

Abstract
The mitochondrial theory of aging proposes that reactive oxygen species (ROS) generated inside the cell will lead, with time, to increasing amounts of oxidative damage to various cell components. The main site for ROS production is the respiratory chain inside the mitochondria and accumulation of mtDNA mutations, and impaired respiratory chain function have been associated with degenerative diseases and aging. The theory predicts that impaired respiratory chain function will augment ROS production and thereby increase the rate of mtDNA mutation accumulation, which, in turn, will further compromise respiratory chain function. Previously, we reported that mice expressing an error-prone version of the catalytic subunit of mtDNA polymerase accumulate a substantial burden of somatic mtDNA mutations, associated with premature aging phenotypes and reduced lifespan. Here we show that these mtDNA mutator mice accumulate mtDNA mutations in an approximately linear manner. The amount of ROS produced was normal, and no increased sensitivity to oxidative stress-induced cell death was observed in mouse embryonic fibroblasts from mtDNA mutator mice, despite the presence of a severe respiratory chain dysfunction. Expression levels of antioxidant defense enzymes, protein carbonylation levels, and aconitase enzyme activity measurements indicated no or only minor oxidative stress in tissues from mtDNA mutator mice. The premature aging phenotypes in mtDNA mutator mice are thus not generated by a vicious cycle of massively increased oxidative stress accompanied by exponential accumulation of mtDNA mutations. We propose instead that respiratory chain dysfunction per se is the primary inducer of premature aging in mtDNA mutator mice.
Keywords: mitochondria, mtDNA mutator mice
The free radical theory of aging, formulated 50 years ago by Harman (1), proposes that aging and associated degenerative diseases can be attributed to deleterious effects of reactive oxygen species (ROS). Intracellular ROS are primarily generated by the mitochondrial electron transport chain, making the mitochondrial network a prime target of oxidative damage. Building on this, the mitochondrial theory of aging predicts that a vicious cycle contributes to the aging process. (i) Normal metabolism causes ROS production by the electron transport chain. (ii) ROS production induces damage to lipids, proteins, and nucleic acids in mitochondria. (iii) ROS-induced mtDNA mutations lead to the synthesis of functionally impaired respiratory chain subunits, causing respiratory chain dysfunction and augmented ROS production (2). (iv) This vicious cycle is proposed to cause an exponential increase of mtDNA mutations over time, resulting in aging and associated degenerative diseases. A substantial amount of correlative data from morphological, bioenergetic, biochemical, and genetic studies of mammalian tissues supports this theory (3). Mitochondria are larger and fewer in older individuals, and mitochondrial abnormalities such as vacuoles, abnormal cristae, and paracrystalline inclusions are more often found (4). In addition, mammalian aging has been associated with increased oxidative damage to proteins (5) and accumulation of somatic mtDNA mutations (6–8), and there is also an emerging consensus that there is an age-associated decline of respiratory chain function with increasing age (9). Furthermore, several studies have demonstrated a correlation between the rate of ROS formation and maximal lifespan in various animal species (10). Caloric restriction, shown to prolong lifespan in all studied organisms, reduces ROS production and induces antioxidant defenses (11). However, most of the available data are merely correlative and therefore do not exclude the possibility that mitochondrial damage and ROS production are consequences rather than driving forces of aging. We have recently developed a mouse model that provides experimental evidence for a causative link between mtDNA mutations and aging phenotypes in mammals (12). We created homozygous knock-in mice expressing an error-prone version of mtDNA polymerase γ (PolgAmut) (12). These mtDNA mutator mice accumulate a substantial burden of somatic mtDNA mutations, associated with premature aging and reduced lifespan. The mtDNA mutator mice display an apparently normal phenotype at birth and during early adolescence. Aging phenotypes such as weight loss, reduced s.c. fat, alopecia, kyphosis, osteoporosis, anemia, reduced fertility, and heart hypertrophy develop from 25 weeks of age (12).
We have now further characterized the patterns of mutation accumulation and cellular metabolism in mtDNA mutator mice and surprisingly found normal ROS production and only a very minor increase in oxidative damage, despite profound respiratory chain deficiency. Mutations were found to accumulate in an approximately linear manner over the mouse lifetime. The profound aging phenotypes generated by accumulation of somatic mtDNA mutations are thus not mediated by dramatically increased ROS production.
Materials and Methods
Genotyping. Standard Southern blot protocols were used for PolgA genotyping (12). Genotyping was also performed by PCR with primers (forward, 5′-gcc atc tca cca gcc cgt at-3′; reverse, 5′-gtg agg aga gtg gcc gct aa-3′) as described (12). Animal studies were approved by the animal welfare ethics committee and performed in compliance with Swedish law.
Analysis of mtDNA Mutations. Total DNA was extracted from upper parts of heads of embryonic day 13.5 (E13.5) embryos and brains of 10- to 11-month-old wild-type and mtDNA mutator mice. The somatic mtDNA mutation load was determined by PCR, cloning, and sequencing, by using primers that specifically amplified the cytochrome b gene (nucleotide position 14093–14880) and noncoding control region (nucleotide position 15378–117) of mouse mtDNA as described (12).
Isolation and Culturing of Mouse Embryonic Fibroblasts (MEFs). Primary MEF cultures were isolated from individual E13.5 embryos obtained from an intercross of heterozygous mtDNA mutator mice. The MEFs were expanded for two passages and genotyped by PCR analysis with total DNA. MEFs were cultured in complete DMEM with high glucose containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg of streptomycin. We used three wild-type and four mtDNA mutator MEF primary cultures to establish immortalized cell lines according to the 3T3 protocol (13). We transferred 3 × 105 cells into new 60-mm dishes every 3 days. All cell lines were continuously cultured without cloning, and immortalized MEF cell lines were obtained.
Measurements of Intracellular ROS. MEFs were harvested by centrifugation, washed twice with ice-cold phosphate-buffered saline (pH 7.4), and suspended in 1 ml of phosphate-buffered saline. The cells were then loaded with either 10 μM2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA) (Invitrogen) or 10 μM dihydroethidium (Invitrogen) and incubated at 37°C for 30 or 120 min.
We used an FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ) to measure ROS generation by the fluorescence intensity of 10,000 cells. The results were analyzed with the cell quest program (BD Biosciences).
Oxygen Consumption Studies. Polarographic analyses were adapted from Rustin et al. (14). In summary, 5 × 106 to 2 × 107 cells (80% confluent) were harvested and transferred to 1 ml of respiratory buffer A (10 mM Hepes, pH 7.4/225 mM mannitol/75 mM sucrose/10 mM KCl/10 mM KH2PO4/5 mM MgCl2/1 mg/ml BSA) in a polarography cuvette. Baseline respiration was recorded for 2 min before 0.5-μl aliquots of 0.1 mM rotenone were added until total inhibition of complex I was obtained. Cell membrane integrity was controlled by measuring succinate-driven respiration (10 μl of 0.1 M ADP plus 20 μl of 1 M succinate, pH 7.4) before and after digitonin permeabilization. Cells were permeabilized by using 1.1 μl of 5 mg/ml digitonin per 1 × 106 cells. Maximum uncoupled respiration rate was obtained by the addition of 1 μl of 2.5 mM carbonyl cyanide m-chlorophenylhydrazone (CCCP). Finally, inhibited respiration was measured after addition of 2.5 μl of 200 mM KCN. A two-way ANOVA was used to calculate statistical significance.
Quantification of Protein Carbonyls. Liver mitochondria were isolated by differential centrifugation as described (15). Total protein extracts and mitochondrial extracts were isolated as described (16). Protein carbonyl groups were quantified with the OxyBlot protein oxidation detection kit (Chemicon, Temecula, CA). In brief, proteins (20 μg) were incubated with 2,4-dinitrophenylhydrazine to form 2,4-dinitrophenyl hydrazone derivatives. 2,4-dinitrophenyl-derivatized proteins were blotted to nitrocellulose membranes (Hybond-C+; Amersham Biosciences, Arlington Heights, IL) in serial dilutions by using the Hybri.dot 96-well filtration manifold system (Thistle Scientific, Glasgow, U.K.). Proteins were then immunostained by using rabbit anti-2,4-dinitrophenyl antiserum (1:250) and goat anti-rabbit IgG conjugated to horseradish peroxidase (1:1,000; Amersham Biosciences). The oxidized proteins were detected by chemiluminescence (Amersham Biosciences), followed by exposure on x-ray films (Eastman Kodak, Rochester, NY). The films were scanned and then analyzed with image master 1d elite software (Amersham Biosciences). The blots were subsequently immunostained with an anti-actin antibody (Sigma) for standardization of total protein levels. The total amount of protein in all samples was also controlled by SDS/PAGE. An aliquot of 20 μgof each protein sample was separated on a 12% polyacrylamide gel and stained by Coomassie blue. The gels were scanned, and the signal in each lane was quantified with image master 1d elite software.
Aconitase Activity. Isolated mitochondria were diluted in a solution consisting of 0.1% Triton X-100, 50 mM KH2PO4, and 1 mM EDTA at pH 7.5, and aconitase activity was determined as described (17). The aconitase activity was expressed as units per unit of citrate synthase activity in the isolated mitochondria (18).
RNA Isolation and Northern Blot Analysis. Total RNA was isolated from heart by using Trizol Reagent (Invitrogen) following the instructions of the manufacturer. RT-PCR products were separated on gels, purified with the QIAEX II gel extraction kit (Qiagen, Hilden, Germany), radiolabeled with [α-32P]dCTP, and used as probes to detect glutathione peroxidase 1 (GPX1) and superoxide dismutase 2 (SOD2) transcripts (19). The signal intensities were recorded with a FUJIX Bio-Imaging Analyzer BAS 1000 (Fuji), and data were analyzed with the image gauge 3.3 program (Fuji). The RNA loading was normalized to 18S rRNA.
Western Blot Analysis. Total liver and heart homogenates and liver mitochondrial preparations were suspended in protein buffer [equal parts of suspension buffer [100 mM NaCl/10 mM Tris·HCl (pH 7.6)/1 mM EDTA (pH 8.0)/1% aprotinin/1% phenylmethylsulfonyl fluoride] and loading buffer [100 mM Tris·HCl (pH 6.8)/4% SDS/20% glycerol/200 mM DTT]. Each sample was boiled for 10 min, followed by sonication and differential centrifugation for 10 min at 10,000 × g. The supernatants containing the proteins were collected and analyzed by Western blot to determine protein levels of mouse SOD2 as described (20). We used 1:1,000 dilutions of a polyclonal antibody raised against the human recombinant SOD2 protein that also recognized mouse, bovine, and rat protein (Upstate Biotechnology, Lake Placid, NY).
H2O2 Treatments and Analysis of Apoptosis. Cells growing in flasks (70–80% confluent) were incubated for 1 h in DMEM with high glucose and with the addition of 0, 0.5, 2, and 10 mM H2O2. Treated cells were harvested and stained with annexin V and propidium iodide included in the Vybrant apoptosis assay kit 2 (Invitrogen), according to the supplied protocol. Flow cytometric analyses were performed on a BD Biosciences flow cytometer (FACScan), and results were analyzed with the cell quest program (BD Biosciences).
Results
Lifetime Increase of mtDNA Mutation Load in Mutator Mice. We measured the accumulation of somatic mtDNA mutations by sequencing multiple cloned mtDNA fragments from embryo heads and/or adult mouse brain. We found 1.5 ± 0.9 mutations/10 kilobase pairs (mut/10 kb) in wild-type embryos (n = 3) and 7.8 ± 0.4 mut/10 kb in mtDNA mutator embryos (n = 3) at E13.5 (Fig. 1A). The mutation levels were 2.3 ± 1.1 mut/10 kb in wild-type and 10.1 ± 1.6 mut/10 kb in mtDNA mutator mice at 25 weeks of age (12), whereas mutation levels were 5.3 ± 1.7 mut/10 kb in wild-type and 15.7 ± 1.8/10 kb in mtDNA mutator mice at 40 weeks of age (Fig. 1A). The load of somatic mtDNA mutations is thus substantial in midgestation mtDNA mutator embryos, and the levels increased further in an approximately linear manner during postnatal life. The same trend was evident in the noncoding control region of mtDNA (data not shown), although mutation levels were lower than in the cytochrome b gene, as seen in ref. 12.
Fig. 1.

Analysis of levels of mtDNA mutations in tissues and characterization of ROS production and respiratory chain function in MEFs. (A) The levels of mtDNA mutations (mut; cytochrome b region) in wild-type (wt; blue bars) and mtDNA mutator (red bars) embryo heads and mouse brains at different time points. Bars show mean values, and error bars indicate the SD. The low background level of PCR-induced mutations has been subtracted. (B) FACS analysis of 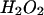 production after 30 min (thin lines) and 120 min (thick lines) incubation with dihydroethidium in primary cultures of MEFs from wild-type (blue lines) and mtDNA mutator (red lines) embryos. (C) FACS analysis of H2O2 production after 30 min incubation with carboxy-H2DCFDA in wild-type (blue line) and mtDNA mutator (red line) primary MEF cultures. (D) FACS analyses of
production after 30 min (thin lines) and 120 min (thick lines) incubation with dihydroethidium in primary cultures of MEFs from wild-type (blue lines) and mtDNA mutator (red lines) embryos. (C) FACS analysis of H2O2 production after 30 min incubation with carboxy-H2DCFDA in wild-type (blue line) and mtDNA mutator (red line) primary MEF cultures. (D) FACS analyses of  production after 30 min incubation with dihydroethidium in wild-type (blue line) and mtDNA mutator (red line) immortalized MEFs and primary MEFs from mtDNA mutator embryos (yellow line). The black line in all FACS analyses indicate negative control, i.e., wild-type cells analyzed without previous treatment with dihydroethidium or H2DCFDA. FL1-H and FL2-H, fluorescence intensity in log scale (green and red fluorescence, respectively). (E) Polarographic investigation of respiratory chain function in immortalized MEFs from wild-type (blue bars) and mtDNA mutator (red bars) embryos. Measurements (mean values ± SD) were performed without addition of exogenous substrate (rotenone-sensitive respiration) and with addition of succinate, with or without uncoupler. Asterisks indicate level of statistical significance: *, P < 0.05; **, P < 0.01.
production after 30 min incubation with dihydroethidium in wild-type (blue line) and mtDNA mutator (red line) immortalized MEFs and primary MEFs from mtDNA mutator embryos (yellow line). The black line in all FACS analyses indicate negative control, i.e., wild-type cells analyzed without previous treatment with dihydroethidium or H2DCFDA. FL1-H and FL2-H, fluorescence intensity in log scale (green and red fluorescence, respectively). (E) Polarographic investigation of respiratory chain function in immortalized MEFs from wild-type (blue bars) and mtDNA mutator (red bars) embryos. Measurements (mean values ± SD) were performed without addition of exogenous substrate (rotenone-sensitive respiration) and with addition of succinate, with or without uncoupler. Asterisks indicate level of statistical significance: *, P < 0.05; **, P < 0.01.
Normal Superoxide and Hydrogen Peroxide Levels in mtDNA Mutator MEFs. We quantified 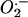 and H2O2 production in E13.5 MEFs by using intracellular probes and FACS analysis. The generation of
and H2O2 production in E13.5 MEFs by using intracellular probes and FACS analysis. The generation of 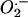 was monitored with dihydroethidium, which is oxidized by
was monitored with dihydroethidium, which is oxidized by 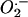 and/or OH– to yield fluorescent ethidium. We found no significant difference in
and/or OH– to yield fluorescent ethidium. We found no significant difference in 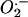 levels between wild-type and mtDNA mutator MEFs (Fig. 1B). It has been observed that very long incubation with dihydroethidium may be required to detect small alterations in ROS production (21). Despite extending the incubation time from 30 to 120 min, the
levels between wild-type and mtDNA mutator MEFs (Fig. 1B). It has been observed that very long incubation with dihydroethidium may be required to detect small alterations in ROS production (21). Despite extending the incubation time from 30 to 120 min, the 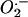 levels remained similar in wild-type and mtDNA mutator MEFs (Fig. 1B). We assessed H2O2 production with the carboxy-H2DCFDA probe. The acetate group of this probe is cleaved by esterases upon cell entry, leading to intracellular trapping of the nonfluorescent 2′,7′-dichlorofluorescin. Subsequent oxidation by ROS, particularly H2O2 and hydroxyl radical, yields the fluorescent product DCF. We found no difference in DCF formation between mtDNA mutator and wild-type MEFs when incubated with carboxy-H2DCFDA for 30 min (Fig. 1C) or 120 min (data not shown).
levels remained similar in wild-type and mtDNA mutator MEFs (Fig. 1B). We assessed H2O2 production with the carboxy-H2DCFDA probe. The acetate group of this probe is cleaved by esterases upon cell entry, leading to intracellular trapping of the nonfluorescent 2′,7′-dichlorofluorescin. Subsequent oxidation by ROS, particularly H2O2 and hydroxyl radical, yields the fluorescent product DCF. We found no difference in DCF formation between mtDNA mutator and wild-type MEFs when incubated with carboxy-H2DCFDA for 30 min (Fig. 1C) or 120 min (data not shown).
We used the standard 3T3 protocol (13) to immortalize MEFs. The mtDNA mutator and wild-type MEFs were subject to 30 passages, corresponding to approximately 40–50 cell divisions, during the immortalization procedure. We hypothesized that this large number of cell divisions might lead to enhanced ROS production in mtDNA mutator cells. However, the 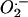 (Fig. 1D) and H2O2 (data not shown) production was similar in immortalized wild-type and mtDNA mutator MEFs.
(Fig. 1D) and H2O2 (data not shown) production was similar in immortalized wild-type and mtDNA mutator MEFs.
Respiration Is Severely Impaired in mtDNA Mutator MEFs. Next, we investigated whether the absence of ROS accumulation could be explained by mitochondrial respiratory function remaining normal despite the accumulation of mtDNA mutations. We cultivated MEFs in high glucose media (4,500 mg per liter) to 70–100% confluency and observed that the pH became acidic after 1–3 days when mtDNA mutator MEFs were cultured. The medium of wild-type MEFs remained neutral even after 1 week of culturing under the same conditions. This finding is highly suggestive of increased dependence on glycolysis and correspondingly decreased oxidative phosphorylation in the mtDNA mutator MEFs. We therefore evaluated respiratory chain function by polarographic measurements of O2 consumption. The respiration of the mtDNA mutator MEFs was <5% that of wild-type MEFs in the absence of exogenously added respiratory substrate (Fig. 1E). We assessed the function of the respiratory chain by permeabilization of the MEFs and subsequent addition of succinate. We found markedly reduced respiration in the presence of succinate and rotenone in both coupled and uncoupled conditions in mtDNA mutator MEFs (Fig. 1E). The finding of impaired respiratory chain function in mtDNA mutator MEFs is in agreement with our previous report that high levels of somatic mtDNA mutations in the heart impair the activities of individual respiratory chain enzyme complexes as well as the mitochondrial ATP production rate (12).
No Increase in Hydrogen Peroxide-Induced Cell Death in mtDNA Mutator MEFs. We tested whether mtDNA mutator MEFs were prone to undergo apoptosis in response to oxidative stress by adding increasing levels of H2O2 to the culture medium. Treated cells were incubated with fluorescently labeled annexin V, which binds to phosphatidyl serine exposed on the cell surface during apoptosis, and propidium iodide, which accumulates in necrotic cells. This approach allows assessment of cell viability by FACS analysis because live cells do not retain propidium iodide and do not bind annexin V on the cell surface. We treated wild-type and mtDNA mutator MEFs with 0.5 mM H2O2 (Fig. 2A) or 2 mM H2O2 for 1 h (Fig. 2B) and observed no difference in the fraction of necrotic or apoptotic cells. We also incubated cells with 10 mM H2O2 for 1 h (Fig. 2C) and observed no difference in the fraction of apoptotic cells, whereas the fraction of necrotic cells was lower in treated mtDNA mutator MEFs (63.0 ± 3.9%) compared with wild-type MEFs (78.5 ± 4.9%). We also performed similar analyses in immortalized MEFs and found no difference in the proportion of necrotic and apoptotic cells when comparing wild-type and mtDNA mutator cells (data not shown). We also extended the H2O2 treatment to 24 h in primary culture MEFs and observed similar fractions of necrotic and apoptotic cells irrespective of the genotype (data not shown). The mtDNA mutator MEFs are thus no more prone to undergo necrosis and apoptosis in response to oxidative stress than wild-type MEFs.
Fig. 2.

H2O2 induced necrosis and apoptosis in wild-type (blue bars) and mtDNA mutator (red bars) primary MEF cultures. Cells were stained with propidium iodide and annexin V and assayed for viability (mean values ± SD) by FACS analysis after 1 h incubation in culture media containing 0.5 mM (A), 2 mM (B), and 10 mM (C) H2O2 or in medium without H2O2 (D). Asterisks indicate level of statistical significance: *, P < 0.05; **, P < 0.01.
No Difference in Oxidative Damage to Proteins. To determine the level of oxidative damage to proteins, we measured the amount of carbonyl groups and found no difference in total protein extracts from hearts of 40-week-old mtDNA mutator and wild-type mice (Fig. 3A). However, a small increase in oxidative damage was found in total liver protein extracts from mtDNA mutator mice (relative levels, 139 ± 21%) compared with wild-type mice (100 ± 23%). Surprisingly, carbonyl levels in protein extracts from liver mitochondria were not significantly different between mtDNA mutator (107 ± 19%) and wild-type mice (100 ± 18%), suggesting that the increase in total liver extracts was because of extra mitochondrial protein carbonylation in mtDNA mutator mice.
Fig. 3.

Oxidative damage to proteins in tissues of wild-type (blue bars) and mtDNA mutator (red bars) mice. (A) Relative amounts of carbonyl groups in liver total protein extracts, liver mitochondrial protein extracts, and heart total protein extracts at 40 weeks of age. (B) Aconitase enzyme activity in hearts of 12-, 25-, and 40-week-old mice. Bars show mean values, and error bars indicate SD. Asterisks indicate level of statistical significance: *, P < 0.05.
Iron–sulfur clusters are particularly sensitive to ROS damage, and proteins harboring such clusters may lose activity in response to oxidative stress (22). The activity of mitochondrial aconitase is critically dependent on an iron–sulfur cluster, and this enzyme activity is, therefore, often used as a marker of oxidative damage. We found normal aconitase activities in heart mitochondria from 12-, 25-, and 40-week-old mtDNA mutator mice (Fig. 3B). Hence, no evidence of increased protein oxidation was found in mitochondria of the prematurely aging mtDNA mutator mice.
No Increased Expression of Antioxidant Defense Enzymes. Increased ROS levels will induce the expression of antioxidant defense enzymes, and increased ROS scavenging could potentially explain the normal ROS levels and absence of oxidative damage in cells and tissues from mtDNA mutator mice. We performed Northern blot analyses and found normal levels of manganese-dependent SOD (or SOD2) transcripts (Fig. 4A) and only a minor (1.5-fold, nonsignificant) increase in glutathione peroxidase 1 (GPX1) transcript levels in heart of 25- and 40-week-old mtDNA mutator mice (Fig. 4A). We also analyzed SOD2 protein levels in total protein extracts from heart and liver and liver mitochondrial protein extracts (Fig. 4B) and found no difference between wild-type and mtDNA mutator mice at age 40 weeks. The minimal induction of expression of SOD2 and GPX1, two ROS scavenging enzymes located in mitochondria, suggests that there is no major increase in oxidative stress in mtDNA mutator mice.
Fig. 4.

Expression of antioxidant defense enzymes. (A) Northern blot analyses showing transcript levels of GPX1 and SOD2 in hearts of 25- and 40-week-old mice. The levels of 18S ribosomal RNA was used as loading control. (B) Western blot analysis showing SOD2 protein levels in total protein extracts from heart, liver, and purified liver mitochondria at 40 weeks of age. Mut, mutants; wt, wild types.
Discussion
We demonstrate that increased ROS formation and oxidative damage are unlikely to be major pathophysiological events in the premature aging process induced by elevated levels of mtDNA mutations. These results are in agreement with findings from a similar mtDNA mutator mouse model that was described recently (23). We have previously studied mitochondrial transcription factor A(Tfam) conditional knockout mice and demonstrated that loss of this protein causes severe mtDNA depletion and a profound respiratory chain deficiency in affected tissues (19, 20, 24–26). Loss of Tfam leads to considerable cell death by apoptotic and/or necrotic pathways in embryos (19, 20) and differentiated tissues such as heart (19, 24), neurons (25), and insulin-secreting β-cells (26). However, in Tfam knockout mice, no or minor induction in the expression of enzymes involved in ROS scavenging was found, suggesting that oxidative stress is not an important pathophysiological mechanism in these animals. Loss of TFAM protein should lead to a global decrease of all 13 mtDNA-encoded respiratory chain subunits, resulting in an essentially quantitative defect of respiratory chain function. In contrast, the mtDNA mutator mice acquire an apparently random set of mtDNA point mutations, which would be expected to result in both quantitative and qualitative effects on respiratory chain function. It can be anticipated that at least some of the point mutations should result in increased ROS production in mtDNA mutator mice. It is therefore surprising that we found no increase of ROS production and no or minor increase of oxidative damage in cultured cells and tissues from mtDNA mutator mice at different ages.
The mitochondrial theory of aging predicts that levels of mtDNA mutations should increase exponentially as a consequence of a vicious cycle by accelerating oxidative stress. However, we found an approximately linear increase of mtDNA mutation levels from midgestation to late adult life in mtDNA mutator mice, implying that there is no vicious cycle. It should be noted that our measurements of mtDNA mutation load might be an underestimate because cells with the highest levels of deleterious mutations may be lost because of cell death and/or replicative disadvantage. Point mutations in protein coding genes affecting codon positions 1 and 2 are more likely to result in amino acid replacements and respiratory chain dysfunction than mutations affecting codon position 3. It can therefore be anticipated that point mutations resulting in a large amount of cell death would lead to preferential loss of cells with mutations at codon positions 1 and 2. In contrast to this prediction, the spectrum of mtDNA mutations in protein-coding genes in mtDNA mutator mice is apparently random with respect to codon position (12), even in the oldest animals studied (data not shown).
Formation of 8-oxo-2′-deoxyguanosine (oxo8-dG) is the most common consequence of oxidative damage to DNA in a cell (6). It has been reported that aging human brain exhibits an increase of oxo8-dG in mtDNA and that this increase correlates with mitochondrial dysfunction (27). It has since been suggested that accumulation of oxo8-dG to high levels leads to increased levels of mtDNA mutations, triggering mtDNA instability, reduced rates of mitochondrial protein synthesis, and production of mutant polypeptides that compromise mitochondrial respiration (for review, see ref. 28). However, a recent study failed to find evidence of mitochondrial respiratory dysfunction even when levels of oxo8-dG are highly elevated (29). Mice deficient for oxoguanine DNA glycosylase (OGG1), an enzyme responsible for oxo8-dG removal, have a 20-fold increase in oxo8-dG levels compared with wild-type mice, yet their mitochondria are functionally normal. The authors found no differences in ATP synthesis rate or maximal activities of complexes I and IV and no indication of increased oxidative stress in mitochondria from OGG1–/– mice, as measured by protein carbonyl content (29). Such findings support the idea that oxidative stress may not be a major inducer of mtDNA mutations in mammals.
Numerous investigators have reported a positive correlation between increased ROS production/oxidative damage and age (3, 30–33). Furthermore, various manipulations that increase lifespan also diminish the age-related increase in oxidatively damaged molecules (3, 34–36). Support for ROS involvement in aging has been obtained from experiments on caloric restricted rodents (37–41) and on different genetically modified flies and worms (36, 42–45). However, most of the evidence that is consistent with the free radical theory of aging is indirect, and some results contradict the theory. Overexpression of cytosolic Cu–Zn SOD (or SOD1) and catalase in long-lived strains of Drosophila melanogaster produced no beneficial effects on survival in the mutant flies (46). The introduction of transgenes encoding SOD2 or thioredoxin reductase in the same genetic background also failed to extend lifespan (46). However, others have shown that overexpression of Cu–Zn SOD in the fly resulted in an increased lifespan of up to 48%, with no additional benefit of overexpressing catalase in the same model (47). Experimental results obtained from genetic mouse models, in which expression levels of different antioxidant enzymes have been altered, are even more confusing. Overexpression of Cu–Zn SOD in mice resulted in animals that were more resistant to cerebral ischemia but had the same lifespan as wild-type mice (48). Conversely, overexpression of human catalase targeted to the mitochondria resulted in extension of median and maximum lifespan in transgenic mice (39). In these animals, H2O2 production and H2O2-induced aconitase inactivation were attenuated, the oxidative damage and the development of mitochondrial deletions were decreased, and the development of cataract and cardiac pathology was delayed. In contrast, targeting human catalase to peroxisomes or nucleus did not extend the lifespan of transgenic mice (39). A mouse heterozygous for a null mutation of SOD2 displayed 30–80% reduced activity of this enzyme in a variety of tissues (37, 40). This resulted in increased oxidative damage in all examined tissues of SOD2+/– mice compared with wild-type mice and higher incidence of cancer (37, 41). However, no difference in either mean or maximal lifespan was evident (37). Furthermore, mice deficient for the both of the major mitochondrial antioxidant enzymes SOD2 and GPX1 (SOD2+/–/GPX1–/–) are extremely sensitive to oxidative stress, yet appear phenotypically normal, and can reproduce and have normal lifespan (38). It should be emphasized that the interpretation of the above-mentioned experiments in model organisms is complicated by the fact that ROS do not only cause oxidative damage but also have important roles in cell signaling (49).
There is clearly an intense, ongoing debate about the role of ROS and oxidative stress in the aging process. The present study shows that there is no direct connection between increased levels of mtDNA mutations and elevated ROS production and argues against any direct role of oxidative stress in the aging process. Because mtDNA mutator mice develop aging-related phenotypes without evidence of increased oxidative stress, but nevertheless do exhibit respiratory chain deficiency, the latter would seem to be a better candidate for the primary inducer of premature aging. Respiratory chain dysfunction may accelerate aging by provoking bioenergy deficit in physiologically crucial cells, by decreasing the signal threshold for cell death, by causing replicative senescence in stem cells, or by some other mechanism. The mtDNA mutator mouse will be an invaluable resource to dissect these pathways further.
Acknowledgments
A.T. is supported by Funds of Karolinska Institutet and Gun and Bertil Sthones Stiftelse and a fellowship from Loo and Hans Ostermans Stiftelse. N.-G.L. is supported by the Swedish Research Council, the Torsten and Ragnar Söderbergs Foundation, the Swedish Heart and Lung Foundation, the Swedish Foundation for Strategic Research (Functional Genomics and INGVAR), the European Union (EUMITOCOMBAT project), and Funds of Karolinska Institutet. J.N.S. and H.T.J. are supported by the Academy of Finland, the Juselius Foundation, the European Union, and the Tampere University Hospital Medical Research Fund. R.W. is supported by Karolinska Institutet fonder and Children Living with Inherited Metabolic Diseases (United Kingdom).
Author contributions: A.T., J.N.S., R.W., and N.-G.L. designed research; A.T., A.H., A.W., A.T.R., E.D., I.K., and R.W. performed research; A.T., H.T.J., and N.-G.L. analyzed data; and A.T., A.H., H.T.J., and N.-G.L. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: En, embryonic day n; GPX1, glutathione peroxidase; H2-DCFDA, 2′,7′-dichlorodihydrofluorescein diacetate; MEF, mouse embryonic fibroblasts; mut/10 kb, mutations/10 kilobase pairs; oxo8-dG, 8-oxo-2′-deoxyguanosine; ROS, reactive oxygen species; SOD, superoxide dismutase; Tfam, transcription factor A.
References
- 1.Harman, D. (1956) J. Gerontol. 11, 298–300. [DOI] [PubMed] [Google Scholar]
- 2.Harman, D. (1972) J. Am. Geriatr. Soc. 20, 145–147. [DOI] [PubMed] [Google Scholar]
- 3.Balaban, R. S., Nemoto, S. & Finkel, T. (2005) Cell 120, 483–495. [DOI] [PubMed] [Google Scholar]
- 4.Frenzel, H. & Feimann, J. (1984) Mech. Aging Dev. 27, 29–41. [DOI] [PubMed] [Google Scholar]
- 5.Oliver, C. N., Ahn, B. W., Moerman, E. J., Goldstein, S. & Stadtman, E. R. (1987) J. Biol. Chem. 262, 5488–5491. [PubMed] [Google Scholar]
- 6.Hayakawa, M., Torii, K., Sugiyama, S., Tanaka, M. & Ozawa, T. (1991) Biochem. Biophys. Res. Commun. 179, 1023–1029. [DOI] [PubMed] [Google Scholar]
- 7.Hayakawa, M., Hattori, K., Sugiyama, S. & Ozawa, T. (1992) Biochem. Biophys. Res. Commun. 189, 979–985. [DOI] [PubMed] [Google Scholar]
- 8.Mecocci, P., MacGarvey, U., Kaufman, A. E., Koontz, D., Shoffner, J. M., Wallace, D. C. & Beal, M. F. (1993) Ann. Neurol. 34, 609–616. [DOI] [PubMed] [Google Scholar]
- 9.Cottrell, D. A. & Turnbull, D. M. (2000) Curr. Opin. Clin. Nutr. Metab. Care 3, 473–478. [DOI] [PubMed] [Google Scholar]
- 10.Perez-Campo, R., Lopez-Torres, M., Cadenas, S., Rojas, C. & Barja, G. (1998) J. Comp. Physiol. B 168, 149–158. [DOI] [PubMed] [Google Scholar]
- 11.Barja, G. (2004) Biol. Rev. Camb. Philos. Soc. 79, 235–251. [DOI] [PubMed] [Google Scholar]
- 12.Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly, Y. M., Gidlof, S., Oldfors, A., Wibom, R., et al. (2004) Nature 429, 417–423. [DOI] [PubMed] [Google Scholar]
- 13.Meek, R. L., Bowman, P. D. & Daniel, C. W. (1977) Exp. Cell Res. 107, 277–284. [DOI] [PubMed] [Google Scholar]
- 14.Rustin, P., Chretien, D., Bourgeron, T., Gerard, B., Rotig, A., Saudubray, J. M. & Munnich, A. (1994) Clin. Chim. Acta 228, 35–51. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Vizarra, E., Lopez-Perez, M. J. & Enriquez, J. A. (2002) Methods 26, 292–297. [DOI] [PubMed] [Google Scholar]
- 16.Ekstrand, M. I., Falkenberg, M., Rantanen, A., Park, C. B., Gaspari, M., Hultenby, K., Rustin, P., Gustafsson, C. M. & Larsson, N. G. (2004) Hum. Mol. Genet. 13, 935–944. [DOI] [PubMed] [Google Scholar]
- 17.Robinson J. B., Sumegi, B. L. & Srere P. A. (1987) in Mitochondria: A Practical Approach, eds. Darley-Usmar, V. M., Rickwood, D. & Wilson, M. T. (IRL, Oxford), pp. 153–170.
- 18.Wibom, R., Hagenfeldt, L. & von Dobeln, U. (2002) Anal. Biochem. 311, 139–151. [DOI] [PubMed] [Google Scholar]
- 19.Wang, J., Silva, J. P., Gustafsson, C. M., Rustin, P. & Larsson, N. G. (2001) Proc. Natl. Acad. Sci. USA 98, 4038–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsson, N. G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., Barsh, G. S. & Clayton, D. A. (1998) Nat. Genet. 18, 231–236. [DOI] [PubMed] [Google Scholar]
- 21.Dufour, E., Boulay, J., Rincheval, V. & Sainsard-Chanet, A. (2000) Proc. Natl. Acad. Sci. USA 97, 4138–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flint, D. H., Tuminello, J. F. & Emptage, M. H. (1993) J. Biol. Chem. 268, 22369–22376. [PubMed] [Google Scholar]
- 23.Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., Hofer, T., Seo, A. Y., Sullivan, R., Jobling, W. A., et al. (2005) Science 309, 481–484. [DOI] [PubMed] [Google Scholar]
- 24.Wang, J., Wilhelmsson, H., Graff, C., Li, H., Oldfors, A., Rustin, P., Bruning, J. C., Kahn, C. R., Clayton, D. A., Barsh, G. S., et al. (1999) Nat. Genet. 21, 133–137. [DOI] [PubMed] [Google Scholar]
- 25.Sorensen, L., Ekstrand, M., Silva, J. P., Lindqvist, E., Xu, B., Rustin, P., Olson, L. & Larsson, N. G. (2001) J. Neurosci. 21, 8082–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silva, J. P., Kohler, M., Graff, C., Oldfors, A., Magnuson, M. A., Berggren, P. O. & Larsson, N. G. (2000) Nat. Genet. 26, 336–340. [DOI] [PubMed] [Google Scholar]
- 27.Richter, C., Park, J. W. & Ames, B. N. (1988) Proc. Natl. Acad. Sci. USA 85, 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bohr, V. A. (2002) Free Radical Biol. Med. 32, 804–812. [DOI] [PubMed] [Google Scholar]
- 29.Stuart, J. A., Bourque, B. M., de Souza-Pinto, N. C. & Bohr, V. A. (2005) Free Radical Biol. Med. 38, 737–745. [DOI] [PubMed] [Google Scholar]
- 30.Ku, H. H., Brunk, U. T. & Sohal, R. S. (1993) Free Radical Biol. Med. 15, 621–627. [DOI] [PubMed] [Google Scholar]
- 31.Sohal, R. S., Toy, P. L. & Allen, R. G. (1986) Mech. Aging Dev. 36, 71–77. [DOI] [PubMed] [Google Scholar]
- 32.Sharma, S. P., Sharma, M. & Kakkar, R. (1995) Gerontology 41, 86–93. [DOI] [PubMed] [Google Scholar]
- 33.Munkres, K. & Rana, R. S. (1978) Mech. Aging Dev. 7, 407–415. [DOI] [PubMed] [Google Scholar]
- 34.Melov, S., Ravenscroft, J., Malik, S., Gill, M. S., Walker, D. W., Clayton, P. E., Wallace, D. C., Malfroy, B., Doctrow, S. R. & Lithgow, G. J. (2000) Science 289, 1567–1569. [DOI] [PubMed] [Google Scholar]
- 35.Migliaccio, E., Giorgio, M., Mele, S., Pelicci, G., Reboldi, P., Pandolfi, P. P., Lanfrancone, L. & Pelicci, P. G. (1999) Nature 402, 309–313. [DOI] [PubMed] [Google Scholar]
- 36.Orr, W. C. & Sohal, R. S. (1994) Science 263, 1128–1130. [DOI] [PubMed] [Google Scholar]
- 37.Van Remmen, H., Ikeno, Y., Hamilton, M., Pahlavani, M., Wolf, N., Thorpe, S. R., Alderson, N. L., Baynes, J. W., Epstein, C. J., Huang, T. T., et al. (2003) Physiol. Genomics 16, 29–37. [DOI] [PubMed] [Google Scholar]
- 38.Van Remmen, H., Qi, W., Sabia, M., Freeman, G., Estlack, L., Yang, H., Mao Guo, Z., Huang, T. T., Strong, R., Lee, S., Epstein, C. J. & Richardson, A. (2004) Free Radical Biol. Med. 36, 1625–1634. [DOI] [PubMed] [Google Scholar]
- 39.Schriner, S. E., Linford, N. J., Martin, G. M., Treuting, P., Ogburn, C. E., Emond, M., Coskun, P. E., Ladiges, W., Wolf, N., Van Remmen, H., et al. (2005) Science 308, 1909–1911. [DOI] [PubMed] [Google Scholar]
- 40.Li, Y., Huang, T. T., Carlson, E. J., Melov, S., Ursell, P. C., Olson, J. L., Noble, L. J., Yoshimura, M. P., Berger, C., Chan, P. H., et al. (1995) Nat. Genet. 11, 376–381. [DOI] [PubMed] [Google Scholar]
- 41.Melov, S., Coskun, P., Patel, M., Tuinstra, R., Cottrell, B., Jun, A. S., Zastawny, T. H., Dizdaroglu, M., Goodman, S. I., Huang, T. T., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy, C. T., McCarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J., Li, H. & Kenyon, C. (2003) Nature 424, 277–283. [DOI] [PubMed] [Google Scholar]
- 43.Johnson, T. E., de Castro, E., Hegi de Castro, S., Cypser, J., Henderson, S. & Tedesco, P. (2001) Exp. Gerontol. 36, 1609–1617. [DOI] [PubMed] [Google Scholar]
- 44.Lee, S. S., Kennedy, S., Tolonen, A. C. & Ruvkun, G. (2003) Science 300, 644–647. [DOI] [PubMed] [Google Scholar]
- 45.Rogina, B., Reenan, R. A., Nilsen, S. P. & Helfand, S. L. (2000) Science 290, 2137–2140. [DOI] [PubMed] [Google Scholar]
- 46.Orr, W. C., Mockett, R. J., Benes, J. J. & Sohal, R. S. (2003) J. Biol. Chem. 278, 26418–26422. [DOI] [PubMed] [Google Scholar]
- 47.Parkes, T. L., Elia, A. J., Dickinson, D., Hilliker, A. J., Phillips, J. P. & Boulianne, G. L. (1998) Nat. Genet. 19, 171–174. [DOI] [PubMed] [Google Scholar]
- 48.Gallagher, I. M., Jenner, P., Glover, V. & Clow, A. (2000) Neurosci. Lett. 289, 221–223. [DOI] [PubMed] [Google Scholar]
- 49.Finkel, T. (2003) Curr. Opin. Cell Biol. 15, 247–254. [DOI] [PubMed] [Google Scholar]