

Abstract
Cytochrome P450 is a family of isozymes responsible for the biotransformation of several drugs. Drug metabolism via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug interactions that can result in drug toxicities, reduced pharmacological effect, and adverse drug reactions. Recognizing whether the drugs involved act as enzyme substrates, inducers, or inhibitors can prevent clinically significant interactions from occurring. Avoiding coadministration or anticipating potential problems and adjusting a patient's drug regimen early in the course of therapy can provide optimal response with minimal adverse effects.
Drug metabolism via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug-drug interactions. A greater degree of interaction predictability has been achieved through the identification of P450 isozymes and some of the drugs that share them. Six different P450 isozymes—CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP2E1, and CYP3A4—that play important roles in drug metabolism have been identified (1, 2). Of these 6 isozymes, shared metabolism by the CYP3A4 isozyme has resulted in several clinically significant drug-drug interactions. More information about the effects of certain drugs on enzyme-mediated biotransformation has led to identification of enzyme inducers and inhibitors, providing even greater insight into the nature of the interactions.
Cytochrome P450 represents a family of isozymes responsible for biotransformation of many drugs via oxidation. The enzymes are heme-containing membrane proteins, which are located in the smooth endoplasmic reticulum of several tissues. Although a majority of the isozymes are located in the liver, extrahepatic metabolism also occurs in the kidneys, skin, gastrointestinal tract, and lungs. Significant inactivation of some orally administered drugs is due to the extensive first-pass metabolism in the gastrointestinal tract by the CYP3A4 isozyme (3).
FACTORS AFFECTING BIOTRANSFORMATION
Numerous factors affect drug biotransformation. Enzyme induction is the process by which exposure to certain substrates (e.g., drugs, environmental pollutants) results in accelerated biotransformation with a corresponding reduction in unmetabolized drug. Most drugs can exhibit decreased efficacy due to rapid metabolism, but drugs with active metabolites can display increased drug effect and/or toxicity due to enzyme induction. Enzyme inhibition occurs when 2 drugs sharing metabolism via the same isozyme compete for the same enzyme receptor site. The more potent inhibitor will predominate, resulting in decreased metabolism of the competing drug. For most drugs, this can lead to increased serum levels of the unmetabolized entity, leading to a greater potential for toxicity. For drugs whose pharmacological activity requires biotransformation from a pro-drug form, inhibition can lead to decreased efficacy.
Factors contributing to interpatient variability in biotransformation include genetic polymorphism, disease, age, and gender. The 2 isozymes most affected by genetic control are CYP2C19 and CYP2D6 (4). Individuals lacking the gene for these isozymes are poor metabolizers; those possessing it are capable of normal drug metabolism and are considered extensive metabolizers (1). Disease states affecting metabolism are hepatic disease, which affects organ function, and congestive heart failure, which causes decreased blood flow to the liver. The cytochrome P450 monooxygenase system is more affected by aging than any other metabolic pathway (3). Decreased biotransformation occurs in newborns due to underdevelopment of hepatic microsomal components (5). In the elderly, decreases in hepatic blood flow, enzyme activity, and liver mass result in reduced metabolic activity.
CYP3A4 ISOZYME INTERACTIONS
Studies on the CYP3A4 isozyme and drug-drug/drug-food interactions are becoming an integral part of drug research. Recent case reports of serious, sometimes fatal reactions due to concomitant administration of certain drugs require careful consideration. Drug prescribing for patients on multidrug regimens warrants thorough review of the patient's current therapy with respect to drug biotransformation.
For CYP3A4-metabolized drugs that require periodic monitoring of serum levels, the interaction of another CYP3A4- metabolized drug can be controlled by dosage adjustments to maintain appropriate levels of the monitored drug. Cyclosporine (CYA), tacrolimus, and carbamazepine are all substrates of CYP3A4. Coadministration of cyclosporine with a CYP3A4 inhibitor decreases an individual's CYA dosage requirement. Drinking grapefruit juice may be an inexpensive way to reduce cyclosporine dosages, but the unpredictable nature of the inhibition of CYA metabolism has not vindicated this practice. Ketoconazole and diltiazem, purer entities of CYP3A4 inhibitors, have been used successfully in this respect. Patients unable to obtain therapeutic CYA levels with orally administered cyclosporine due to inadequate absorption can been placed on either of these agents to achieve this goal.
The real problem with prescribing drugs that share the CYP3A4 pathway has been seen with drugs whose levels are not measured. When the serum levels of these drugs reach a toxic state, the toxicity can manifest itself with serious medical consequences. The pro-arrhythmic effects from high serum levels of the nonsedating antihistamines terfenadine and astemizole have severely limited their usefulness and led to the development of newer agents to take their place. Mibefradil (Posicor), a potent inhibitor of CYP3A4, was withdrawn from the market after numerous reports of serious drug-drug interactions.
Another drug class of note in this category is the 3-hydroxy- 3-methylglutaryl-coenzyme A (HMG CoA) reductase inhibitors. High serum concentrations of some of these agents are strongly linked to the development of rhabdomyolysis. Adding a CYP3A4 inhibitor to a drug regimen that includes certain HMG CoA reductase inhibitors greatly increases the patient's risk of developing rhabdomyolysis. One advantage of recognizing this drug interaction has been the subsequent studies conducted to identify which agents can be used safely in multidrug combinations. Research focusing on CYP3A4 inhibitors and HMG CoA reductase inhibitors has found that pravastatin and fluvastatin can be coadministered with itraconazole, a potent CYP3A4 inhibitor, without significant changes in maximum serum concentrations (6, 7).
CONCLUSION
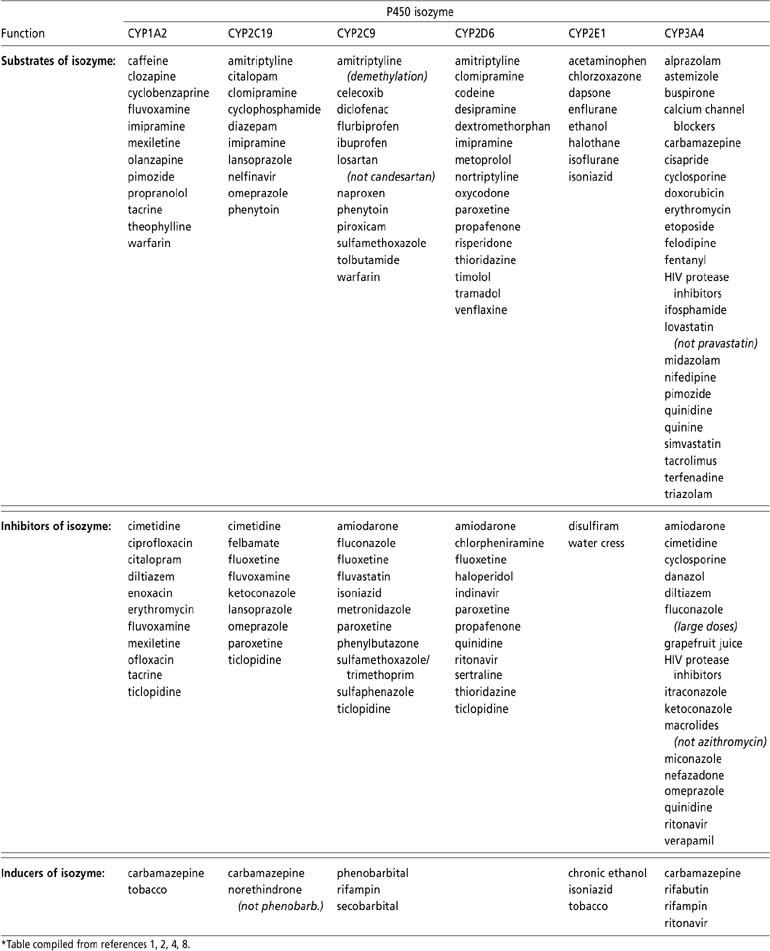
The Table has been provided to identify those drugs that share the CYP3A4 isozyme. Some drugs are metabolized by more than one isozyme, and because they possess a dual pathway of metabolism, their use may not be precluded after risk/benefit analysis. Further studies on cytochrome P450 metabolism will continue to provide clinicians with guidelines for appropriate agents to use when circumstances arise warranting the use of multiple-drug regimens.
Table.
Cytochrome P450 drug metabolism*
Acknowledgment
Special thanks to Dr. Carlos E. Velasco for his assistance in the preparation of this manuscript.
References
- 1.DiPiro JT, editor. Pharmacotherapy: A Pathophysiologic Approach. 4th ed. Stamford, Conn: Appleton & Lange; 1999. pp. 29–30. [Google Scholar]
- 2.Cupp MJ, Tracy TS. Cytochrome P450: new nomenclature and clinical implications. Am Fam Physician. 1998;57:107–116. [PubMed] [Google Scholar]
- 3.Goodman LS, Limbird LE, Milnoff PB, Gilman AG, Hardman JG, editors. Goodman & Gilman's: The Pharmacological Basis of Therapeutics. 9th ed. New York: McGraw-Hill; 1996. pp. 12–16. [Google Scholar]
- 4.Belpaire FM, Bogaert MG. Cytochrome P450: genetic polymorphism and drug interactions. Acta Clinica Belgica. 1996;51:254–260. doi: 10.1080/22953337.1996.11718518. [DOI] [PubMed] [Google Scholar]
- 5.Nelson WE, editor. Textbook of Pediatrics. 15th ed. Philadelphia: WB Saunders Co; 1996. p. 1127. [Google Scholar]
- 6.Neuvonen PJ, Kantola T, Kivisto KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998;63:332–341. doi: 10.1016/S0009-9236(98)90165-5. [DOI] [PubMed] [Google Scholar]
- 7.Kivisto KT, Kantola T, Neuvonen PJ. Different effects of itraconazole on the pharmacokinetics of fluvastatin and lovastatin. Br J Clin Pharmacol. 1998;46:49–53. doi: 10.1046/j.1365-2125.1998.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abramowicz A, editor. The Medical Letter. 1999;41(July):61–62. [Google Scholar]