

Abstract
Development of spatial patterns in multicellular organisms depends on gradients in the concentration of signaling molecules that control gene expression. In the Drosophila embryo, Bicoid (Bcd) morphogen controls cell fate along 70% of the anteroposterior axis but is translated from mRNA localized at the anterior pole. Gradients of Bcd and other morphogens are thought to arise through diffusion, but this basic assumption has never been rigorously tested in living embryos. Furthermore, because diffusion sets a relationship between length and time scales, it is hard to see how patterns of gene expression established by diffusion would scale proportionately as egg size changes during evolution. Here, we show that the motion of inert molecules through the embryo is well described by the diffusion equation on the relevant length and time scales, and that effective diffusion constants are essentially the same in closely related dipteran species with embryos of very different size. Nonetheless, patterns of gene expression in these different species scale with egg length. We show that this scaling can be traced back to scaling of the Bcd gradient itself. Our results, together with constraints imposed by the time scales of development, suggest that the mechanism for scaling is a species-specific adaptation of the Bcd lifetime.
Keywords: bicoid, morphogen, dipteran evolution
One of the classic challenges for models of embryonic development is the problem of scaling: Although organisms vary substantially in size, the variations in proportion are much less significant, and in cases where the fully developed organism has a segmented structure the number of segments often is invariant across a wide range of sizes. In well studied systems such as Drosophila, segmented structures have their origin in spatially periodic patterns of gene expression that are visible at early stages of embryonic development, and the emergence of these patterns has been described, at varying levels of detail, in terms of the diffusion and interaction of the relevant “morphogen” molecules (1–3). In nonbiological systems governed by similar equations, the spatial scale of patterns is set by local parameters analogous to the diffusion constants and reaction rates; hence, when we change the size of the system, the dimensions of the stripes remain fixed and the number of stripes changes (4). How then are we to understand the scaling of segment size and the conservation of their number in the biological case? Here, we use Drosophila melanogaster and a set of closely related dipteran species as model systems in which to address this problem.
Anteroposterior patterning in the Drosophila embryo is controlled by gradients in the concentration of maternal gene products that arise soon after fertilization. These protein molecules establish broad domains of gene expression that interact to establish final segmentation. One of the best studied maternal determinants in Drosophila is bicoid (bcd) (5). Bcd RNA is deposited during oogenesis at the anterior pole of the egg. After fertilization, the RNA is translated and a Bcd protein gradient is established along the anteroposterior axis of the egg. Subsequently, Bcd acts as a transcription factor, regulating genes such as hunchback, krüppel, and even-skipped in a concentration-dependent manner (6), and these gene products feed into a regulatory circuit that generates striped patterns of expression of the gap and pair rule genes (cf. Fig. 2).
Fig. 2.

Immunofluorescence stainings for products of the gap and pair-rule genes in higher diptera. (A) Immunofluorescence staining of L. sericata (upper embryos) and D. melanogaster (lower embryos) for Hunchback (green) and Giant (red) in the left column, and for Paired (green) and Runt (red) in the right column. (B) Anti-Hunchback (green) and anti-Runt (red) immunofluorescence staining of D. melanogaster (upper embryo) and D. busckii (lower embryo). (Scale bars: 100 μm.)
The Bcd gradient is established in a syncytium, where nuclei replicate without intervening cell division (7), and the size of the embryo stays constant, so that pattern formation is independent of growth. The absence of cell membranes would seem to provide an ideal environment for diffusion-based gradient formation. On the other hand, visual observation of the developing embryo reveals contractions of the egg cortex and large-scale cytoplasmic motions occurring with each mitotic cycle, raising questions about whether the relevant movement of any molecule can be described by simple diffusion.
Parallel to the question of whether diffusion is a good description of molecular motion in the embryo is the question of scaling. Certainly, related species of flies have similarly scaled proportions in their body plans, but relatively little is known about the underlying pattern of gene expression in the embryo. One can imagine, for example, scaling of body plan without scaling of the Bcd gradient itself, by changing the rules that the downstream genes use in responding to this gradient.
Here, we address both questions. To test the validity of the diffusion model, we use microinjections of inert, fluorescent molecules to probe and measure diffusive properties of embryonic cytoplasm of various dipteran species. To test for scaling, we use immunofluorescence stainings to compare Bcd gene expression profiles in those species. Our findings show that these different measurements, taken together, strongly constrain models for the formation and readout of the Bcd gradient.
Methods
Antibody Staining. Embryos were selected from early interphase of cell cycle 14, i.e., before significant membrane invagination, and the images were focused at mid-embryo to avoid geometric distortion. All species were stained with antibodies raised against D. melanogaster (8). For the Lucilia sericata and D. melanogaster embryos in Fig. 2 A, we used rabbit anti-paired, guinea pig anti-runt, guinea pig anti-hunchback, and rat anti-giant. For the D. melanogaster and Drosophila busckii embryos in Fig. 2B, we used rat anti-hunchback and guinea pig anti-runt. Images were taken with a Zeiss LSM510 confocal microscope, with Zeiss ×10 (N.A. 0.45) and ×20 (N.A. 0.6) air objectives.
Diffusion Measurements. Dextran particles were purchased from Molecular Probes (10, 40, and 70 kDa) and Sigma-Aldrich (150 kDa). Corresponding hydrodynamic radii, rs, were taken from ref. 9 and ref. 10, respectively. Calibrated volumes of ≈4–5 pl were pressure-injected with pulled borosilicate capillaries (back-filled with 10 μM solutions of dextran powder dissolved in dH2O) at the anterior pole of dechorionated halocarbon oil 27-embedded (Sigma-Aldrich) embryos. To maintain natural embryonic viscosity, the embryos were not desiccated before microinjection. Of the successfully injected embryos, 90% survived at least through gastrulation, and only these have been incorporated in our analysis.
Two-photon microscopy (11) was performed by using a custom-built microscope similar in design to that of ref. 12. Time-lapse image sequences were taken with a Zeiss ×25 (N.A. 0.8) oil-immersion objective and an excitation wavelength of 900–920 nm. Average laser power at the specimen was 15–25 mW.
To analyze these data, we solved numerically the discretized 3D diffusion equation on a grid. Grid points were separated by a linear distance of 1/100 of the embryo length. Boundary conditions were reflecting at the egg's outer membrane, which was determined by 3D reconstruction from a stack of two-photon images. The initial condition was uniform zero concentration throughout the embryo except for unit concentration at a point inside the embryo corresponding to the tip of the injection needle, i.e., ≈25 μm posterior of the anterior tip of the embryo. A nonlinear fitting routine (Nelder–Mead) (13) was used to adjust the diffusion coefficient to best match the experimental time courses at 18 sampled spatial positions. For the fit, we chose a single free parameter, the diffusion constant, that had to fit all 18 positions at once. To account for differences in optical depth, our fitting procedure allowed a different time-independent value of background fluorescence in each of the three focal planes. Furthermore, each sampled position was allowed a normalization parameter (again, independent of time) to account for optical unevenness. We did not notice statistically significant differences of diffusion constants between data sets of embryos of different pregastrula ages, or between data sets of fertilized versus unfertilized eggs (data not shown).
Bcd Gradient Quantification. Bcd protein profiles were extracted from digital images of stained embryos by using software routines (matlab, MathWorks, Natick, MA) that allowed a circular window of the size of a nucleus to be systematically moved along the outer edge of the embryo (14). At each position, the average pixel intensity within the window was plotted versus the projection of the window center along the anteroposterior axis of the embryo. Measurements of the Bcd concentration were made separately along the dorsal and ventral sides of the embryo; for consistency, we compared only dorsal profiles. All embryos were prepared, and images were taken under the same conditions: (i) all embryos were formaldehyde fixed for 20 min, (ii) embryos were stained and washed together in the same tube, and (iii) all images were taken with the same microscope settings in a single acquisition cycle.
In the simplest model (14), Bcd protein diffuses through the embryo and decays with a lifetime τ. The spatiotemporal dynamics of the concentration profile are determined by
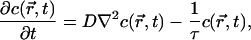 |
[1] |
where D is the diffusion constant. The steady state therefore is determined by
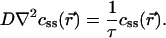 |
[2] |
If there is a source (translation of maternal RNA) at x = 0 and no variations along the dorsal–ventral direction, then the solution, projected along the anteroposterior axis, is css(x) = A exp(–x/λ), where 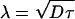 , and the constant A is set to match the diffusive flux to the translation rate at x = 0; this solution is valid if L/λ ≫ 1, as observed. Identifying staining intensity as proportional to concentration, and allowing for background fluorescence B, the raw data of Bcd immunofluorescence intensities were fitted by I = A exp(–x/λ) + B for abscissae x ∈ 15–85% egg length. A nonlinear Nelder–Mead fitting procedure was used to estimate the parameters A, B, and λ for each embryo (13).
, and the constant A is set to match the diffusive flux to the translation rate at x = 0; this solution is valid if L/λ ≫ 1, as observed. Identifying staining intensity as proportional to concentration, and allowing for background fluorescence B, the raw data of Bcd immunofluorescence intensities were fitted by I = A exp(–x/λ) + B for abscissae x ∈ 15–85% egg length. A nonlinear Nelder–Mead fitting procedure was used to estimate the parameters A, B, and λ for each embryo (13).
Results and Discussion
Testing the Diffusion Model. We measured the behavior of biologically inert, fluorescently labeled dextran molecules in living D. melanogaster embryos (see Methods). Dextran was injected into the embryo, and the spreading pattern of fluorescence was observed by using two-photon microscopy (11, 12) (see Fig. 1A). These movies provide measurements of concentration vs. time at many spatially separated locations. If the underlying molecular motion is in fact diffusive, then these dynamics at each location will be fit by the solution of the diffusion equation, with only a single free parameter (the diffusion constant itself) that can be chosen to fit all of the data. Although the data were roughly consistent with analytic predictions for diffusion along one dimension, for quantitative analysis we used numerical calculations in realistic 3D geometries to more accurately model the expected concentration dynamics (see Methods). Fig. 1B indicates that concentration changes on the length and time scales relevant for development are well described by the diffusion equation and hence that the molecular motion can be approximated by random walks.
Fig. 1.

Diffusion of inert molecules in the Drosophila embryo. (A) Two-photon image of a wild-type D. melanogaster embryo 8 min after injection of fluorescently labeled dextran molecules at the mid-plane of the embryo. The tip of the glass micropipette used for the injection is located at the anterior pole (black asterisk on the left side of the image). Colored discs show areas where fluorescence intensity was analyzed. (B) Changes in the fluorescence intensity with time for the six color-corresponding discs in A, extracted from a time series of images taken with a frame rate of 8 s. Solid lines represent the time courses computed from the best fit of a numerical 3D diffusion model. Note that 18 curves (6 per focal plane) are fit by the solutions of the same diffusion equation, with only a single free parameter, the diffusion constant D. (C) Diffusion coefficients of dextran molecules of different hydrodynamic radii (red dots). The solid line represents diffusion coefficients expected from the modified Stokes–Einstein relation (10), D = kBT/(6π ηR) + b, with a viscosity η = 4.1 ± 0.4 cP and b = 6.2 ± 1.0 μm2/s; dashed line is at the value of b.
If random molecular movement is due to Brownian motion (passive diffusion), then it is governed by the Stokes–Einstein relationship: diffusion coefficients decrease inversely with increasing molecular radius. To test this relationship, we measured diffusion constants for dextran molecules of four different nominal molecular masses (Table 1). Fig. 1C shows a good fit of the Stokes–Einstein relation to our data with an effective cytoplasmic viscosity of 4.2 cP (1 P = 0.1 kg·m–1·s–1), four times higher than water. This is well within the range of viscosities reported in other systems (15, 16). We also observe a constant, radius-independent contribution to the diffusion constant (the parameter b in the legend to Fig. 1C), as noted previously (10). This is consistent with a random “stirring” of the cytoplasm and is ≈25% of the total at molecular masses of 55 kDa, the molecular mass of Bcd. This would represent an active, and hence biologically controllable, contribution to the dynamics of molecular motion. Although this enhances the effective diffusion constant, our experiments show that it does not invalidate the description of the dynamics by the diffusion equation.
Table 1. Effective diffusion constants, D of dextran molecules of different sizes in D. melanogaster.
| Molecular mass, kDa | rs, nm | N | D, μm2/s |
|---|---|---|---|
| 10 | 2.3 | 11 | 29.1 ± 4.2 |
| 40 | 4.5 | 20 | 17.6 ± 1.8 |
| 70 | 5.9 | 8 | 15.3 ± 1.4 |
| 150 | 9.0 | 5 | 12.9 ± 3.4 |
The sample size N refers to the number of diffusion experiments analyzed.
Scaling of Gene Expression Profiles. The above results make plausible that spreading of Bcd from its localized source, and hence the generation of the primary anteroposterior gradient, will be described by the diffusion equation. However, diffusion-based models provide no natural mechanism for generating spatial patterns that scale with the size of the egg. Specifically, in systems where patterns emerge through a combination of diffusion and biochemical reactions, the diffusion constant and reaction rates determine an absolute length scale. Thus, when the size of the system changes, the spacing of the pattern elements would remain fixed (4). Although Bcd is conserved across >100 million years of dipteran evolution (17), the eggs of closely related species vary over at least a factor of five in length (Table 2). Despite these changes in size, the stripe-like patterns of gap and pair-rule genes scale with egg length, as is clear qualitatively in Fig. 2. As a quantitative example of this scaling, the point of half maximal hunchback expression is at 45 ± 6% egg length in L. sericata and at 48 ± 3% in D. melanogaster, so that the absolute positions of this boundary are changing in proportion to egg length over a nearly threefold range.
Table 2. Effective diffusion constants of 40-kDa dextran molecules in dipteran species.
| Species (mean egg length) | N | D, μm2/s |
|---|---|---|
| D. busckii (344 μm) | 8 | 14.5 ± 3.8 |
| D. melanogaster (485 μm) | 20 | 17.6 ± 1.8 |
| L. sericata (1,170 μm) | 6 | 22.8 ± 1.5 |
| C. vicina (1,420 μm) | 4 | 20.3 ± 1.3 |
In D. melanogaster, the expression patterns illustrated in Fig. 2 reflect and depend on the underlying distribution of Bcd (17). We can envision two very different mechanisms for generating scaled versions of these profiles in the species with larger embryos. First, the Bcd gradient could stay the same, and the cis-acting control sites of downstream genes could have adapted over evolution so that specific genes are activated by lower concentrations of Bcd in species with larger eggs. Alternatively, the Bcd gradient itself could scale, while the readout mechanisms encoded in the control sites of downstream genes are conserved across species.
To distinguish between these possibilities, we examined Bcd protein profiles from images of immunofluorescently stained embryos in L. sericata, D. melanogaster, and D. busckii embryos (Fig. 3A; see Methods). In Fig. 3B Upper, we show Bcd profiles from multiple embryos of each species, and in Fig. 3B Lower, we show the same data but with the x axis normalized by embryo length for each individual. Bcd protein extends farther in the larger eggs; however, when scaled to egg size, the Bcd gradients for the different species overlay one another.
Fig. 3.

Scaling of Bcd profiles. (A) Typical confocal images of Bcd immunofluorescence staining for L. sericata (top), D. melanogaster (middle), and D. busckii (bottom). The focal plane is at mid-embryo and top-embryo in the left and right columns, respectively. (Scale bar: 100 μm.) (B) Intensity profiles of Bcd fluorescence of 27 L. sericata (blue), 35 D. melanogaster (red), and 18 D. busckii (green) embryos. The abscissa in Upper is absolute; the abscissa in Lower is relative to egg length. (C) Length constants λ as a function of egg length for L. sericata (blue), D. melanogaster (red), and D. busckii (green). (D) Cumulative probability distributions of length constants λ for L. sericata (blue), D. melanogaster (red), and D. busckii (green). Asterisks indicate the means of the three distributions.
For each embryo in all species studied, the apparent concentration of Bcd vs. position has an exponential form, c(x) ∝ exp(–x/λ), which is consistent with the simplest model of diffusion and degradation (see Methods). Here, λ is a characteristic length; rapidly (slowly) decaying gradients have a short (long) λ. In a scatter plot of λ vs. egg length (Fig. 3C), we see that the large variations of egg length across species are associated with changes in absolute values of λ. Within each species, we observe significant embryo-to-embryo variability, as reported previously for D. melanogaster (14), indicating that individual egg size does not feed back on the shape of the individual gradients. The adjustment in average λ across species, however, achieves almost perfect scaling. The distributions of length constants in units of embryo length are nearly identical in all species (Fig. 3D), and in particular the mean values for these distributions agree within 2%. Thus, we conclude that the scaling of zygotic gene expression (Fig. 2) has its origins in scaling of the primary Bcd gradient.
Mechanisms of Scaling. How is scaling of the Bicoid gradient achieved? In the simplest model, the length constant 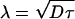 , where τ is the protein lifetime (see Methods). The active contribution to the effective diffusion constant D that we have identified above raises the possibility that total effective diffusive transport itself can be adjusted across species. To test this possibility, we injected 40-kDa dextran molecules into eggs of D. busckii, L. sericata, and Calliphora vicina. Table 2 shows a summary of our results: the diffusion constants in the different species vary only slightly. There is a tendency for increased diffusivity and decreased variability with increasing egg length, but the increase does not scale with egg size.
, where τ is the protein lifetime (see Methods). The active contribution to the effective diffusion constant D that we have identified above raises the possibility that total effective diffusive transport itself can be adjusted across species. To test this possibility, we injected 40-kDa dextran molecules into eggs of D. busckii, L. sericata, and Calliphora vicina. Table 2 shows a summary of our results: the diffusion constants in the different species vary only slightly. There is a tendency for increased diffusivity and decreased variability with increasing egg length, but the increase does not scale with egg size.
Given our diffusion and length constant measurements, τ has to scale across species, and hence the Bcd lifetime would range from τ = 3 min in D. busckii to τ = 32 min in L. sericata. These values represent a lower bound on τ, because the real diffusion constant of Bcd protein could still be modulated across species in ways that would not be detected in experiments with inert molecules, e.g., by binding to immobile proteins. But such mechanisms usually are associated with a slowing down of diffusion (18), and this is problematic: lower diffusion constants require longer protein lifetimes to achieve the same values for λ. Because relaxation to steady state requires a time ≫τ, large eggs would need more time to produce stable gradients.
To test the plausibility of these time scales, we observed the developmental sequence in all these species. The number of nuclei, Nnuc, is roughly constant across species, log2 Nnuc = 12.8 ± 0.2 (mean ± SD), implying that all species undergo 13 nuclear divisions after fertilization. We found that they show remarkably similar time courses, with 9- to 20-min cleavage cycles, a pause to cellularize at 2 h postfertilization, and gastrulation after 3 h at 25°C, as is well documented for D. melanogaster (7). Thus, the Bcd lifetimes required to generate scaling of λ are near the limit of what is possible for the larger embryos, even assuming that diffusion is unhindered. Within the simplest model, then, essentially unhindered diffusion with a species-specific adaptation of the Bcd lifetime seems to be the only viable explanation of scaling.
Bcd lifetimes could be adjusted in several different ways. First, the different sequences of Bcd in different species could directly influence the susceptibility of the protein to degradation. Second, different species could adjust the activity of the degradation machinery so as to modulate the Bcd lifetime. Finally, degradation could be occurring in a significantly nonuniform fashion, so that the effective Bcd lifetime becomes sensitive to the embryo geometry. As an example, if degradation occurs dominantly within nuclei, then the effective lifetime depends on the density of nuclei, and our observation that the number of nuclei is fixed across species implies that this density will scale with embryo size.
Conclusion
Our results indicate that scaling of body plans during the evolution of higher diptera can be traced back to scaling of spatial patterns in the expression of morphogens and to the primary anteroposterior gradient in Bcd itself. This systematic scaling across species stands in contrast to the absence of scaling of the Bcd gradient among individual D. melanogaster eggs of different sizes. Passive diffusion constants for Bcd-sized molecules do not vary significantly across species, nor do the time scales of development. Indeed, given these time scales, pattern formation based on diffusible morphogens would be physically impossible in embryos much larger than C. vicina. Of the many possible mechanisms for scaling, the only one that is consistent with our data is variation in the effective lifetime of the Bcd protein itself.
Acknowledgments
All antibodies were gifts from J. Reinitz (Stony Brook University, Stony Brook, NY). We thank G. Deshpande, A. McGregor, and R. Samanta. This work was supported, in part, by the Materials Research Science and Engineering Centers Program of the National Science Foundation under Award DMR-0213706 and by National Institutes of Health Grant P50 GM071508.
Author contributions: T.G., W.B., R.R.d.R.v.S., D.W.T., and E.F.W. designed research; T.G., W.B., R.R.d.R.v.S., D.W.T., and E.F.W. performed research; T.G., W.B., R.R.d.R.v.S., D.W.T., and E.F.W. contributed new reagents/analytic tools; T.G., W.B., R.R.d.R.v.S., D.W.T., and E.F.W. analyzed data; and T.G., W.B., D.W.T., and E.F.W. wrote the paper.
Conflict of interest statement: No conflicts declared.
References
- 1.Wolpert, L. (1969) J. Theor. Biol. 25, 1–47. [DOI] [PubMed] [Google Scholar]
- 2.Crick, F. (1970) Nature 225, 420–422. [DOI] [PubMed] [Google Scholar]
- 3.Ephrussi, A. & St. Johnston, D. (2004) Cell 116, 143–152. [DOI] [PubMed] [Google Scholar]
- 4.Cross, M. C. & Hohenberg, P. C. (1993) Rev. Mod. Phys. 65, 851–1112. [Google Scholar]
- 5.Driever, W. & Nüsslein-Volhard, C. (1988) Cell 54, 83–94. [DOI] [PubMed] [Google Scholar]
- 6.Driever, W. & Nüsslein-Volhard, C. (1988) Cell 54, 95–104. [DOI] [PubMed] [Google Scholar]
- 7.Foe, V. E., Odell, G. M. & Edgar, B. A. (1993) in The Development of Drosophila melanogaster, eds. Bate, M. & Martinez Arias, A. (Cold Spring Harbor Lab. Press, Cold Spring Harbor, NY), pp. 149–300.
- 8.Kossman, D., Small, S. & Reinitz, J. (1988) Dev. Genes Evol. 208, 290–298. [DOI] [PubMed] [Google Scholar]
- 9.Nicholson, C. & Tao, L. (1993) Biophys. J. 65, 2277–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang, I., Scholz, M. & Peters, R. (1986) J. Cell Biol. 102, 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denk, W., Strickler, J. H. & Webb, W. W. (1990) Science 248, 73–76. [DOI] [PubMed] [Google Scholar]
- 12.Svoboda, K., Denk, W., Kleinfeld, D. & Tank, D. W. (1997) Nature 385, 161–165. [DOI] [PubMed] [Google Scholar]
- 13.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. (1992) Numerical Recipes in Fortran (Cambridge Univ. Press, Cambridge, U.K.).
- 14.Houchmandzadeh, B., Wieschaus, E. & Leibler, S. (2002) Nature 415, 798–802. [DOI] [PubMed] [Google Scholar]
- 15.Luby-Phelps, K., Taylor, D. & Lanni, F. (1986) J. Cell Biol. 102, 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potma, E., de Boeij, W., Bosgraaf, L., Roelofs, J., van Haastert, P. & Wiersma, D. (2001) Biophys. J. 81, 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGregor, A. P. (2005) BioEssays 27, 904–913. [DOI] [PubMed] [Google Scholar]
- 18.Hodgkin, A. L. & Keynes, R. D. (1957) J. Physiol. 138, 253–281. [DOI] [PMC free article] [PubMed] [Google Scholar]