested with an unprecedented level of precision, representing a range of tissues and time points, with an emphasis on early development. The association of expressed genes with such carefully collected tissue samples greatly enhances the potential for functional characterization of the genes and should be useful for studies aimed at bioinformatic and biochemical characterization of gene-expression regulation. The data, among the most comprehensive currently available for mouse development, represent a significant addition to available mouse genomic resources. All data, tag-to-gene mappings, and software tools for data analysis are available at www.mouseatlas.org. The data and other software tools, including mouse sage genie, are available from http://cgap.nci.nih.gov/SAGE.
Supplementary Material
Acknowledgments
We thank the staff at Canada's Michael Smith Genome Sciences Centre for expert technical, computational, and administrative support; Mehrdad Oveisi (Canada's Michael Smith Genome Sciences Centre) for providing software; Brent Gowan, Jason Y. Y. Wong, Earnest H. Leung, and Rachel Montpetit for laboratory assistance; Robyn Hanson for expert project management; and Adrian Burke (Genome BC) for administrative assistance. This work was supported by Genome Canada; the British Columbia Cancer Foundation; and the National Cancer Institute, National Institutes of Health, under Contract No. N01-C0-12400. E.M.S. holds a Canada Research Chair in Genetics and Behavior. M.A.M., S.J.M.J., R.A.H., C.D.H., and P.A.H. are Scholars of the Michael Smith Foundation for Health Research. P.A.H. is a Canadian Institute for Health Research New Investigator. M.A.M. is a National Cancer Institute of Canada Terry Fox Young Investigator.
Author contributions: A.S.S., J.K., A.D.D., B.H., J.L.R., Y.-Y.X., M.H., S.J.M.J., C.D.H., E.M.S., P.A.H., and M.A.M. designed research; A.S.S., J.K., A.D.D., Y.Z., C.A., J.A., R.B., S. Barber, J.B., S. Bohacec, M.B.-J., S.C., D.C., A.M.C., R.C., N.D., R.F., D.S.G., B.H., R.A.H., J.H., B.Y.L.K., L.L.C.L., S.L., D.L., K.M., C.M., M.M., H.M., A.-l.P., P.P., T.R.d.A., J.L.R., D.S., J.S., M.T., R.V., P.V., D.W., M.K.W., Y.-Y.X., G.Y., I.Z., and M.H. performed research; A.S.S., J.K., A.D.D., Y.Z., J.A., N.D., S.L., D.L., K.M., H.M., A.-l.P., P.P., G.J.R., R.V., and M.A.M. contributed new reagents/analytic tools; A.S.S. and A.D.D. analyzed data; and A.S.S. and M.A.M. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: MGC, Mammalian Gene Collection; SAGE, serial analysis of gene expression.
References
- 1.Saha, S., Sparks, A. B., Rago, C., Akmaev, V., Wang, C. J., Vogelstein, B., Kinzler, K. W. & Velculescu, V. E. (2002) Nat. Biotechnol. 20, 508-512. [DOI] [PubMed] [Google Scholar]
- 2.Velculescu, V. E., Vogelstein, B. & Kinzler, K. W. (2000) Trends Genet. 16, 423-425. [DOI] [PubMed] [Google Scholar]
- 3.Lu, J., Lal, A., Merriman, B., Nelson, S. & Riggins, G. (2004) Genomics 84, 631-636. [DOI] [PubMed] [Google Scholar]
- 4.Su, A. I., Cooke, M. P., Ching, K. A., Hakak, Y., Walker, J. R., Wiltshire, T., Orth, A. P., Vega, R. G., Sapinoso, L. M., Moqrich, A., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 4465-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang, W., Morris, Q. D., Chang, R., Shai, O., Bakowski, M. A., Mitsakakis, N., Mohammad, N., Robinson, M. D., Zirngibl, R., Somogyi, E., et al. (2004) J. Biol. 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bono, H., Yagi, K., Kasukawa, T., Nikaido, I., Tominaga, N., Miki, R., Mizuno, Y., Tomaru, Y., Goto, H., Nitanda, H., et al. (2003) Genome Res. 13, 1318-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang, G. S., Stott, J. M., Smailus, D., Barber, S. A., Balasundaram, M., Marra, M. A. & Holt, R. A. (2005) BMC Genomics 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters, D. G., Kassam, A. B., Yonas, H., O'Hare, E. H., Ferrell, R. E. & Brufsky, A. M. (1999) Nucleic Acids Res. 27, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewing, B. & Green, P. (1998) Genome Res. 8, 186-194. [PubMed] [Google Scholar]
- 10.Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. (2002) Nature 420, 520-562. [DOI] [PubMed] [Google Scholar]
- 11.Stabenau, A., McVicker, G., Melsopp, C., Proctor, G., Clamp, M. & Birney, E. (2004) Genome Res. 14, 929-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M. & Haussler, D. (2002) Genome Res. 12, 996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403-410. [DOI] [PubMed] [Google Scholar]
- 14.Rozen, S. & Skaletsky, H. (2000) Methods Mol. Biol. 132, 365-386. [DOI] [PubMed] [Google Scholar]
- 15.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. (2000) Nat. Genet. 25, 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messina, D. N., Glasscock, J., Gish, W. & Lovett, M. (2004) Genome Res. 14, 2041-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strausberg, R. L., Feingold, E. A., Klausner, R. D. & Collins, F. S. (1999) Science 286, 455-457. [DOI] [PubMed] [Google Scholar]
- 18.Strausberg, R. L., Feingold, E. A., Grouse, L. H., Derge, J. G., Klausner, R. D., Collins, F. S., Wagner, L., Shenmen, C. M., Schuler, G. D., Altschul, S. F., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 16899-16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerhard, D. S., Wagner, L., Feingold, E. A., Shenmen, C. M., Grouse, L. H., Schuler, G., Klein, S. L., Old, S., Rasooly, R., Good, P., et al. (2004) Genome Res. 14, 2121-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pruitt, K. D., Tatusova, T. & Maglott, D. R. (2003) Nucleic Acids Res. 31, 34-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruitt, K. D. & Maglott, D. R. (2001) Nucleic Acids Res. 29, 137-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler, D. L., Church, D. M., Edgar, R., Federhen, S., Helmberg, W., Madden, T. L., Pontius, J. U., Schuler, G. D., Schriml, L. M., Sequeira, E., et al. (2004) Nucleic Acids Res. 32, D35-D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carninci, P., Waki, K., Shiraki, T., Konno, H., Shibata, K., Itoh, M., Aizawa, K., Arakawa, T., Ishii, Y., Sasaki, D., et al. (2003) Genome Res. 13, 1273-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eyras, E., Caccamo, M., Curwen, V. & Clamp, M. (2004) Genome Res. 14, 976-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birney, E., Andrews, T. D., Bevan, P., Caccamo, M., Chen, Y., Clarke, L., Coates, G., Cuff, J., Curwen, V., Cutts, T., et al. (2004) Genome Res. 14, 925-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curwen, V., Eyras, E., Andrews, T. D., Clarke, L., Mongin, E., Searle, S. M. & Clamp, M. (2004) Genome Res. 14, 942-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stalker, J., Gibbins, B., Meidl, P., Smith, J., Spooner, W., Hotz, H. R. & Cox, A. V. (2004) Genome Res. 14, 951-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International Human Genome Sequencing Consortium (2004) Nature 431, 931-945. [DOI] [PubMed] [Google Scholar]
- 29.Chen, J., Sun, M., Kent, W. J., Huang, X., Xie, H., Wang, W., Zhou, G., Shi, R. Z. & Rowley, J. D. (2004) Nucleic Acids Res. 32, 4812-4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quere, R., Manchon, L., Lejeune, M., Clement, O., Pierrat, F., Bonafoux, B., Commes, T., Piquemal, D. & Marti, J. (2004) Nucleic Acids Res. 32, e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cawley, S., Bekiranov, S., Ng, H. H., Kapranov, P., Sekinger, E. A., Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A. J., et al. (2004) Cell 116, 499-509. [DOI] [PubMed] [Google Scholar]
- 32.Maniatis, T. & Tasic, B. (2002) Nature 418, 236-243. [DOI] [PubMed] [Google Scholar]
- 33.Zhao, J., Hyman, L. & Moore, C. (1999) Microbiol. Mol. Biol. Rev. 63, 405-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zavolan, M., Kondo, S., Schonbach, C., Adachi, J., Hume, D. A., Hayashizaki, Y. & Gaasterland, T. (2003) Genome Res. 13, 1290-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Audic, S. & Claverie, J. M. (1997) Genome Res. 7, 986-995. [DOI] [PubMed] [Google Scholar]
- 36.Kiyosawa, H., Mise, N., Iwase, S., Hayashizaki, Y. & Abe, K. (2005) Genome Res. 15, 463-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}