

Abstract
Neurotransmitters such as acetylcholine (ACh) and glycine mediate fast synaptic neurotransmission by activating pentameric ligand-gated ion channels (LGICs). These receptors are allosteric transmembrane proteins that rapidly convert chemical messages into electrical signals. Neurotransmitters activate LGICs by interacting with an extracellular agonist-binding domain (ECD), triggering a tertiary/quaternary conformational change in the protein that results in the fast opening of an ion pore domain (IPD). However, the molecular mechanism that determines the fast opening of LGICs remains elusive. Here, we show by combining whole-cell and single-channel recordings of recombinant chimeras between the ECD of α7 nicotinic receptor (nAChR) and the IPD of the glycine receptor (GlyR) that only two GlyR amino acid residues of loop 7 (Cys-loop) from the ECD and at most five α7 nAChR amino acid residues of the M2-M3 loop (2-3L) from the IPD control the fast activation rates of the α7/Gly chimera and WT GlyR. Mutual interactions of these residues at a critical pivot point between the agonist-binding site and the ion channel fine-tune the intrinsic opening and closing rates of the receptor through stabilization of the transition state of activation. These data provide a structural basis for the fast opening of pentameric LGICs.
Keywords: allosteric proteins, chimeric receptor, Cys-loop receptor, transition state
Pentameric ligand-gated ion channels (LGICs), such as the cationic nicotinic acetylcholine receptor (nAChR) and the anionic glycine receptor (GlyR), mediate fast excitatory or inhibitory chemical neurotransmission between neurons (1-6). A unique feature of these receptors is that they activate the ion channel, a process known as gating, in less than a ms. For nicotinic receptors, a detailed single-channel analysis has recently established a speed limit for the opening of the ion channel in the μs time range (7). Perturbations of this rapid transmission pathway by natural mutants lead to severe diseases such as congenital myasthenic syndromes (8), hereditary hyperekplexia (9), or epileptic disorders (10).
Pentameric LGICs, or Cys-loop receptors, are composed of five homologous subunits, sharing a common structural organization, arranged (pseudo)symmetrically around the central ion pore (1, 2). All subunits are made of two distinct topological domains: the extracellular (ECD) and the ion pore domains (IPD). First, the ECD is folded into a twisted β-sandwich core, as revealed by x-ray crystallographic studies of the mollusk acetylcholine-binding protein (AChBP), a soluble pentameric protein homologous to the extracellular domain of LGICs (11-14). Second, electron microscopy images of Torpedo nAChR at 4-Å resolution revealed that the four transmembrane segments (M1 to M4) of the IPD are folded into α-helices joined by linking loops of variable lengths (15). By combining these structural data, we built a 3D model of the full α7 nAChR (16). In this model, the coupling zone located at the interface between the two domains is framed by flexible loops. As noted earlier for GABAA receptors (17), loops 2 and 7 (the so-called Cys-loop) from the ECD are close (<5 Å) to the C-terminal end of α-helix M2 from the IPD. Even though structural information is now available for the linking of the ECD and IPD, the molecular mechanism coupling the agonist-binding site and ion channel resulting in the fast opening of the ion channel remains elusive.
In this article, we show, by combining whole-cell and single-channel recordings of recombinant chimeras between the ECD of the homomeric α7 nicotinic receptor (nAChR) and the IPD of the homomeric α1 glycine receptor (GlyR), that only two GlyR amino acid residues of the Cys-loop from the ECD and at most five α7 nAChR amino acid residues of the M2-M3 loop (2-3L) from the IPD fine-tune the fast activation rates of the α7/Gly chimera and WT GlyR. We propose that mutual interactions of these residues with their WT counterparts in the LGICs result in the acceleration of both the intrinsic opening and closing rate constants through stabilization of the transition state of activation. These data provide a structural basis for the fast opening of pentameric LGICs.
Methods
Mutagenesis. Chick α7 cDNA encoding the ECD of α7 nAChR was fused with the human α1 cDNA encoding the IPD of α1 glycine receptor. Chimeric cDNAs were subcloned into pMT3 containing the peptide signal sequence of the α7 subunit. Mutants of the Cys-loop region were performed on the synthetic gene of the α7 subunit as described (18), whereas, for the 2-3L region and WT GlyR, mutations were created by using the QuikChange kit (Stratagene) according to the manufacturer's instructions. All mutant and chimeric cDNAs were verified by restriction enzyme analysis and sequencing.
Whole-Cell Recordings. Human embryonic kidney cells (HEK 293) were grown and transiently transfected as described (18). Recordings were made 48-72 h after transfection at room temperature (25°C ± 3°C), by using the whole-cell patch-clamp technique (19). Cells were maintained at a holding potential of -60 mV. External solution contained 140 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM Glucose, and 10 mM Hepes-NaOH (pH 7.3); Ca2+ and Mg2+ were removed just after the whole-cell configuration was obtained. To determine chloride permeability of the channel, extracellular chloride was reduced through total replacement of NaCl of the external solution by 140 mM sodium isethionate. Patch pipette (1-2 MΩ) solutions contained 140 mM CsCl2, 2 mM NaATP, 10 mM Hepes-CsOH, and 10 mM EGTA (pH 7.3). Current-voltage relationships were determined by the application of two inverted voltage ramps (+50 to -100 mV in 200 ms, from an initial holding potential of -60 mV) during the steady-state phase of an ACh-evoked response. Leak currents were subtracted. External solutions containing the agonist (± antagonist) were delivered through three parallel tubes placed immediately above the cell. These tubes are displaced horizontally with the aid of a computer-driven system (SF 77A Perfusion fast step, Warner) that ensured a 10-90% solution exchange in 5-10 ms, as measured by changes of tip potential of an open electrode in external solution and in 1:10 dilution of external solution with water. The perfusion system thus determines accurately apparent on-rates kapp ≤ 1,000 s-1. Macroscopic currents were analyzed by using pulsefit software (HEKA Electronics, Lambrecht/Pfalz, Germany). The following single exponential function was fitted to the initial rising (10-90%) agonist-evoked currents: I(t) = I0 + I∞[1 - exp(-kapp × t)] where I0 and I∞ are the initial and steady-state currents, respectively, and kapp is the apparent constant of activation. From the simple conformational change that follows binding reactions between the agonist and its receptor, one can derive kapp as a function of agonist concentration from:
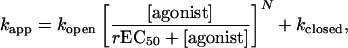 |
[1] |
where kopen is an approximation of the opening rate constant, kclosed is an approximation of the closing rate constant, rEC50 is the concentration of agonist that gives half of the theoretical opening rate constant, and N the actual number of binding sites. For all α7/Gly chimeras, N was fixed to five as previously shown for WT α7 nAChR (20). Dose-response curves were obtained by measuring the steady-state amplitude of the currents evoked by different concentrations of agonist, and data were fitted with the Hill equation to yield EC50 and Hill coefficient, nH. All data reported are means ± SD. Statistical differences were determined by using the Mann-Whitney U test with statview 5.0.
Single-Channel Recordings. Recordings were obtained in the cell-attached configuration at 25°C ± 3°C at a holding potential of -60 mV. Extracellular and patch pipette (around 5 MΩ) solutions contained 140 mM NaCl, 2.8 mM KCl, 10 mM glucose, and 10 mM Hepes-NaOH (pH 7.3). Single-channel currents were recorded by using an EPC-10 amplifier (HEKA), sampled at 100-μs intervals, recorded to the computer hard disk, and detected by the half-amplitude threshold criterion by using the program pulsetools (HEKA) at a final bandwidth of 1 kHz. Open-time histograms were plotted by using a logarithmic abscissa and a square-root ordinate (21) and fitted to the sum of exponentials with an imposed time resolution of 200 μs.
Binding. ACh competition measurements against the initial rate of α-125I-bungarotoxin binding were as determined previously (18, 22), and data were fitted with the Hill equation.
Results
Design of α7/Gly Chimeras. To unravel the molecular basis of the activation mechanism of LGICs, we constructed recombinant chimeric subunits containing the two topological distinct domains: the ECD of the nicotinic homomeric α7 nAChR and the IPD of the homomeric α1 GlyR. These homomeric α7/Gly chimeras, homologous to the previously described α7/5HT3 chimera (23), are novel because the ECD of a cationic receptor is coupled to an anionic channel. Three different cDNA constructs were made and transfected in HEK 293 cells that resulted in cell-surface expression of functional receptors. Only one chimera, named α7/Gly, is detailed in this article (for details, see Supporting Materials and Methods and Fig. 4, which are published as supporting information on the PNAS web site).
α7/Gly Chimera Is Gated by ACh and Selective to Chloride. The α7/Gly chimera was gated by ACh, producing large whole-cell inward currents (1-5 nA) with very little, if any, desensitization (Fig. 1a). This chimeric receptor displayed the pharmacological properties, as well as the allosteric site for calcium potentiation (24), expected from its α7 nAChR ECD and was selective for chloride as expected from its GlyR ion channel.
Fig. 1.

Functional characterization of α7/Gly chimera. (a) Whole-cell current (1 mM ACh) through α7/Gly and its reversible inhibition by 300 μM dihydro-β-erythroidine (DHβE). (b) Current potentiation by 4 mM Ca2+ during a 10-mM ACh application. Recorded from a different cell. (c) Dose-response curves in the absence (open circles) or presence (filled circles) of 4 mM Ca2+. Currents are normalized to 10 mM ACh-evoked currents. (d) Current-voltage relationships. External sodium chloride (control condition) substituted by sodium isethionate (Ise condition). (e) Typical currents evoked by 100 μM (bottom trace), 1 mM (middle trace), or 3 mM (top trace) ACh concentrations. kapp, derived from single exponential functions, are indicated. (f) kapp plotted as function of ACh concentrations. Data were fitted with Eq. 1. For clarity, only positive error bars are represented.
First, ACh-evoked current amplitudes increased in a dose-dependent manner (Fig. 1c); the apparent affinity (effective concentration for half maximal response, EC50 = 300 μM; see Table 1, which is published as supporting information on the PNAS web site) was in the same range as described for α7/5HT3 chimeric receptor (22). Second, the competitive α7 receptor antagonist dihydro-β-erythroidine (DHβE) reversibly inhibited responses evoked by ACh (Fig. 1a). Third, in cells expressing α7/Gly, external application of 4 mM Ca2+ potentiated the response evoked by a saturating ACh concentration (Fig. 1b). This calcium potentiation was observed at all concentrations of ACh, as described for α7/5HT3 chimera (24) (Fig. 1c). Finally, substitution of external sodium chloride by sodium isethionate resulted in the expected right-shift of reversal potential indicative of a chloride permeability (Fig. 1d). These results demonstrate the functional coupling of a cationic ECD to an anionic IPD and strongly suggest that a common activation or gating mechanism is shared by all members of the pentameric LGIC superfamily.
α7/Gly Displays Slow Activation Rates. Interestingly, applications longer than 1 s were necessary to reach a steady-state response even at saturating ACh concentrations (Fig. 1 a, b, and e). This slow activation contrasts with that of LGICs that activate in less than a ms. We fitted the time course of the currents with single exponential functions (Fig. 1e and Methods). The apparent activation rates (kapp) increased in a dose-dependent manner and reached limiting values at around 3 mM (Fig. 1f). No linear dependency was observed, ruling out a simple bimolecular reaction between ACh and the chimera. The existence of limiting kapp values for high concentrations indicates a more complex reaction. A model in which a slow conformational transition follows the rapid binding of ACh described the data (Fig. 1f and Methods), yielding initial estimates of the channel opening (kopen = 2.3 ± 0.6 s-1) and closing rates (kclosed = 0.20 ± 0.07 s-1; see Table 1). This model was successfully used to describe kinetic properties of the Torpedo nAChR (25) and α1 GlyR (26). The kopen values derived for α7/Gly were ≈400-fold slower than the time resolution of our perfusion system (≈1,000 s-1; see Methods) excluding possible limitations due to solution exchange. Therefore, these results suggest that the apparent kinetics of α7/Gly activation are specifically altered.
The Cys-Loop Plays a Major Role in Accelerating the Kinetics of Activation. Given that both WT α7 nAChR and GlyR are fast gating receptors, we hypothesized that a poor coupling between the two domains resulted in the slow activation kinetics in α7/Gly chimera. We built a molecular model of the α7/Gly chimera by homology modeling using the previously described α7 model (16). The model shows that the Cys-loop from the ECD plunges into the extracellular face of the IPD close to the four M1-M4 α-helices (Fig. 2a). Comparison of sequences shows that some residues are not conserved in the Cys-loop between nAChR and GlyR (see Fig. 5, which is published as supporting information on the PNAS web site). Thus, an attractive hypothesis is that in the α7/Gly chimera interactions between Cys-loop residues from α7 and IPD residues from GlyR might not be optimal, conferring a poor coupling between the two domains. To test this hypothesis, we replaced in α7/Gly its chick α7 Cys-loop by the homologous human α1 GlyR Cys-loop. This substitution resulted in a new chimera α7(Cys-L)/Gly that displayed enhanced activation rates at saturating concentrations of ACh concomitant with an increase of the apparent desensitization rate (Fig. 2b). These data demonstrate that the Cys-loop plays a major role in the fast activation rate in the α7(Cys-L)/Gly chimera.
Fig. 2.

Kinetic characterization of α7(Cys-L)/Gly chimera. (a) 3D model of the α7/Gly interface (one subunit shown) with α7 ECD (pale yellow) and GlyR IPD (blue marine). α7 Cys-loop and numbers are highlighted in red. (b) Normalized whole-cell currents (10 mM ACh) through α7/Gly (gray trace) and α7(Cys-L)/Gly (dark trace). (c) Dose-response curves for α7/Gly (open circles) and α7(Cys-L)/Gly (filled triangles). Whole-cell currents are normalized to 10 mM ACh-evoked currents. (d) ACh competition against α-125I-bungarotoxin binding to α7/Gly (open circles, Kp = 540 μM) and α7(Cys-L)/Gly (filled triangles, Kp = 600 μM). Maximal binding (Bmax) is defined without ACh. (e) Concentration dependency of kapp for α7/Gly (open circles) and α7(Cys-L)/Gly (filled triangles). Data are fitted with Eq. 1. For α7/Gly, data are the same as for Fig. 1f. For clarity, only positive error bars are represented. (f) Single-channel currents through α7/Gly (gray trace) and α7(Cys-L)/Gly (dark trace) from cell-attached patches (10 μM ACh). Currents are displayed at 1-kHz bandwidth, with channel openings as downward deflections. (g) Open time histograms. Simulated probability density functions are indicated for each unliganded (A0) and liganded open state (A1,...,A5). The sum of these functions is indicated in green traces (for details, see Supporting Materials and Methods). (h) Comparison of computed (sim) and experimental (exp) whole-cell currents (10 mM ACh) through α7/Gly (gray traces) and α7(Cys-L)/Gly (dark traces). (i) Gating free energy diagram for α7/Gly (gray trace) and α7(Cys-L)/Gly (dark trace). ΔΔGclosed is arbitrarily set to zero, and vertical bar represents 10 kcal/mol.
The Cys-Loop Stabilizes the Transition State of Activation. These results might be explained by changes in agonist association rates, channel-gating rates, and/or conductance levels. The following observations support a change in the channel-gating rates only.
First, whole-cell dose-response curves revealed no significant differences of EC50 values between α7/Gly and α7(Cys-L)/Gly chimeras (Fig. 2c). Furthermore, no differences were noticed in the apparent dissociation constants of ACh as measured by competition against α-125I-bungarotoxin binding to intact cells (Fig. 2d). Second, compared with α7/Gly, a parallel increase of kapp was observed for α7(Cys-L)/Gly (Fig. 2e). As a result, significant increases in both kopen (16.4 ± 3.9 s-1) and kclosed (1.77 ± 0.68 s-1) were observed (7-fold for kopen and 9-fold for kclosed, P < 0.01; for other kinetic details, see Supporting Materials and Methods). Third, single-channel recordings revealed that 10 μM ACh elicited shorter openings in α7(Cys-L)/Gly as compared with α7/Gly chimera (Fig. 2f). Indeed, open-time histograms were fitted with three exponential components for α7(Cys-L)/Gly (τ1 = 0.89 ms, a1 = 0.35; τ2 = 8.5 ms, a2 = 0.35; τ3 = 65 ms, a3 = 0.30) and with four components for α7/Gly (τ1 = 0.75 ms, a1 = 0.20; τ2 = 5.9 ms, a2 = 0.21; τ3 = 42 ms, a3 = 0.28; τ4 = 193 ms, a4 = 0.31; Fig. 2g). A left-shift to shorter open-time values of all components was observed for α7(Cys-L)/Gly compared with α7/Gly that resulted in a 3-fold decrease of the mean open time (τmean = 23 ms for α7(Cys-L)/Gly versus τmean = 73 ms for α7/Gly). This decrease implies that the shortest component for α7(Cys-L)/Gly equivalent to that observed for α7/Gly is likely to be missed, explaining that only three exponential components were sufficient to fit the data. Because the number of channels in patches is unknown, comparison of the closed time histograms between chimeras was judged to be uninformative and was not included. Fourth, no significant modifications on the conductance levels were observed for both chimeras (data not shown). Finally, to help illustrate the experimental data, a set of theoretical parameters derived from a kinetic-based allosteric model (see Fig. 6, which is published as supporting information on the PNAS web site) (27) simulated adequately the macroscopic currents (Fig. 2h) and open-time distribution of α7/Gly (Fig. 2g; for modeling details, see Supporting Materials and Methods). It should be noted that these simulations illustrate only the functional results and do not result from a fitting procedure through the data. Simulations show that changing only the intrinsic opening and closing rates in α7(Cys-L)/Gly resulted in the expected increase of the activation rate observed in whole-cell recordings (Fig. 2h) and decrease of the mean open time observed in single-channel recordings (Fig. 2g). In term of free energy, simulations predicted a stabilization of the transition state between the closed and open conformations (ΔΔG‡)by -1.36 kcal/mol (Fig. 2i). We concluded from these data that the Cys-loop substitution increases the intrinsic opening and closing rates by stabilization of the transition state, without modifying substantially the ACh binding or the ion conduction properties.
Only Two Residues from the Cys-Loop Fine-Tune the Kinetics of Activation in α7/Gly and WT GlyR. We next investigated which amino acids within the Cys-loop are responsible for the fast activation rate. A systematic analysis of the Cys-loop was undertaken by introducing microcassettes or single residues of the α1 GlyR Cys-loop into α7/Gly. For each construction, kopen and kclosed rates were as previously derived (for a detailed description of each construction, see Fig. 7, which is published as supporting information on the PNAS web site, and Supporting Materials and Methods). This study showed that the double mutation V131L/W133N was sufficient to confer a fast activation rate, similar to that of α7(Cys-L)/Gly (Fig. 3a). Then, to test the reciprocity of the mutation, we produced the converse double mutant L142V/N144W in the WT GlyR. This mutant displayed responses to glycine that were significantly slower (7-fold, P < 0.05) than those of the WT GlyR (Fig. 3 a and b). This difference was in the same range (≈10-fold) as observed after the Cys-loop exchange in α7/Gly chimera (Fig. 3a). Consistent with the fact that α7(Cys-L)/Gly was not as fast as the WT GlyR, the double mutant L142V/N144W in GlyR remained faster than α7/Gly chimera (Fig. 3a). Overall, our results highlight the functional importance of the two residues from the Cys-loop in tuning the kinetics of activation in both the chimeric and WT receptors and suggest that other regions from the ECD might also contribute to the fast activation rate.
Fig. 3.

Tuning the activation kinetics in LGICs. (a) kopen bar plot of the indicated constructions. Sequences of the Cys-loop and 2-3L region are indicated. Residues from α7 nAChR are indicated in gray and GlyR in bold-type characters. (b) Normalized whole-cell currents (10 mM glycine) through GlyR WT (gray trace) and L142V/N144W mutant (dark trace). (c) Normalized whole-cell currents (10 mM ACh) through α7/Gly (gray trace) and α7/(2-3L)Gly (dark trace). (d) Mapping of the identified residues on the α7/Gly 3D model (red spheres for Cys-L and cyan for 2-3L). The inner and outer blocks are colored in orange and green, respectively. For clarity, only four subunits are represented. (Inset) Enlarged view of the coupling zone.
The M2-M3 Loop also Plays a Major Role in Accelerating the Kinetics of Activation. Finally, the 3D model of α7/Gly showed that the side chains of the two identified residues of the Cys-loop are in close contact with those of M2-M3 loop (2-3L) (Fig. 3d). This loop links the two α-helices M2/M3 and is not conserved between nAChRs and GlyRs/GABAA receptors (Fig. 5). We reasoned that replacing the human α1 GlyR residues of 2-3L in α7/Gly (Y279-A282) by the homologous residues of chick α7 nAChR (D265-L269) should restore a homogeneous coupling between the Cys-loop and 2-3L. Therefore, we produced the new chimera α7/(2-3L)Gly, which indeed displayed a significantly accelerated activation of the current evoked by saturating concentration of ACh, yielding kopen values close to those of α7(Cys-L)/Gly (Fig. 3 a and c). These data show that the introduction of at most five α7 nAChR residues in the 2-3L is sufficient to rescue a homogeneous coupling in α7/Gly chimera, as did the exchange of two α1 GlyR amino acids in the Cys-loop. Thus, our results demonstrate that both the Cys-loop and the 2-3L region mediate bidirectional allosteric coupling between the agonist-binding and the ion channel domains in LGICs.
Discussion
In the present study, we demonstrate that the Cys-loop and 2-3L region fine-tune the speed of the signal transduction in LGICs. We reached this conclusion by designing a chimeric receptor made from the ECD of α7 nAChR and the IPD of GlyR. This chimera is functional but displays abnormally slow activation rates. By swapping either the Cys-loop or the 2-3L region with their WT counterparts, we succeeded in accelerating the slow activation rate of the α7/Gly chimera. A mutational analysis indicated that only a double mutation in the Cys-loop (V131L/W133N) suffices to restore the fast activation rate in the chimera. We then succeeded in slowing the WT GlyR by producing the converse double mutant (L142V/N144W). Therefore, the Cys-loop and 2-3L play critical roles in the fine-tuning of fast gating in LGICs. Our results also show that the mutated α7/Gly chimeras were still not as fast as the WT GlyR and that the mutated GlyR was not as slow as α7/Gly, suggesting that other regions, yet unidentified (possibly loops 2, 9, and pre-M1, and the C-terminal end of M4), might contribute to the control of the fast gating.
The Cys-loop (28-32) and 2-3L region (17) have been previously identified as important loops for the allosteric coupling; our present work demonstrates their role in the control of the fast opening of the ion channel. Furthermore, sequence analysis shows that the 2-3L region identified here differs from that found earlier in the GABAA receptor (17). This discrepancy may be explained by local changes in the structure of the 2-3L region in the GABAA receptor or alternatively by differences in the construction of the 3D model. Indeed, on the basis of the recent EM images of the ion channel, the M2 segment was interpreted as a 40-Å-long α-helix (15), longer than it was commonly assumed. This extension inevitably brings the 2-3L loop to a more C-terminal region that corresponds to the region we identified in the present study.
Our results, supported by the 3D model of the receptor, suggest that the Cys-loop and 2-3L region interact cooperatively to produce fast gating (Fig. 3d). Three general conclusions can be drawn about their mutual interactions in the gating process.
First, although a common activation mechanism is shared by cationic and anionic LGICs, fine structural complementarity at the interface region is likely to be a general rule for optimal allosteric coupling, as already suggested by chimeras made from the acetylcholine-binding protein (AChBP) and the 5HT3 receptor (18, 32). Indeed, for GlyR coupling, fast gating is mediated by only two nonconserved amino acid residues from the Cys-loop whereas for α7 nAChR fast coupling occurs through at most five nonconserved residues from the 2-3L region. Therefore, optimal coupling in LGICs is mediated by specific interactions, which depend on residues found specifically in anionic versus cationic receptors. The specific coupling interactions between GlyR residues depicted in Fig. 3d should therefore be different from those of α7 nAChR. Further experiments are needed to identify the amino acids from the GlyR 2-3L region that interact with the two Cys-loop residues and the amino acids from the α7 nAChR Cys-loop that interact with the five 2-3L residues, to assign more precisely the minimal cluster of residues that mediate fast gating in the LGICs.
Second, the complementary interaction between the Cys-loop and 2-3L results probably in the stabilization of the transition state of gating. In turn, this stabilization allows the fast opening of the ion channel necessary to mediate fast synaptic neurotransmission. For such large multimeric proteins, subtle changes in the transition state energy barrier (≈1.36 kcal/mol) might explain their swiftness of activation. We propose that fast gating of LGICs is thus achieved by the interactions of some critical residues that lower the energy barrier by a few kcal/mol.
Third, the interaction of the Cys-loop with 2-3L region facilitates the physical opening of the ion pore. This last conclusion is supported by several structural gating mechanisms (15, 16, 33). In particular, normal mode analysis recently proposed that the conformational transition that causes the opening of the ion pore is essentially a quaternary twist motion of the protein accompanied by discrete tertiary changes of each subunit (16). These tertiary changes correspond to the relative motions of two rigid blocks (Fig. 3d). One of these, the inner block, is essentially composed of the inner β-sheet of the ECD that carries the agonist-binding site and α-helix M2 that frames the ion channel wall (16). Therefore, signal transmission between the agonist-binding site and the ion channel is mediated by the intrinsic concerted movement of the rigid inner block. The two flexible loops, the Cys-loop and 2-3L region, undergo discrete conformational changes, enabling the concerted motions of the two rigid blocks associated with a minimal energy cost, facilitating the rapid twist of all five α-helix M2 necessary to open the ion channel (see Movies 1 and 2, which are published as supporting information on the PNAS web site). According to this structural gating mechanism, the Cys-loop and 2-3L region are critically positioned at a pivot point between the ACh-binding site and the ion channel (Fig. 3d and Fig. 8, which is published as supporting information on the PNAS web site).
Finally, a natural mutation that causes severe myasthenia was recently found within the Cys-loop in the nAChR α-subunit (34). The mutation, homologous to the V131L mutation described in the current work, displayed abnormal kinetics of activation. Therefore, beyond the basic understanding of the molecular details of fast activation processes of LGICs, the identified region may be viewed as a potential target for new therapeutic agents that could act as allosteric modulators able to rescue abnormal activation occurring in natural mutants.
Supplementary Material
Acknowledgments
We thank B. Molles for critical reading of the manuscript, A. Marty for fruitful discussion on single-channel analysis, H. Betz (Max-Planck-Institute for Brain Research, Frankfurt, Germany) for providing the human α1 GlyR clone, and M. Soudan and Y. Archambeau for technical assistance. This work was supported by the Association Française Contre les Myopathies, the Commission of the European Communities, the Association pour la Recherche Contre le Cancer, the Collège de France, and the Centre National de la Recherche Scientifique.
Author contributions: T.G., L.P.d.C., and J.-P.C. designed research; T.G., L.P.d.C., V.D., and A.T. performed research; T.G., L.P.d.C., and S.J.E. analyzed data; and T.G. and L.P.d.C. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: LGIC, ligand-gated ion channel; nAChR, nicotinic acetylcholine receptor; GlyR, glycine receptor; ECD, extracellular agonist-binding domain; IPD, ion pore domain; 2-3L, M2-M3 loop.
References
- 1.Corringer, P. J., Le Novère, N. & Changeux, J. P. (2000) Annu. Rev. Pharmacol. 40, 431-458. [DOI] [PubMed] [Google Scholar]
- 2.Karlin, A. (2002) Nat. Rev. Neurosci. 3, 102-114. [DOI] [PubMed] [Google Scholar]
- 3.Changeux, J. P. & Edelstein, S. J. (2005) Science 308, 1424-1428. [DOI] [PubMed] [Google Scholar]
- 4.Lester, H. A., Dibas, M. I., Dahan, D. S., Leite, J. F. & Dougherty, D. A. (2004) Trends Neurosci. 27, 329-336. [DOI] [PubMed] [Google Scholar]
- 5.Connolly, C. N. & Wafford, K. A. (2004) Biochem. Soc. Trans. 32, 529-534. [DOI] [PubMed] [Google Scholar]
- 6.Absalom, N. L., Lewis, T. M. & Schofield, P. R. (2004) Exp. Physiol. 89, 145-153. [DOI] [PubMed] [Google Scholar]
- 7.Chakrapani, S. & Auerbach, A. (2005) Proc. Natl. Acad. Sci. USA 102, 87-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engel, A. G., Ohno, K. & Sine, S. M. (2003) Nat. Rev. Neurosci. 4, 339-352. [DOI] [PubMed] [Google Scholar]
- 9.Laube, B., Maksay, G., Schemm, R. & Betz, H. (2002) Trends Pharmacol. Sci. 23, 519-527. [DOI] [PubMed] [Google Scholar]
- 10.Bertrand, D., Picard, F., Le Hellard, S., Weiland, S., Favre, I., Phillips, H., Bertrand, S., Berkovic, S. F., Malafosse, A. & Mulley, J. (2002) Epilepsia 43, 112-122. [DOI] [PubMed] [Google Scholar]
- 11.Smit, A. B., Syed, N. I., Schaap, D., van Minnen, J., Klumperman, J., Kits, K. S., Lodder, H., van der Schors, R. C., van Elk, R., Sorgedrager, B., et al. (2001) Nature 411, 261-268. [DOI] [PubMed] [Google Scholar]
- 12.Brejc, K., van Dijk, W. J., Klaassen, R. V., Schuurmans, M., van der Oost, J., Smit, A. B. & Sixma, T. K. (2001) Nature 411, 269-276. [DOI] [PubMed] [Google Scholar]
- 13.Celie, P. H. N., van Rossum-Fikkert, S. E., van Dijk, W. J., Brejc, K., Smit, A. B. & Sixma, T. K. (2004) Neuron 41, 907-914. [DOI] [PubMed] [Google Scholar]
- 14.Unwin, N. (2005) J. Mol. Biol. 346, 967-989. [DOI] [PubMed] [Google Scholar]
- 15.Miyazawa, A., Fujiyoshi, Y. & Unwin, N. (2003) Nature 423, 949-955. [DOI] [PubMed] [Google Scholar]
- 16.Taly, A., Delarue, M., Grutter, T., Nilges, M., Le Novere, N., Corringer, P. J. & Changeux, J. P. (2005) Biophys. J. 88, 3954-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kash, T. L., Jenkins, A., Kelley, J. C., Trudell, J. R. & Harrison, N. L. (2003) Nature 421, 272-275. [DOI] [PubMed] [Google Scholar]
- 18.Grutter, T., Prado de Carvalho, L., Dufresne, V., Taly, A., Fischer, M. & Changeux, J. P. (2005) C. R. Biol. 328, 223-234. [DOI] [PubMed] [Google Scholar]
- 19.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflügers Arch. 391, 85-100. [DOI] [PubMed] [Google Scholar]
- 20.Palma, E., Bertrand, S., Binzoni, T. & Bertrand, D. (1996) J. Physiol. (London) 491, 151-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sigworth, F. J. & Sine, S. M. (1987) Biophys. J. 52, 1047-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grutter, T., Prado de Carvalho, L., Le Novère, N., Corringer, P. J., Edelstein, S. & Changeux, J. P. (2003) EMBO J. 22, 1990-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eiselé, J. L., Bertrand, S., Galzi, J. L., Devillers-Thiéry, A., Changeux, J. P. & Bertrand, D. (1993) Nature 366, 479-483. [DOI] [PubMed] [Google Scholar]
- 24.Galzi, J. L., Bertrand, S., Corringer, P. J., Changeux, J. P. & Bertrand, D. (1996) EMBO J. 15, 5824-5832. [PMC free article] [PubMed] [Google Scholar]
- 25.Heidmann, T. & Changeux, J. P. (1980) Biochem. Biophys. Res. Commun. 97, 889-896. [DOI] [PubMed] [Google Scholar]
- 26.Grewer, C. (1999) Biophys. J. 77, 727-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelstein, S. J., Schaad, O., Henry, E., Bertrand, D. & Changeux, J. P. (1996) Biol. Cybern. 75, 361-379. [DOI] [PubMed] [Google Scholar]
- 28.Sala, F., Mulet, J., Sala, S., Gerber, S. & Criado, M. (2005) J. Biol. Chem. 280, 6642-6647. [DOI] [PubMed] [Google Scholar]
- 29.Absalom, N. L., Lewis, T. M., Kaplan, W., Pierce, K. D. & Schofield, P. R. (2003) J. Biol. Chem. 278, 50151-50157. [DOI] [PubMed] [Google Scholar]
- 30.Schofield, C. M., Jenkins, A. & Harrison, N. L. (2003) J. Biol. Chem. 278, 34079-34083. [DOI] [PubMed] [Google Scholar]
- 31.Schofield, C. M., Trudell, J. R. & Harrison, N. L. (2004) Biochemistry 43, 10058-10063. [DOI] [PubMed] [Google Scholar]
- 32.Bouzat, C., Gumilar, F., Spitzmaul, G., Wang, H. L., Rayes, D., Hansen, S. B., Taylor, P. & Sine, S. M. (2004) Nature 430, 896-900. [DOI] [PubMed] [Google Scholar]
- 33.Law, R. J., Henchman, R. H. & McCammon, J. A. (2005) Proc. Natl. Acad. Sci. USA 102, 6813-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen, X. M., Ohno, K. J., Tsujino, A., Brengman, J. M., Gingold, M., Sine, S. M. & Engel, A. G. (2003) J. Clin. Invest. 111, 497-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}