

Abstract
Among the tRNA population of the archaeal parasite Nanoarchaeum equitans are five species assembled from separate 5′ and 3′ tRNA halves and four species derived from tRNA precursors containing introns. In both groups an intervening sequence element must be removed during tRNA maturation. A bulge–helix–bulge (BHB) motif is the hallmark structure required by the archaeal splicing endonuclease for recognition and excision of all introns. BHB motifs are recognizable at the joining sites of all five noncontinuous tRNA species, although deviations from the canonical BHB motif are clearly present in at least two of them. Here, we show that the N. equitans splicing endonuclease cleaves tRNA precursors containing normal introns, as well as all five noncontinuous precursor tRNAs, at the predicted splice sites, indicating the enzyme's dual role in the removal of tRNA introns and processing of tRNA halves to be joined in trans. The cleavage activity on a set of synthetic canonical and noncanonical BHB constructs showed that the N. equitans splicing endonuclease accepts a broader range of substrates than the homodimeric Archaeoglobus fulgidus enzyme. In contrast to the A. fulgidus endonuclease, the N. equitans splicing enzyme possesses two different subunits. This heteromeric endonuclease type, found in N. equitans, in all Crenarchaeota, and in Methanopyrus kandleri, is able to act on the noncanonical tRNA introns present only in these organisms, which suggests coevolution of enzyme and substrate.
Keywords: tRNA processing
Many tRNA genes in Eukarya and Archaea contain intron sequences that must be removed during tRNA processing to generate mature, functional tRNAs. The first step of tRNA splicing is catalyzed by a specific endonuclease that excises the intron from the precursor tRNA (pre-tRNA), yielding two tRNA half-molecules, a 5′ half-exon ending with a 2′,3′-cyclic phosphate, and a 3′ half-exon with a 5′-hydroxyl terminus (1, 2). This step is followed by the joining of the tRNA exons catalyzed by a tRNA splicing-specific ligase in Eukarya; however, little is known about the tRNA ligation mechanism in Archaea (3). The archaeal splicing endonuclease recognizes a consensus bulge–helix–bulge (BHB) structure that is found at the intron–exon junctions of intron-containing tRNA (4), rRNA (5, 6), and mRNA (7). The BHB motif exhibits pseudo-twofold symmetry with the two 3-nt bulges present on the same minor groove face of the central 4-bp helix, termed the 3-4-3 BHB (8). This structure makes the splice sites well positioned for attack by the splicing endonuclease. Two archaeal endonuclease families are found in Euryarchaeota: (i) α2 homodimeric enzymes of 288–353 aa in length or (ii) α4 homotetrameric enzymes of <200 aa in length (1, 9). These endonucleases seem to have a strict requirement for the pseudosymmetric 3-4-3 BHB RNA (4). Recently, a third family of splicing endonucleases was discovered in the Crenarchaeon Sulfolobus solfataricus; the enzyme has a heteromeric subunit composition (10). Multiple sequence alignments and blast searches suggest that this splicing endonuclease family is present in all sequenced Crenarchaeota. Interestingly, the occurrence of this endonuclease family correlates with the observation that this archaeal kingdom displays a wide variety of noncanonical BHB motifs and nonstandard locations (between positions 37 and 38) of tRNA introns (11).
Examination of the genome sequence of the hyperthermophilic archaeon Nanoarchaeum equitans, the only known representative of the new archaeal phylum Nanoarchaeota, and subsequent analysis of the total tRNA molecules led to the discovery of a unique process in which full-length tRNAs are assembled from separate genes encoding 5′ and 3′ tRNA halves (12–14) (Fig. 1). The tRNA halves form a 12- to 14-nt GC-rich RNA duplex between the end of the 5′ tRNA half and the beginning of the 3′ tRNA half and contain BHB motifs similar to those found in crenarchaeal intron-containing tRNAs. It has been shown that the Methanocaldococcus jannaschii splicing endonuclease is able to recognize designed BHB motifs formed in trans in reporter target mRNAs in mouse cells (15). However, N. equitans is the only organism known to date to require tRNA trans-splicing in vivo to generate a full, functional set of tRNA species. Here, we identify the N. equitans splicing endonuclease and demonstrate its ability to cleave both canonical and noncanonical BHB motifs formed in trans during the assembly of tRNA halves.
Fig. 1.

Schematic representation of cis- and trans-splicing of N. equitans tRNAs. During conventional tRNA cis-splicing, the archaeal splicing machinery recognizes and cleaves a BHB motif in the pre-tRNA, leading to the excision of the intron. The initial step of the proposed trans-splicing mechanism requires the annealing of the primary transcripts of a 5′ tRNA-half gene and a 3′ tRNA-half gene. The intervening reverse complementary sequences (gray) form a duplex that facilitates the joining and folding of the tRNA body (black). Noncanonical BHB motifs are accommodated at the junctions suitable for recognition by the splicing endonuclease.
Materials and Methods
General. Oligonucleotide synthesis and DNA sequencing were performed at the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University. Synthetic RNA oligonucleotides were synthesized by Dharmacon (Lafayette, CO). [α-32P]ATP [6,000 Ci/mmol (1 Ci = 37 GBq)] was from Amersham Pharmacia Biosciences. N. equitans cells were a gift of K. O. Stetter and M. Thomm (University of Regensburg, Regensburg, Germany).
Production of Recombinant Splicing Endonuclease. The N. equitans genes NEQ205 and NEQ261 were PCR-amplified from N. equitans genomic DNA by using the Expand High Fidelity PCR System (Roche Molecular Biochemicals). Both PCR products were cloned together into the pETDuet-1 vector (Novagen) to facilitate coexpression of both genes. NEQ205 was cloned into the NcoI/HindIII sites, and NEQ261 was cloned into the NdeI/AvrII sites of the pETDuet-1 plasmid. Both genes were also cloned individually into the NdeI and BamHI sites of the pET15b vector (Novagen). The identity of the clones was determined by DNA sequencing. The individual vectors were transformed into Escherichia coli BL21-CodonPlus (DE3)-RIL (Stratagene). The transformants were grown at 37°C in Luria–Bertani medium supplemented with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml), and protein expression was auto-induced by using the Overnight Express Autoinduction System (Novagen) according to the manufacturer's instructions. Cells were resuspended in buffer containing 50 mM Tris·HCl (pH 7.5), 0.5 M NaCl, and 3 mM DTT and broken by sonication. S-100 fractions were extensively flocculated at 80°C for 30 min and then centrifuged for 30 min at 20,000 × g. The supernatant was immediately loaded onto a Superdex 200 HR 10/30 gel filtration column (Amersham Pharmacia), equilibrated in 20 mM Na Hepes (pH 7.5) containing 150 mM NaCl and 5 mM DTT at a flow rate of 0.5 ml/min. Active fractions were pooled, concentrated by ultrafiltration (Ultrafree-15, molecular weight cutoff of 10,000 Da; Amicon, Witten, Germany), and stored in aliquots at –80°C. The splicing endonuclease of Archaeoglobus fulgidus was prepared as described (9).
Preparation and Purification of RNA Substrates. The individual RNA constructs were cloned by annealing two complementary DNA oligonucleotides containing either the entire intron-containing pre-tRNA [tRNAHis and initiator tRNAMet (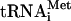 )] or the BHB portion (tRNALys, tRNAGln, and tRNAGlu) downstream of a T7 RNA polymerase promoter and direct ligation into the BamHI and HindIII restriction sites of plasmid pUC19. The BHB portions were based on the predicted BHB motifs (14) closed by a UAAUA loop. The 3′ end of the transcription template was digested overnight with NsiI or HindIII, respectively. The tRNA molecules and BHB constructs were synthesized by in vitro T7 RNA polymerase runoff transcription (16) in the presence of [α-32P]GTP. The labeled RNA molecules were purified by electrophoresis on 8 M urea/12% polyacrylamide gels and elution from the gel. The eluates were filtered by using Sephadex G25 spin columns (Amersham Pharmacia) and were ethanol-precipitated. All RNA constructs were heated to 75°C in the presence of 1 mM EDTA and, after the addition of 10 mM MgCl2, were cooled to room temperature to facilitate refolding of the tRNA body. Synthetic RNA oligonucleotides were deprotected/desalted according to the manufacturer's protocols. The oligonucleotide sequences were 5′-A AGCGACCGACCAUAGCUGCA for strand A and 5′-AAUGCAGCGGUCAAAGGUCGC for strand B. The italicized triplets are the bulges for the canonical BHB synthetic substrate. For the noncanonical substrates, an adenosine was added to or subtracted from either the 3′ or 5′ bulge, which contained only adenosine. RNAs were phosphorylated by using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (MP Biomedical, Irvine, CA). Paired strands at 2 μM each were annealed in buffer containing 30 mM cacodylic acid, 2.5 mM MgCl2, and 1 mM spermidine. The annealing reactions were incubated for 15 min at 70°C followed by cooling on ice for 15 min.
)] or the BHB portion (tRNALys, tRNAGln, and tRNAGlu) downstream of a T7 RNA polymerase promoter and direct ligation into the BamHI and HindIII restriction sites of plasmid pUC19. The BHB portions were based on the predicted BHB motifs (14) closed by a UAAUA loop. The 3′ end of the transcription template was digested overnight with NsiI or HindIII, respectively. The tRNA molecules and BHB constructs were synthesized by in vitro T7 RNA polymerase runoff transcription (16) in the presence of [α-32P]GTP. The labeled RNA molecules were purified by electrophoresis on 8 M urea/12% polyacrylamide gels and elution from the gel. The eluates were filtered by using Sephadex G25 spin columns (Amersham Pharmacia) and were ethanol-precipitated. All RNA constructs were heated to 75°C in the presence of 1 mM EDTA and, after the addition of 10 mM MgCl2, were cooled to room temperature to facilitate refolding of the tRNA body. Synthetic RNA oligonucleotides were deprotected/desalted according to the manufacturer's protocols. The oligonucleotide sequences were 5′-A AGCGACCGACCAUAGCUGCA for strand A and 5′-AAUGCAGCGGUCAAAGGUCGC for strand B. The italicized triplets are the bulges for the canonical BHB synthetic substrate. For the noncanonical substrates, an adenosine was added to or subtracted from either the 3′ or 5′ bulge, which contained only adenosine. RNAs were phosphorylated by using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (MP Biomedical, Irvine, CA). Paired strands at 2 μM each were annealed in buffer containing 30 mM cacodylic acid, 2.5 mM MgCl2, and 1 mM spermidine. The annealing reactions were incubated for 15 min at 70°C followed by cooling on ice for 15 min.
Splicing Endonuclease Assay. The radioactive-labeled tRNA molecules (200 nM) were incubated with 1 μM purified splicing endonucleases in reaction mixtures containing 20 mM Na Hepes (pH 7.5), 10 mM MgCl2, 100 mM NaCl, and 3 mM DTT at 65°C for 20 min. The cleavage pattern of the splicing reactions was analyzed by electrophoresis on denaturing 12% polyacrylamide gels. A typical cleavage assay for synthetic RNA substrates consisted of 200 nM annealed RNA and 1 μM enzyme in 40 mM Tris·HCl (pH 7.8), 2 mM MgCl2, and 1 mM EDTA. The reaction mixture was incubated for 30 min at 60°C, and the reaction products were separated on a denaturing 20% polyacrylamide gel. The gels were exposed to a Fuji BAS-III imaging screen for 2 h and analyzed by using a Molecular Dynamics Model Storm PhosphorImager and imagequant 5.1 software.
Phylogenetic Analysis of Splicing Endonucleases. Splicing endonucleases display two distinct domains that belong to two Pfam categories (PF02778 and PF01974). Newly identified splicing endonucleases from Pyrolobus fumarius and Ignicoccus sp. KIN4 were aligned to other archaeal homologs and representative eukaryotic genes from human and yeast based on hidden Markov model-based profiles (HMM profiles) of the two separate domains by using hmmalign (hmmer 2.3, available at http://hmmer.wustl.edu). The two alignments were concatenated and inspected for accuracy. Regions of the alignment that were considered of low confidence were masked out. The final alignment contained 36 sequences and 101 amino acid positions. A phylogenetic analysis was performed with proml (phylip 3.6 package; proml and seqboot are part of this software package) (17) by using maximum likelihood, the Jones–Taylor–Thornton (JTT) (18) amino acid substitution matrix, five global rearrangements with randomized sequence input, and among-site rate variation modeled with an eight-rate category discrete approximation to a Γ distribution. The model parameters were estimated by using tree-puzzle 5.1 (19). To estimate statistical support, a bootstrapped data set with 100 replicates was generated by using seqboot (phylip 3.6 package). Maximum-likelihood distances were calculated for that data set under the same among-site rate-variation model by using puzzleboot and tree-puzzle 5.1 (19). Consensus neighbor-joining and minimal evolution trees were calculated in phylip.
Results
BLAST analysis of the N. equitans genome sequence revealed two homologs of the archaeal splicing endonuclease. The predicted gene product of ORF NEQ205 is most similar to the Methanocaldococcus jannaschii splicing endonuclease, whereas the product of ORF NEQ261 shows the highest similarity to the N-terminal repeat of the presumably homodimeric Methanosarcina acetivorans endonuclease. Only the NEQ205 protein exhibits the conserved catalytic residues consisting of a histidine, a tyrosine, and a lysine (1). We expressed both genes individually, but neither protein showed activity on any RNA template including constructs containing the canonical BHB motif (data not shown). Therefore, we cloned both ORFs into a vector suitable for coexpression and produced both recombinant proteins simultaneously. Whereas the isolated single proteins were unstable in low-salt conditions or at elevated temperatures (>65°C), coexpression of both proteins increased not only the protein yield but also their thermostability. This allowed for protein purification by heat denaturation at temperatures up to 100°C for 20 min followed by size-exclusion chromatography. This method indicated the presence of an endonuclease complex with a molecular weight according to a tetrameric enzyme (data not shown). The purified N. equitans enzyme preparation contained both subunits in equal amounts judged by a Coomassie blue-stained SDS/polyacrylamide gel (data not shown). This heteromeric N. equitans endonuclease showed RNA cleavage activity on a RNA construct containing the canonical BHB motif derived from Halobacterium volcanii pre-tRNATrp (8) (Fig. 2), demonstrating that this enzyme belongs to the heteromeric family of splicing endonucleases found in Crenarchaeota. NEQ205 most likely encodes the catalytic subunit (named α), and NEQ261 encodes an accessory subunit (named β). As we know for other archaeal endonucleases, the presence of the tRNA body is not required for cleavage activity (2).
Fig. 2.

Splicing endonuclease activity on BHB constructs of joined tRNAGlu halves. The depicted RNA constructs were synthesized by T7 RNA polymerase in the presence of [α-32P]GTP. The canonical BHB motif is derived from H. volcanii pre-tRNATrp (8). Arrows indicate the splice sites. The RNA molecules were incubated in the absence (–) or presence (+) of 1 μM N. equitans splicing endonuclease at 65°C for 20 min. The reaction products were separated by electrophoresis on a denaturing 12% polyacrylamide gel. The 5′ cleavage product (5′half) is indicated, and the excised intervening sequences are marked by *.
The canonical BHB motif has been studied in many tRNAs of different organisms. Much less work has been done in organisms harboring BHB structures that do not conform to the canonical 3-4-3 motif, as is the case for many tRNA introns in Crenarchaeota and N. equitans (11). The junctions of the five noncontiguous tRNAs in N. equitans exemplify an even more interesting example, where noncanonical BHB motifs are formed in trans. Therefore, we asked whether the heteromeric αβ splicing endonuclease would be able to correctly recognize and cleave these motifs. The joined N. equitans pre-tRNAGlu contains a BHB motif where the base pair immediately downstream to the 5′ splice site is a CA mismatch (14). Accordingly, three RNA constructs based on the split tRNAGlu (CUC) gene were produced, with each containing the acceptor stem, the noncanonical BHB motif, and an intervening duplex closed by a loop (UAAUA) (Fig. 2). The length of the GC-rich duplex was varied in three different constructs. In each case the N. equitans splicing endonuclease cleaved the internally labeled RNA substrate. The intervening helix was excised, leading to a cleavage product differing in size according to the length of the duplex (Fig. 2). Cleavage of the minimalist construct lacking this stable duplex, BHBGlu1, led to the accumulation of a minor cleavage product; this suggests that the intervening duplex may play a role in stabilizing the noncanonical BHB structure for cleavage at both sites. In a very recent paper, it was reported that the S. solfataricus endonuclease is also capable of cleaving similar N. equitans tRNAGlu substrates (20).
To show that the N. equitans enzyme acts on all of the predicted BHB motifs (14) formed after annealing the organism's tRNA halves, we produced the appropriate RNA transcripts. In the case of the split tRNAHis and tRNAiMet, the intervening duplex was closed by a loop, leading to continuous intron-containing pre-tRNAs. For the split tRNALys and tRNAGln, only the BHB region was cloned, like in the three tRNAGlu BHB constructs described above. We compared cleavage activities of the dimeric α2 A. fulgidus splicing endonuclease with the heteromeric αβ N. equitans enzyme with these in vitro transcripts. Although both enzymes cleaved the canonical BHB construct to completion, they differed drastically in cleavage efficiency on the N. equitans tRNA substrates containing non-canonical BHB motifs (Fig. 3). Whereas the N. equitans enzyme was able to cleave all pre-tRNA constructs to near completion under the conditions used, the A. fulgidus splicing endonuclease clearly lacked activity toward tRNAGlu, tRNALys, and tRNAGln precursors and had partial activity toward 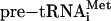 (Fig. 3). This finding is in contrast to the recent demonstration of N. equitans pre-tRNAGlu cleavage by the A. fulgidus enzyme (20). In the reaction with the A. fulgidus endonuclease we observed accumulation of a larger cleavage product for the
(Fig. 3). This finding is in contrast to the recent demonstration of N. equitans pre-tRNAGlu cleavage by the A. fulgidus enzyme (20). In the reaction with the A. fulgidus endonuclease we observed accumulation of a larger cleavage product for the 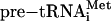 substrate (Fig. 3). Further investigation showed that this RNA band is the product of single cleavage at the 3′ splice site, suggesting that the 5′ terminal bulge of
substrate (Fig. 3). Further investigation showed that this RNA band is the product of single cleavage at the 3′ splice site, suggesting that the 5′ terminal bulge of 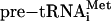 was discriminated by the A. fulgidus endonuclease (data not shown). These observations show that under our conditions only the N. equitans splicing endonuclease is able to cleave the 5′ terminal bulge adjacent to an AG mismatch in the central helix.
was discriminated by the A. fulgidus endonuclease (data not shown). These observations show that under our conditions only the N. equitans splicing endonuclease is able to cleave the 5′ terminal bulge adjacent to an AG mismatch in the central helix.
Fig. 3.

Comparison of splicing activity on joined tRNA substrates. Radiolabeled RNA transcripts of the joined tRNAs of N. equitans were produced (see Materials and Methods) and incubated without enzyme (–) or with either 1 μM splicing endonuclease of N. equitans (Neq) or A. fulgidus (Afu) at 65°C for 20 min. The BHB motifs of the individual substrates are depicted. Arrows indicate the splice sites. The 5′ cleavage product is indicated by 5, and the excised intervening sequences are marked by *. A fragment produced by cleavage of only the 3′ terminal bulge is indicated by “2/3”.
In some cases we observed an increased electrophoretic mobility of the excised intervening duplex. To unambiguously characterize the products of cleavage, we isolated the corresponding fragments and analyzed the RNAs by partial digestion with RNase T1, RNase A, and nuclease S1 (data not shown). We found that the products indeed contained the predicted 3′ terminal bulge cleavage site, and it seemed that its unusual mobility was an artifact of its high thermostability in the gel despite the presence of 8 M urea and extensive heating. Although the nature of the increased stability of this duplex is not known, it is in agreement with its proposed function of joining to RNA molecules in an organism that grows at 90°C. Thus, all of the BHB motifs recently predicted to form during the joining of the individual tRNA halves are indeed substrates for the heteromeric αβ splicing endonuclease. These results support the presence of a tRNA trans-splicing mechanism in N. equitans.
To ensure that the observed discrepancy between the N. equitans and the A. fulgidus endonucleases was not caused by the difference in recognition of their endogenous RNA substrates, we designed a set of synthetic RNA oligonucleotides that, after annealing, form BHB motifs in trans. In this set we varied the number of nucleotides in the bulges from the canonical 3-4-3 BHB motif while maintaining the integrity of the central helix and the two flanking helices. The variable bulges were designed to contain two to four adenosine nucleotides, leading, for example, to 2-4-3 and 4-4-3 BHB motifs. Again, both splicing endonucleases cleaved the canonical substrate but differed in recognition of the substrates with altered bulge length (Fig. 4). Only the N. equitans enzyme was able to cleave both cleavage sites of the 2-4-3 and 4-4-3 BHB motifs efficiently. In one case we could distinguish splice site preferences on an asymmetric BHB motif harboring a 3-nt and a short, 2-nt bulge respectively, the 2-4-3 BHB motif. The N. equitans splicing endonuclease clearly cleaved both sites of the substrate, whereas the A. fulgidus enzyme activity was limited to the standard 3-nt bulge (Fig. 4). Moreover, low activity was shown for the N. equitans, but not A. fulgidus enzyme on the 2-4-4 or 4-4-4 BHB motifs. No activity was observed for the 2-4-2 substrate by either enzyme, suggesting that both enzymes require at least three nucleotides on one bulge. The heterotetrameric S. solfataricus endonuclease was also shown to have broader substrate specificity than the homodimeric A. fulgidus enzyme (21). These results, together with those on pre-tRNAs, obviously imply a difference in recognition of unusual tRNA introns between the homodimeric α2 and the heterotetrameric (αβ)2 splicing endonuclease. Because it was recently suggested that these two endonuclease families have the same cleavage capability on variant substrate structures (20), the degree of permissible variation from the canonical BHB structure remains to be defined.
Fig. 4.

Comparison of splicing activity on BHB motifs with altered bulge length. Noncanonical BHB motifs made from synthetic RNA oligonucleotides in trans were incubated without enzyme (–) or with either 1 μM splicing endonuclease of N. equitans (Neq) or A. fulgidus (Afu) at 60°C for 30 min. Synthetic BHB motifs are comprised of two bulges separated by 4 bp and flanked by 5-bp helices at each end. They are denoted as a pair of numbers that refer to the numbers of nucleotides in each bulge, with * indicating the radioactively labeled strand.
Discussion
The Presence of Unusual tRNA Gene Introns Necessitates the Existence of the Heteromeric Splicing Endonuclease. Most archaeal tRNA introns are located one nucleotide 3′ to the anticodon and are characterized by the canonical 3-4-3 BHB motif first defined by studies on the H. volcanii intron-containing pre-tRNATrp (4). However, the sequencing of archaeal genomes led to the identification of a large number of tRNA genes with introns that use noncanonical recognition motifs or are positioned at different locations (11). tRNA genes were found with introns in the acceptor stem, D loop, D stem, T loop, variable loop, and anticodon stem. Introns at these unusual positions most often display only one 3-nt bulge flanking the characteristic 4-bp helix and another degenerated bulge. This is the case for N. equitans tRNAGlu, which has a mismatch in the stem adjacent to the 5′ splice site, and for 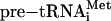 , which has a mismatch in the central helix. Interestingly, the same central helix mismatch (5′-CUCA-3′ base-paired with 5′-GGGG-3′) is also found in Sulfolobus tokodaii tRNAMet and tRNALeu. Despite these variations, the 3′ terminal bulge structure in all archaeal tRNA introns present in the anticodon seems to be well conserved with a clear preference for a purine in the first bulge position (it is A in N. equitans introns). The archaeal splicing endonuclease may use the 3′ terminal bulge as its primary recognition site.
, which has a mismatch in the central helix. Interestingly, the same central helix mismatch (5′-CUCA-3′ base-paired with 5′-GGGG-3′) is also found in Sulfolobus tokodaii tRNAMet and tRNALeu. Despite these variations, the 3′ terminal bulge structure in all archaeal tRNA introns present in the anticodon seems to be well conserved with a clear preference for a purine in the first bulge position (it is A in N. equitans introns). The archaeal splicing endonuclease may use the 3′ terminal bulge as its primary recognition site.
It is interesting to note that tRNA genes with such variations of the canonical splicing motif are concentrated in one archaeal kingdom, the Crenarchaeota. Examples include Pyrobaculum aerophilum, which contains 19 unusually located tRNA introns, and S. tokodaii, whose tRNA introns display 15 relaxed BHB motifs. A phylogenetic analysis of the distribution of splicing endonucleases in archaeal and eukaryal genomes shows a conserved family of endo β subunits in all crenarchaeal genomes as well as in Methanopyrus kandleri and N. equitans (Fig. 5). In Fig. 5, the two catalytic subunits SEN2 and SEN34 of the eukaryal heterotetrameric splicing endonuclease are included for comparison. Obviously, the occurrence of unusual intron positions and structures in Crenarchaeota correlates with the new heteromeric αβ splicing endonuclease family (Fig. 5). The N. equitans enzyme belongs in this group and possesses relaxed substrate recognition features compared with the homodimeric A. fulgidus enzyme (see Figs. 3 and 4), suggesting a direct link between RNA specificity and subunit composition.
Fig. 5.

Phylogenetic analysis of splicing endonucleases. A phylogenetic tree for the splicing endonucleases was produced as detailed in Materials and Methods. The catalytic subunits (endo α) are easily distinguishable from the structural subunits (endo β) that are present in N. equitans (red), M. kandleri, and in all known Crenarchaeota (green). Catalytic subunits of Euryarchaeota are shown in blue, and eukaryal subunits are shown in black. The bar represents the number of changes per position for a unit branch length. The bootstrap value was computed with puzzleboot from 100 replications.
A Structural Model of N. equitans Endonuclease Displays Different Functional Roles for the Two Subunits. The archaeal tRNA endonucleases fall into three families with homotetrameric, homodimeric, and heterotetrameric subunit composition (20, 21). To gain insight into the structural basis of the broad RNA specificity of the N. equitans endonuclease, we constructed a 3D model of the heterodimeric N. equitans enzyme based on previously known structures of the Methanocaldococcus jannaschii and A. fulgidus RNA splicing endonucleases (1, 9). The latter enzymes have a conserved “four-unit” architecture in which one structural unit is dimerized (α4) or covalently linked to one catalytic unit (α2); two such pairs come together to precisely orient the catalytic sites for interaction with the BHB RNA. The N. equitans endonuclease is different, with an αβ structure; the enzyme exhibits efficient catalytic activity toward both canonical and noncanonical BHB motifs. Based on our experimental results, it is plausible that the N. equitans endonuclease arranges its two active sites in an orientation similar to that seen in the α4 and α2 endonucleases. NEQ205, the α-subunit, is presumed to be the catalytic subunit because it exhibits strong sequence homology to the Methanocaldococcus jannaschii endonuclease and contains the three residues of the catalytic triad (1). NEQ261, the β-subunit, has a weak overall homology to the Methanocaldococcus jannaschii endonuclease but maintains all structural elements including (i) the β-strand β10, required for dimerization with the catalytic subunit, and (ii) loop L10, required for tetramerization of the enzyme into the (αβ)2 configuration (Fig. 6). In contrast, the NEQ205 protein lacks the loop L10 conserved sequences; therefore, it will most likely not participate in tetramerization. Thus, the most sensible prediction of the N. equitans endonuclease structure is a symmetric assembly of the four subunits. In this model, two NEQ205 molecules participate as catalytic units, while two NEQ261 subunits function as structural units along the long diagonal of the four-unit parallelogram. To visualize this arrangement, we modeled the individual N. equitans endonuclease subunit structures by using a threading server (22) and assembled the holoenzyme structure based on that of the Methanocaldococcus jannaschii endonuclease (Fig. 6). The L10 loop of each NEQ261 subunit protrudes into the space formed between the C- and N-terminal domains of NEQ205, acting like glue to bring two catalytic subunits together. The active sites marked “His102” are separated by ≈28 Å, in accordance with the distance between the two scissile phosphates of a canonical BHB RNA (8).
Fig. 6.

Modeled N. equitans endonuclease structure in two orthogonal views. His-102 is the conserved catalytic histidine in all endonucleases and denotes the two active sites. The loop, L10, of the structural subunit is responsible for tetramerization, and the β-strands, β10, of both catalytic and structural subunits are responsible for heterodimerization. Both secondary structural elements are labeled.
What Is the Mechanism of Relaxed vs. Canonical BHB Recognition? Answers to this question require identification of elements in the enzyme that interact with BHB RNA. In the absence of such information and a crystal structure of an RNA/endonuclease complex, one can speculate on several possible scenarios based on the differences of enzyme assembly and predicted RNA binding surface between the (αβ)2 and α4 or α2 families of endonucleases. The bulge-binding sites within N. equitans endonuclease may be more flexible and thus may adapt to variable numbers of bulge nucleotides. Moreover, the β-subunit may contribute to substrate binding differently than the structural subunits in the α4 and α2 endonucleases. Alternatively, the (αβ)2 endonucleases may have a different dynamic behavior that contributes to their broader substrate specificity. Structural and biochemical studies are required to verify these mechanisms.
However, the presence of an endonuclease that recognizes and removes all sorts of BHB structures may extend the cleavage activities of splicing endonucleases from tRNAs to a larger pool of mRNAs and rRNAs. Exemplary are BHB-like introns found to be excised from the mRNA of archaeal homologs of the centromere-binding factor 5 in the Crenarchaea Aeropyrum pernix, S. solfataricus, and S. tokodaii and the Euryarchaeon Methanopyrus kandleri (7), which remarkably all possess the αβ splicing endonuclease type.
The specific recognition of a variety of noncanonical BHB motifs by the heteromeric splicing endonuclease highlights yet another example of proteins required for the sequence-independent recognition of small RNA molecules. A different example is found in the mechanism of gene silencing by RNA interference (RNAi) mediated by short interfering RNAs and micro-RNAs. Here is raised the question of how several components of the RNAi pathway selectively bind the short (19–21 bp) interfering RNA ligands characterized by 2-nt, single-stranded 3′ overhangs and 5′ monophosphate groups (23). Furthermore, multiple protein families involved in IFN-controlled antiviral response and pre-mRNA editing possess double-stranded RNA binding motifs (24). One key challenge for future research in this area will be to define the basis for the observed binding selectivity of these proteins. Further insights are available for a class of RNA binding zinc finger proteins that recognize specific AU-rich sequences through backbone atom interaction with the Watson–Crick edges of the adenine and uracil bases (25). The zinc finger scaffold was found to be involved in backbone recognition of the major groove of a double-stranded RNA region and displays an almost customized RNA base and loop recognition (26). It will be interesting to see how these proteins achieve substrate specificity and how these mechanisms relate to the substrate recognition mediated by the splicing endonuclease.
Acknowledgments
We thank S. Herring, D. Jahn, and A. Kohlway for help and encouragement and M. Thomm and K. Stetter for providing biological materials. This work was supported by grants from the National Institute of General Medical Sciences (to D.S.), National Science Foundation (to H.L.), and the Department of Energy (to D.S.).
Author contributions: L.R., K.C., M.H., and J.Y. performed research; M.P. and H.L. analyzed data; and D.S. designed research.
Conflict of interest statement: No conflicts declared.
Abbreviations: BHB, bulge–helix–bulge; pre-tRNA, precursor tRNA.
References
- 1.Li, H., Trotta, C. R. & Abelson, J. (1998) Science 280, 279–284. [DOI] [PubMed] [Google Scholar]
- 2.Abelson, J., Trotta, C. R. & Li, H. (1998) J. Biol. Chem. 273, 12685–12688. [DOI] [PubMed] [Google Scholar]
- 3.Englert, M. & Beier, H. (2005) Nucleic Acids Res. 33, 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson, L. D. & Daniels, C. J. (1990) J. Biol. Chem. 265, 18104–18111. [PubMed] [Google Scholar]
- 5.Tang, T. H., Rozhdestvensky, T. S., d'Orval, B. C., Bortolin, M. L., Huber, H., Charpentier, B., Branlant, C., Bachellerie, J. P., Brosius, J. & Hüttenhofer, A. (2002) Nucleic Acids Res. 30, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kjems, J. & Garrett, R. A. (1988) Cell 54, 693–703. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe, Y., Yokobori, S., Inaba, T., Yamagishi, A., Oshima, T., Kawarabayasi, Y., Kikuchi, H. & Kita, K. (2002) FEBS Lett. 510, 27–30. [DOI] [PubMed] [Google Scholar]
- 8.Diener, J. L. & Moore, P. B. (1998) Mol. Cell 1, 883–894. [PubMed] [Google Scholar]
- 9.Li, H. & Abelson, J. (2000) J. Mol. Biol. 302, 639–648. [DOI] [PubMed] [Google Scholar]
- 10.Tocchini-Valentini, G. D., Fruscoloni, P. & Tocchini-Valentini, G. P. (2005) Proc. Natl. Acad. Sci. USA 102, 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marck, C. & Grosjean, H. (2003) RNA 9, 1516–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waters, E., Hohn, M. J., Ahel, I., Graham, D. E., Adams, M. D., Barnstead, M., Beeson, K. Y., Bibbs, L., Bolanos, R., Keller, M., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 12984–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randau, L., Münch, R., Hohn, M. J., Jahn, D. & Söll, D. (2005) Nature 433, 537–541. [DOI] [PubMed] [Google Scholar]
- 14.Randau, L., Pearson, M. & Söll, D. (2005) FEBS Lett. 579, 2945–2947. [DOI] [PubMed] [Google Scholar]
- 15.Deidda, G., Rossi, N. & Tocchini-Valentini, G. P. (2003) Nat. Biotechnol. 21, 1499–1504. [DOI] [PubMed] [Google Scholar]
- 16.Sampson, J. R. & Uhlenbeck, O. C. (1988) Proc. Natl. Acad. Sci. USA 85, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenstein, J. (1989) Cladistics 5, 164–166. [Google Scholar]
- 18.Jones, D. T., Taylor, W. R. & Thornton, J. M. (1992) Comput. Appl. Biosci. 8, 275–282. [DOI] [PubMed]
- 19.Schmidt, H. A., Strimmer, K., Vingron, M. & von Haeseler, A. (2002) Bioinformatics 18, 502–504. [DOI] [PubMed] [Google Scholar]
- 20.Tocchini-Valentini, G. D., Fruscoloni, P. & Tocchini-Valentini, G. P. (2005) Proc. Natl. Acad. Sci. USA 102, 15418–15422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calvin, K., Hall, M. D., Xu, F., Xue, S. & Li, H. (2005) J. Mol. Biol. 353, 952–960. [DOI] [PubMed] [Google Scholar]
- 22.Meller, J. & Elber, R. (2001) Proteins 45, 241–261. [DOI] [PubMed] [Google Scholar]
- 23.Lingel, A. & Sattler, M. (2005) Curr. Opin. Struct. Biol. 15, 107–115. [DOI] [PubMed] [Google Scholar]
- 24.Carlson, C. B., Stephens, O. M. & Beal, P. A. (2003) Biopolymers 70, 86–102. [DOI] [PubMed] [Google Scholar]
- 25.Hall, T. M. (2005) Curr. Opin. Struct. Biol. 15, 367–373. [DOI] [PubMed] [Google Scholar]
- 26.Theunissen, O., Rudt, F. & Pieler, T. (1998) Eur. J. Biochem. 258, 758–767. [DOI] [PubMed] [Google Scholar]