

Abstract
HIV-1 envelope glycoprotein-mediated fusion is driven by the concerted coalescence of the HIV-1 gp41 N- and C-helical regions, which results in the formation of 6-helix bundles. These two regions are considered prime targets for peptides and antibodies that inhibit HIV-1 entry. However, the parameters that govern this inhibition have yet to be elucidated. We address this issue by monitoring the temporal sequence of conformational states of HIV-1 gp41 during the course of HIV-1-mediated cell–cell fusion by quantitative video microscopy using reagents that bind to N- and C-helical regions, respectively. Env-expressing cells were primed by incubation with target cells at different times at 37 °C followed by washing. The reactivity of triggered gp41 to the NC-1 monoclonal antibody, which we demonstrate here to bind to N-helical gp41 trimers, increased rapidly to a maximal level in the primed state but decreased once stable fusion junctions had formed. In contrast, reactivity with 5-helix, which binds to the C-helical region of gp41, increased continuously as a function of time following the priming. The peptide N36Mut(e,g) reduced NC-1 monoclonal antibody binding and enhanced 5-helix binding, consistent with the notion that this molecule promotes dissociation of gp41 trimers. This inactivation pathway may be important for the design of entry inhibitors and vaccine candidates.
Membrane fusion mediated by human immunodeficiency virus (HIV)-11 envelope glycoproteins (gp120–gp41) is a critical step in the entry of the virus into susceptible cells (1). The current model of HIV viral entry involves the binding of the trimeric viral envelope glycoprotein gp120/gp41 to cell-surface receptor CD4 and chemokine coreceptor CXCR4 or CCR5 (2, 3), which triggers conformational changes in the envelope proteins (4). gp120 then disengages from gp41, allowing for the fusion peptide to be inserted into the target membrane and the prehairpin configuration of the ectodomain to form (5–7). The C-terminal heptad repeat region (C-helical region) and the leucine/isoleucine zipper region (N-helical region)2 then form the thermostable 6-helix bundle (8–11), which drives the membrane merger and eventual fusion (12). In addition to structural information, a wealth of HIV Env-mediated fusion data, which includes inhibition by peptides that mimic the sequences of the N-and C-helical regions (13–21) and fusion kinetics (22), has lent credence to this model. In the absence of complete structural information, some of the details of the HIV-1 Env-mediated fusion reaction have been inferred from immunochemical, biochemical, and mutagenic analysis. However, conclusions reached from these studies are controversial with regard to the timing and nature of conformational changes in HIV-1 gp41 that lead to fusion.
A number of regions of HIV-1 gp41 have been found to be particularly immunogenic in vivo. One of these regions spans amino acid residues 598–604 (cluster I) and includes a nonhelical hydrophilic region that forms a disulfide loop in the 6-helix bundle (23). The other region (cluster II) includes residues 644–663 and comprises a portion of the C-helix. In the absence of triggering by HIV receptors, antibodies to these epitope clusters react minimally with HIV-1 Env. After the interaction with sCD4 or cell surface-bound CD4, a substantial increase in immunoreactivity to these antibodies is observed, probably reflecting conformational changes in gp41 (24). The increased reactivity has been observed in the absence of gp120 dissociation (25), indicating that subunit dissociation is not absolutely required for these CD4-induced conformational changes to occur. The sensitivity of HIV-1 Env primed by sCD4 or cell-surface-bound CD4 (6, 12, 22, 26) to N- or C-helical region peptides leads to the hypothesis that the immunoreactive gp41 antigen is reflecting exposure of the gp41 prehairpin fusion intermediate. While there seems to be general concurrence on the CD4-triggered conformational changes in gp41 that lead to the appearance of prehairpin conformations prior to the fusion event, there is disagreement regarding the formation of the 6-helix bundle relative to fusion.
According to Weiss and colleagues, CD4 is sufficient for triggering Env into the 6-helix bundle (27). This proposition was supported by the observation that the presumed anti-6-helix bundle MAb, NC-1, and rabbit sera raised against 6-helix bundle react with CD4-triggered HIV-1 gp41 (28). If CD4 is sufficient to trigger 6-helix bundle formation, the question arises as to why coreceptors are required for the fusion event. It has been suggested that the coreceptors function by facilitating other events in the fusion process (6). Golding and co-workers (29) proposed that the 6-helix bundles help bring the cellular and viral membranes in close proximity and that subsequent higher order clustering of the 6-helix bundles facilitates membrane fusion. Melikyan and co-workers (12) arrived at entirely opposite conclusions based on their studies of the timing of fusion pore formation, determined by the flow of a fluorescent dye between the effector and target cell, in relationship to the action of HIV gp41-derived peptides that inhibit bundle formation (C34 or N36). They showed that bundle formation is required for the fusion process, but some (if not all) bundle formation occurs after a pore has formed (30).
We examined this issue by monitoring the temporal sequence of conformational states of receptor-triggered HIV-1 gp41 using NC-1, a monoclonal antibody (MAb) (25, 31) that has been shown to react with gp41 following the triggering of Env by CD4 and coreceptors (31). In addition, we used 5-helix, a designed, recombinant protein that consists of three N-helical peptides and two C-helical peptides (18). This protein binds to the C-helical region of gp41 and potently inhibits the fusogenic activity of gp41 at nanomolar concentrations. A tagged 5-helix protein containing the Myc epitope has been shown to immunopreciptate HIV-1 gp41 following its triggering by HIV-1 receptors (19).
Using analytical and quantitative video microscopy, we have monitored conformational changes in gp120–gp41 expressed on cells as a function of time following interactions of Env-expressing cells with target cells (32) using a nonspecific probe. In this paper, we have extended these observations using reagents that are specific for the different conformations of triggered HIV-1 gp41. Our results shed some light on conformations of gp41 trimer that may be targets for neutralization.
EXPERIMENTAL PROCEDURES
Cells Lines, Vaccinia Vectors, Antibodies, 5-Helix Protein, and Peptides
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: NIH 3T3 bearing CD4 and CXCR4 or CCR5 from Dan Littman; Chinese hamster ovary (CHO)-WT from Carol Weiss and Judith White; SupT1 from James Hoxie; Recombinant vaccinia vector vPE16 expressing IIIB/Lai BH8 Env (33) from Pat Earl and Bernard Moss; and Hela cells (ATCC, Manassas, VA) were cultured in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA) (DMEM-10). SupT1 cells were cultured in Roswell Park Memorial Institute medium 1640 (Invitrogen, Carlsbad, CA) with added 10% FBS. CHO cells permanently transfected to express IIIB HIV-1 Env (CHO-WT) (34) were cultured in a custom designed medium formula number 94-0243DJ, lot 1027860 by GibcoBRL (Invitrogen, Carlsbad, CA) supplemented with 350 μM methionine sulfoximine (Sigma, St. Louis, MO). All culture medium contained 100 units/mL penicillin–streptomycin. Conformational specific mouse MAb, NC-1 (31), designed against the 6-helix bundle of gp41 was a gift from Dr. Shibo Jiang. Rabbit C34 antiserum was kindly donated by Carol Weiss. Secondary antibodies, goat anti-mouse IgG (H+L) polyclonal fragment antibodies (Fab), were purchased from Jackson Immunoresearch Laboratories, Inc., West Grove, PA, which also provided Fab conjugated with Cy5-bis-OSU, N,N′-biscarboxypentyl-5,5′-disulfonatoindodicarbocyanine (Cy5). Myc tag MAb, 9E10, was purchased from Abgent, Inc., San Diego, CA. The 5-helix protein, whose N-terminal consists of three N40 segments, two C38 segments, a 6×His tag, and a 3×Myc epitope tag (19), was a gift from David Chan. NCCG–gp41, N36Mut(e,g), and N35CCG–N13 were prepared as described (15–17). The peptide C34, derived from HIV-1 Env (10), was synthesized by SynPep (Dublin, CA).
Expressing HIV-1 Env on HeLa Cells
HeLa cells were plated in 12 well plates, 105 cells per well, 24 h prior to infection. The vaccinia stock (20 μL of approximately 109 plaque forming units/mL) was mixed with 30 μL of 0.25 mg/mL trypsin solution in Dulbecco’s phosphate-buffered saline (D-PBS), incubated for 20 min, and then diluted in 2.4 mL of DMEM. After removal of the media from each well, 200 μL of this vaccinia suspension was added to the plated cells. The cells were incubated for 1 h by shaking every 10 min to avoid drying in the middle of the wells. At the end of the hour, the cells were supplemented with 2 mL/well DMEM-10 and incubated for another 5 h at 37 °C.
Immunofluorescence
The Env-expressing HeLa or CHO-WT were previously stained with the cytoplasmic dye CellTracker Blue CMAC (7-amino-4-chloromethylcoumarin) (Molecular Probes, Inc., Eugene, OR). SupT1 cells were loaded with calcein AM (Molecular Probes, Inc., Eugene, OR), which is cleaved inside the cell by nonspecific esterases, and the colorless calcein AM is converted to fluorescent calcein green. To observe binding of the NC-1 MAb or 5-helix, the Env-expressing cells were plated at the bottom of 35 mm wells (P35Gcol-0-10-C, MatTek Corporation, Ashland, MA) and incubated with target cells at 37 °C and 5% CO2 for a desired time in the range of 0–30 min. After timed incubation, the cells were immediately placed on ice to stop the fusion processes. Cells were washed twice with ice-cold washing buffer (D-PBS with calcium and magnesium, Quality Biological, Inc., number 114-059-101, containing 0.1% bovine serum albumin and 0.02% sodium azide). Nonspecific Fab binding was blocked with blocking solution (2% normal goat serum in D-PBS) containing 1 μg/mL unconjugated goat anti-mouse Fab for 30 min on ice. Then, the cells were washed twice with the washing buffer, once with 2% goat serum, and incubated in 2% goat serum blocking solution supplemented with 1 μg/mL of the NC-1 MAb. Incubation continued for 1 h on ice. The cells were then washed twice with washing buffer, once with blocking solution, and incubated with 1 μg/mL Cy5 conjugated goat anti-mouse Fab in blocking solution. Incubation with the secondary antibody continued for 1 h on ice. The cells were washed 3 times and covered with 1 mL/well washing buffer prior to microscopic observation. To detect the interaction with 5-helix, we had one more step in the immunostaining procedure, i.e., after the initial blocking the cells were incubated with 5-helix for 1 h on ice. Then, the procedure continued by incubating with the primary 9E10 MAb and Cy5-conjugated goat anti-mouse Fab as described above. Cell fusion was monitored using a dye redistribution assay as described previously (35).
Microscopy
All images were collected by using a NIKON 200TE (Melville, NJ) inverted microscope supplied with PlanFluor 20×, ELWD, NA = 0.45 objective. We used single beam-splitter cubes: Nikon B-2E/C, 465–495/505/515–555 for the Calcein staining, Nikon UV-2E/C (DAPI), 340–380/400/435–485 for the CellTracker Blue CMAC, and HQ41008, 590–650/660/663–738 (Chroma Technology Corp., Rockingham, VT) for the Cy5-conjugated Fab. The microscope was automated through the MetaMorph 4.0 software (Universal Imaging Co., Downingtown, PA).
Quantitation of gp41 Epitope Exposure
The stacked images were processed by using the MetaMorph 4.0 software to quantify the relative presence of different conformations of gp41 on the cells by measuring the fluorescent intensity of Cy5 dye, which was covalently bound to the secondary anti-mouse polyclonal fragment antibody. The target cells were added to the plated Env-expressing cells. Stacks of four successive images were collected in the same field using different filter cubes and microscope modes: differential interferometry contrast image, blue fluorescence image for the Env-expressing cells, green fluorescence image for the target cells with eventual fusion events, and red fluorescence image for the Cy5-staining monitoring the gp41 conformational changes. We used two different approaches to quantify the amount of triggered gp41, both on the basis of quantification of the binding of the Cy5-conjugated Fab, which is assumed to be proportional to the amount of the triggered gp41. When the Env expression was relatively high, the fluorescence was spread all over the cell membrane. In this case, we placed a threshold on the blue image showing the area occupied by the Env-expressing cells stained with CMAC, marked this area in regions, switched the stack image to the red one (Cy5), and integrated both the area and the fluorescence intensity within the regions. The ratio between integrated fluorescence intensity and total region area, i.e., intensity per unit cell area, was used as a relative measure for the number of gp41 molecules that have undergone conformational changes. When the Env expression was not very high, aggregates of gp41 molecules were visualized as fluorescent dots on the cell membranes. Even though the number of gp41 molecules in these fluorescent “islands” is not known, we assumed that the number of fluorescent islands is proportional to the number of triggered gp41 molecules. We subtracted the background intensity level and enhanced the contrast by cutting out high-intensity levels. Thus, the resultant image represents a black panel with white dots, which are visualized as aggregates of triggered gp41 molecules. We counted the number of these white dots and divided by the number of the Env-expressing cells.
For a better representation of the immunofluorescence results, we normalized almost all obtained antibody-binding data by using the following equation: B = (I – Imin)/(Imax –Imin), where B is the extent of binding plotted on the graph, I is the measured fluorescence for a graph point, and Imax and Imin are the respective maximum and minimum fluorescence, respectively, obtained in that experimental set.
Western Blotting
Proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis on pre-made 10–20% linear-gradient nonreducing Tris-N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine (Tricine) gels (Invitrogen, Carlsbad, CA) as described (17). The gels were soaked for 10 min and then transferred onto a nitrocellulose membrane (Schleicher and Schuell, Keene, NH) in 25 mM Tris buffer at pH 8, 190 mM glycine, and 20% methanol using a mini-electrophoretic transfer apparatus (Bio-Rad, Hercules, CA). The blots were then processed using a Western Breeze kit, a chromogenic immunogenic system for the detection of rabbit secondary antibodies (Invitrogen, Carlsbad, CA).
RESULTS
To probe conformational changes in gp41, we used the NC-1 MAb, which was generated by immunizing mice with the minimal ectodomain of gp41, comprising the polypeptide N36(L6)C34 that forms 100% 6-helix bundle in physiological solution (31). Figure 1 displays the results of Western blot analysis with the NC-1 MAb and polyclonal anti-C34 sera. The antibodies were probed against four protein and peptide samples characterized by gel electrophoresis under denaturing conditions in Figure 1A: trimeric NCCG–gp41, which comprises the N-helix bundle of HIV gp41 grafted in helical phase onto the N terminus of a minimal thermostable 6-helix bundle of gp41, stabilized by intermolecular disulfide bridges (15) (lane 1); predominantly trimeric N35CCG–N13, comprising only the internal N-helical trimeric coiled-coil of the ectodomain of HIV-1 gp41 (17) with about 6% contaminating C34 (lane 2); a mixture of trimeric (three intermolecular disulfide bonds), dimeric (one or two intermolecular disulfide bonds), and monomeric (no intermolecular disulfide bonds) N35CCG–N13 (lane 3); and C34 (lane 4). Figure 1B displays the results of Western blot analysis against the NC-1 MAb: only the trimeric forms of NCCG–gp41 (lane 1) and N35CCG–N13 (lanes 2 and 3) are recognized by the NC-1 MAb. The NC-1 MAb does not react with the dimeric and monomeric forms of N35CCG–N13 (lane 3) or with C34 (lane 4). Figure 1C displays the results of Western blot analysis against polyclonal anti-C34 serum. A weak band is evident for NCCG–gp41, as expected because this comprises the 6-helix bundle, which includes the C-helical region, as well as the exposed trimeric coiled-coil of N-helices stabilized by intermolecular disulfide bonds. Bands at the position of monomeric C34 are also seen in the N35CCG–N13 sample with 6% C34 present (lane 2) and the pure C34 sample (lane 4). No evidence for the presence of C34 is seen in the N35CCG–N13 sample in lane 3. Because the NC-1 MAb was raised against the minimal ectodomain core of gp41 in which the N- and C-helical regions were linked by a 6-residue linker sequence and because the NC-1 MAb clearly recognizes specifically the trimeric form of N35CCG–N13 (with no C34 present), it follows that the NC-1 MAb is directed against the surface of the N-helices that is exposed between the C-helices in the 6-helix bundle. As seen in Figure 2, the N-helices comprise a relatively wide and shallow groove along the entire length of the 6-helix bundle.
Figure 1.
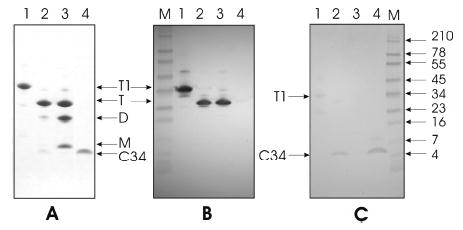
Characterization of the NC-1 MAb specificity. Proteins were electrophoresed on 10–20% gradient Tris-Tricine precast gels and were either Coomassie-stained (A) or subjected to immuno-blotting with NC1 MAb (B; 2 μg/mL) or rabbit C34 antisera (C; 960 at a dilution of 1:100, peptide code P16 from Carol Weiss). Lanes 1–4 represent NCCG-gp41 trimer (35 442 Da), N35CCG-N13 trimer (22 639 Da) and C34 (4289 Da as monomer), a mixture of N35CCG-N13 trimer, dimer, and monomer, and C34, respectively, in A–C. T1 denotes trimeric, disulfide-linked NCCG-gp41, and T, D, and M denote trimeric (three intermolecular disulfide bonds), dimeric (one or two intermolecular disulfide bonds), and monomeric (no intermolecular disulfide bonds) N35CCG-N13, respectively. (A) A total of 4 μg of each protein in lanes 1–3 and 0.5 μg of C34 (lane 4) were subjected to electrophoresis. The sample in lane 2 also contains ~0.25 μg of C34 in addition to N35CCG-N13 trimer. The amount of C34 in lane 2 was estimated by co-electrophoresis of varying amounts of C34 and comparing the intensities after Coomassie staining. The sample in lane 3 intentionally contains a significant amount of the trimeric, dimeric, and monomeric forms of N35CCG-N13 but has no C34. (B and C) A total of 0.5 μg of each protein except for lane 4 (1 μg). M denotes molecular-weight markers [see blue prestained standard in B and C (Invitrogen, Carlsbad, CA)]. Immunoblotting was performed using the chromogenic kit purchased also from Invitrogen.
Figure 2.

Molecular surface of the HIV-1 6-helix bundle. The image is based on the crystal structure of (N36/C34)3 in the heterotrimeric coiled-coil conformation (10) (PDB 1AIK). The internal N-helices (N36, residues 546–579) are colored in blue, while the external C-helices (C34, residues 628–661) are shown in red. Note that a significant portion of the N-helices, constituting a relatively wide and shallow groove along the length of the ectodomain core, is surface-accessible.
Using this information, we monitored conformational changes in gp120–gp41 expressed on cells by immunofluorescence following the interactions with CD4 and core-ceptors on the target cells. After different times of coculture at 37 °C of HeLa cells expressing HIV-1LAI Env with SupT1 cells, the mixture was cooled to 4 °C and immunoassayed with the NC-1 MAb. Images were examined in differential interference contrast (top panels of Figure 3) and in fluorescence for Cy5 staining to measure the NC-1 MAb binding (bottom panels of Figure 3). At zero time, only a few Cy5-stained cells were seen. However, there was a marked increase in the number of Cy5-stained cells after 10 and 15 min of incubation with SupT1 cells. After 20 min of coculture on the other hand, the number of Cy5-stained cells had markedly decreased, indicating that the NC-1 MAb binding was reduced as a result of more prolonged interactions between Env-expressing cells and target cells. Because the immunostaining procedure involves cooling to 4 °C, followed by washing off the SupT1 cells before addition of antibodies, we were not able to determine whether the Cy5 staining does appear at the contact region between target and Env-expressing cells. However, the transient interactions between Env-expressing and target cells at 37 °C leave an imprimatur on the Env-expressing cells, which is revealed by staining with the conformation-specific NC-1 MAb. Figure 3B illustrates the labeling pattern in the course of fusion between HeLa cells expressing high levels of Env and SupT1 cells that were preloaded with the aqueous dye calcein. The figure shows that the calcein is confined in SupT1 cells at 10 min of incubation but redistributes to HeLa cells after 30 min, indicating the appearance of fusion. NC-1 staining, which can be seen 10 min after binding to the target cells, significantly decreases after 30 min.
Figure 3.
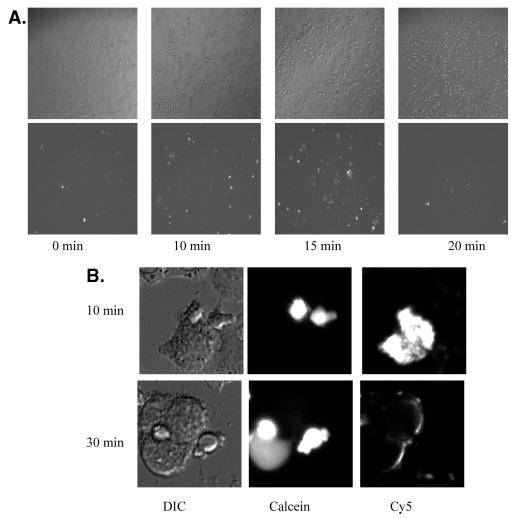
Binding of the NC-1 MAb to triggered HIV-1 gp41. (A) Images are shown of HeLa cells expressing relatively low levels of HIV-1IIIB Env incubated with SupT1 at 37 °C for 0, 10, 15, and 20 min. Differential interference contrast (DIC) images of the cell morphology are seen in the top panels, and Cy5 staining that reveals NC-1 MAb binding is seen in the bottom panels. (B) Images are shown of HeLa cells expressing high levels of HIV-1IIIB Env incubated with calcein-loaded SupT1 at 37 °C for 10 min (top panels) and 30 min (bottom panels). DIC images of the cell morphology are seen in the left panels. The middle panels show the calcein dye, which is confined in SupT1 cells at 10 min of incubation but redistributed to HeLa cells after 30 min, indicating the appearance of fusion. Cy5 staining that reveals NC-1 MAb binding is shown in the right panels.
The above result shows the transient appearance of gp41 conformational intermediates in the fusion process. To examine the kinetics of conformational changes in gp120–gp41 expressed on cells as a function of time following interactions of Env-expressing cells with target cells, we processed the images such as those shown in Figure 3 using the MetaMorph 4.0 software. The relative presence of different conformations of gp41 on the cells was quantified by measuring the fluorescent intensity of the Cy5 dye. HeLa cells expressing HIV-1LAI Env were incubated with SupT1 cells that were preloaded with the aqueous dye calcein. After different times of coculture at 37 °C, the mixture was cooled to 4 °C and examined for calcein staining of the HeLa cells to detect cell–cell fusion and for Cy5 staining to measure NC-1 MAb binding. Figure 4 shows that there was a marked increase in intensity of Cy5 fluorescence in HeLa cells after 10 min of incubation with SupT1 cells in the absence of dye redistribution, i.e., before fusion occurs. Binding of the NC-1 MAb reached a maximum value after 15 min and then declined to the background after about 30 min when fusion started to take off. The fact that the NC-1 MAb binds trimeric N35CCG–N13 (see Figure 1) indicates the appearance by immunostaining (Figures 3 and 4) of an N-terminal trimeric gp41 prehairpin intermediate, which occurs significantly prior to 6-helix bundle formation.
Figure 4.
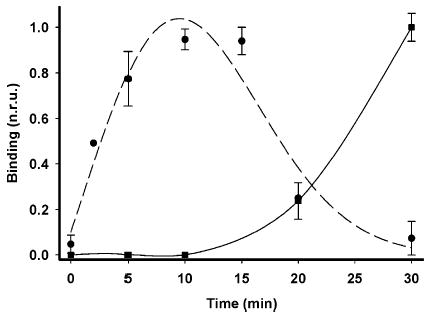
Kinetics of conformational changes in HIV-1 gp41 during the fusion process. HIV-1IIIB Env-expressing CHO cells were incubated with CD4 and CXCR4 bearing SupT1 cells at 37 °C. The appearance of NC-1 MAb binding (circles) and fusion (squares) was quantified at different time points as described in the Experimental Proceduresthods. The normalized data represent averages of five separate experiments with standard deviations indicated by vertical bars. Smoothed curves passing through the data points of the graphs were generated by a cubic spline interpolation (–) or a polynomial function (- - -) using SigmaPlot (SPSS, Inc., Chicago, IL).
Because previous studies indicate that the prehairpin can be triggered by CD4 alone (6, 12, 22), we examined the time course of conformational changes induced by cells expressing CD4. For these experiments, we used NIH3T3 cells that have been engineered to express equal levels of cell surface CD4 as NIH3T3 cells, which express both CD4 and CXCR4. As shown in Figure 5, the kinetics of NC-1 MAb binding to HIV-1LAI Env-expressing cells following incubation with NIH3T3CD4 exhibits a pattern similar to that following incubation with NIH3T3CD4CXCR4. These data confirm that the conformational changes seen by the NC-1 MAb are for the most part triggered by CD4. These conformational changes do not lead to fusion because no dye redistribution is seen when the Env-expressing cells are incubated with cells expressing only CD4. The conformational changes triggered by both CD4 and CXCR4, on the other hand, lead to fusion. In relation to the profile of NC-1 binding to Env-expressing cells with time, the greatest NC-1 binding is observed early in the fusion event (Figure 3). Because the amount of 6-helix bundle formation required for fusion is presumed to be minor at this early stage (1) and likely undetectable by probing with NC-1, it follows that NC-1 binding observed at this point is directed toward the prehairpin intermediate on the cell surface. Furthermore, the decline in NC-1 binding as cell–cell fusion proceeds suggests that any existing 6-helix bundle occurring at the contact region between cells is inaccessible to NC-1.
Figure 5.

Kinetics of conformational changes in HIV-1 gp41 triggered by NIH3T3CD4 or NIH3T3CD4CXCR4 cells. HeLa cells were infected with recombinant vaccinia virus vPE16 encoding HIV-1IIIB Env for 1 h in DMEM at 37 °C. The cells were then supplemented with DMEM-2 and incubated for 5 h at 37 °C and 5% CO2 to express the Env. After the IIIB Env was expressed, the cells were overlaid by NIH3T3CD4 or NIH3T3CD4CXCR4 previously lifted off by using PBS-based cell dissociation buffer (Invitrogen Corp., Carlsbad, CA). The ratio between the overlaid and plated Env-expressing cells was 1:1. The appearance of NC-1 MAb binding and fusion was quantified at different time points as described in the Experimental Procedures for quantification of relatively high Env expression. Smoothed curves passing through the data points of the graphs were generated by a cubic spline interpolation using SigmaPlot (SPSS, Inc., Chicago, IL).
What then is the nature of gp41 molecules whose detection by the NC-1 MAb rapidly declines after 10–15 min of incubation of Env-expressing cells with target cells? To further examine the kinetics of these conformational changes, we used the tagged 5-helix protein containing the Myc epitope. This molecule binds to free C34 and inhibits HIV-1 Env-mediated fusion at nanomolar concentrations (18, 19). Moreover, C34 inhibits the fusion inhibitory activity of 5-helix. Hence, 5-helix interacts with the gp41 C-helical regions when they are not in contact with the N-helical regions as in the case of the prehairpin intermediate. In the 6-helix bundle, these two regions interact, and hence, the C-helical region is not accessible to 5-helix. Figure 6 shows that binding of 5-helix to HIV-1 Env-expressing cells increases steadily as a function of time, even when fusion takes off, indicating that some form of gp41 that is not detected by the NC-1 MAb still remains on the cell surface at later times.
Figure 6.

Binding of 5-helix to triggered HIV-1 gp41 during the fusion process. HIV-1IIIB Env-expressing CHO cells were incubated with CD4 and CXCR4 bearing SupT1 cells at 37 °C. The binding of 5-helix and fusion was quantified at different time points as described in the Experimental Procedures. The data were normalized to B = (I – Imin)/(Imax – Imin), where I represents fusion (○) or 5-helix (▵) binding and Imax and Imin are the respective maximum and minimum fusion or antibody-binding fluorescence obtained in that experimental set. B is the plotted number. The error bars protruding in the negative are due to the normalization procedure, which also includes background subtraction.
We hypothesize that the decline in NC-1 MAb binding and increase in 5-helix binding is due to the dissociation of prehairpin trimers into gp41 monomers (11, 16, 36). To test this hypothesis, we used the peptide N36Mut(e,g), which was designed to completely abolish any N-helical binding to the C-helical region while maintaining the ability to self-associate into well-defined trimers (16). N36Mut(e,g) is assumed to inhibit HIV Env-mediated fusion by shifting the trimeric forms of the gp41 prehairpin to monomeric forms of membrane-bound gp41 (16). In accordance with this hypothesis, we find that, in the presence of N36Mut(e,g), binding of 5-helix is significantly enhanced in comparison to the binding of the NC-1 MAb (Figure 7). Because the immunostaining procedure involves cooling to 4 °C, followed by 2-fold washing with ice-cold buffer before the addition of antibodies, the bound N36Mut(e,g) molecules are most likely dissociated from the heterotrimer by the time binding with the NC-1 MAb or 5-helix is enabled. Therefore, binding of the NC-1 MAb to possible gp41- N36Mut(e,g) heterotrimers is unlikely to be observed, and the predominant gp41 form detected on the cell surface by 5-helix represents gp41 monomers.
Figure 7.

Effect of N36Mut(e,g) on NC-1 MAb and 5-helix binding to triggered HIV-1 gp41. Binding of NC-1 MAb (▵) and 5-helix (•) to HIV-1IIIB Env-expressing CHO cells during incubation with SupT1 cells in the presence of 25 μM N36Mut(e,g) was determined as described in the caption of Figure 3. Smoothed curves passing through the data points of the graphs were generated by a cubic spline interpolation using SigmaPlot (SPSS, Inc., Chicago, IL).
As a control for the N36Mut(e,g) experiment, we further examined the binding of the NC-1 MAb to triggered gp41 in the presence of C34, which potently inhibits fusion by binding to the N-helical trimer in the prehairpin intermediate state, thus preventing 6-helix bundle formation (37). Figure 8 shows that at low concentrations of C34 the NC-1 MAb binding increases with time, consistent with the notion that the N-helical trimer/C34 complex preserves the topology of the prehairpin intermediate, which is accessible for NC-1 MAb binding. Because the binding site for C34 is in the groove formed by the N-helical trimer (37) and is known to inhibit fusion at low nanomolar concnentrations, it presumably binds more tightly to the prehairpin and is not removed by the washing procedure. However, surprisingly, at a concentration of 100 nM C34, NC-1 MAb binding is significantly reduced.
Figure 8.

Effect of C34 on NC-1 MAb binding to triggered HIV-1 gp41. Binding of NC-1 MAb to HIV-1IIIB Env-expressing CHO cells during incubation with SupT1 cells in the presence of 10 nM (•) and 100 nM (▵) C34 was determined as described in the caption of Figure 3. Smoothed curves passing through the data points of the graphs were generated by a cubic spline interpolation using SigmaPlot (SPSS, Inc., Chicago, IL).
DISCUSSION
In this paper, we have addressed questions regarding mechanisms of HIV entry and its inhibition. One question is related to the timing and nature of the conformational changes in HIV-1 gp41 that lead to fusion. In doing so, we had to reevaluate the specificity of the gp41 and NC-1 MAb, which was raised against the 6-helix bundle (31). Because the NC-1 MAb does not bind to N and C peptides but recognizes N-peptide/C-peptide mixtures (31, 38, 39) and binds to the 6-helix bundle formed by N36(L6)C34 but not to domains with mutations in N- and C-helical regions that disrupt the 6-helix bundle formation (31), it has been assumed that the NC-1 MAb uniquely binds to the 6-helix bundle. However, on the basis of the data shown in Figure 1, it is clear that the NC-1 MAb binds to N-helical trimers as well as the 6-helix bundle and not to monomers. We have used the ability of the NC-1 MAb to detect N-helical trimers to monitor conformational changes in gp41 as the reaction proceeds.
The generally accepted model for HIV-1 Env-mediated fusion is diagrammed in Figure 9. Triggering the HIV-1 envelope glycoprotein trimer (E3) by clusters of CD4 on the target membrane (T′) results in a conformational intermediate (E3′) that exposes the coreceptor-binding site on gp120 and forms the gp41 prehairpin intermediate. This event is followed by the engagement of gp120 with clusters of coreceptors on the target membrane (T″), which results in the formation of the gp41 6-helix bundles (E3″) that drive the fusion (1, 5, 7). Presumably, the NC-1 MAb cannot bind in the areas of contact between Env-expressing and target membranes (E3′T′, E3′T″, and E3″T″). However, the transient interactions between Env-expressing effector cells and target cells leaves the conformationally altered Env (E3′) exposed, which is available for NC-1 MAb binding (Figures 3–5). The similar kinetics of Env triggering exhibited by NIH3T-3CD4 and NIH3T3CD4CXCR4 (Figure 5) is consistent with the model. It appears that only a few 6-helix bundles are required for fusion (1). Therefore, the slight increase in potential NC-1 MAb binding sites (in the form of 6-helix bundle) triggered by CD4 and CXCR4 will not contribute to the overall signal. Moreover, these 6-helix bundle binding sites reside in the contact region between cells and are therefore unlikely to be accessible to the NC-1 MAb, which has no detectable inhibitory activity for HIV-1 infection or fusion (31). Because binding to E3′ alone should not affect the fusion reaction, the model also explains why a MAb that binds well to Env is incapable of neutralization (40). The decrease in NC-1 MAb staining after it reaches a peak value at 10–15 min is presumed to be the result of formation of gp41 monomers (E′), a process that is accelerated in the presence of certain entry inhibitors. The time course of NC-1 MAb staining is very similar to that shown by Finnegan et al. (25) using anti-gp41 cluster I and II MAbs.
Figure 9.

Model of intermediates during HIV-1 Env-mediated fusion. The interactions of HIV-1 Env trimers (E3) with CD4 clusters (T′) induce conformational changes that result in the exposure of the binding site on gp120 that can engage CXCR4 and the exposure of the gp41 prehairpin trimer (E3′) in an Env-target membrane complex (E3′T′). This is followed by engagement of CXCR4 clusters (T″) to form an E3′T″ complex that results in 6-helix bundle formation (E3″) and fusion. In a parallel reaction, Env-target complexes dissociate to expose triggered Env (E3′) on the surface, which is susceptible to NC-1 MAb binding. The triggered Env trimer (E3′) can then dissociate to gp41 monomers, a process that is enhanced by entry inhibitors such as N36Mut(e,g). See the text for further explanation.
We surmise that the NC-1 MAb does not inhibit HIV-1 fusion because it has limited access to the contact region between viral and target membranes, although it binds well to the gp41 prehairpin intermediate once the target cells are removed. The notion that the gp41 prehairpin trimer has limited access in the contact region is consistent with a recent study (41) showing that C-terminal peptides attached to cargo proteins of various sizes progressively lose inhibitory potency with increasing cargo size, although they bind with equal potency to a gp41 N-helical trimer construct. On the other hand, certain MAbs directed toward the gp41 membrane proximal external region exhibit broadly neutralizing activity (42). A structural study of the MAb 2F5 bound to its peptide suggests a possible mechanistic explanation for the neutralizing activity of this MAb (43). They discovered a long complementarity determining region loop on the antibody with a hydrophobic patch of amino acids at the end of this loop, which interacts with both the target peptide and adjacent viral membrane. The particular structure of this class of MAb’s may therefore allow accessibility of the MAb to its target site even in the contact region between the virus and cell.
Most studies on the mechanism of HIV fusion have focused on the triggered conformational changes in Env, which eventually result in the formation of gp41 6-helix bundles that drive the fusion between HIV and target membranes (12, 22). The pathway (Figure 9) that involves the dissociation of prehairpin trimers (E3′) to gp41 monomers (E′) had originally been proposed by Clore and co-workers (11) to account for the mode of action of certain peptidic entry inhibitors. Several peptides that mimic the sequence of the C- and N-helical regions of HIV-1 have been found to inhibit fusion either by blocking the interaction between the N- and C-helical regions (37, 44) or by promoting the dissociation of gp41 trimers into monomers and thus preventing the formation of the 6-helix bundle fusogenic state of gp41 (16). C-helical-derived peptides such as C34 inhibit HIV-1 Env-mediated cell fusion in the nanomolar range, through binding to the hydrophobic grooves that line the internal N-terminal trimeric coiled-coil core of the gp41 ectodomain (37). N-helical-derived peptides such as N36 are much less effective inhibitors with IC50 values in the micromolar range (13). To examine the mode of action of this class of peptides, Bewley et al. (16) designed a multisite mutant known as N36Mut(e,g), which completely abolishes any N-helical peptide binding to the C-helical region while maintaining the ability to self-associate into well-defined trimers. N36Mut(e,g) forms a monodisperse, helical trimer in solution, does not interact with C34, and yet inhibits fusion about 50-fold more effectively than the parent N36 peptide (16). Because N36Mut(e,g) forms a well-defined trimeric species that does not interact with either C34 or the chimeric protein NCCG-gp41 (in which the N-helices of the solvent-exposed trimeric coil-coil are covalently linked by interhelical disulfide bonds), it must target the N-helical region of the prehairpin intermediate by forming fusion-incompetent heterotrimers (16). The fusion-inhibitory activity of N36Mut(e,g) therefore indicates the presence of a dynamic equilibrium between monomeric, dimeric, and trimeric forms of membrane-bound gp41 that allows subunit exchange to take place in the prehairpin intermediate state. The rate of exchange between these species must be sufficiently fast to permit efficient heterotrimer formation within the lifetime (~15 min) of the prehairpin intermediate (16). An analysis of the inhibition of HIV Env-mediated cell fusion by N36Mut(e,g) indicates that, at saturating concentrations of N36Mut(e,g), the predominant species is a heterotrimer consisting of one gp41 prehairpin and two N36Mut(e,g) molecules (16).
The fact that the NC-1 MAb binds to trimeric and not monomeric gp41 provided us with an opportunity to monitor gp41 trimer–monomer transitions following the triggering of HIV-1 Env. The enhanced formation of monomer gp41 and decreased formation of trimer observed when the Env-expressing cells were being incubated with target CD4 and coreceptor-expressing cells in the presence of N36Mut(e,g) (Figure 7) is consistent with the notion that N36Mut(e,g) inhibits by shifting the equilibrium of the prehairpin intermediate to a heteromeric state, which eventually results in the appearance of gp41 monomers. On the other hand, a low concentration (10 nM) of C34 appears to prolong NC-1 MAb binding to triggered Env (Figure 8), consistent with the notion that C34 binds to the N-terminal internal trimeric coiled-coil to form a peptide/protein hybrid structure (44). At higher C34 concentrations (100 nM), we do observe a decrease in NC-1 MAb binding (Figure 8), indicating that there is low-affinity binding of C34 to the gp41 monomer (36); at high C34 concentrations, the reaction could then be driven toward the gp41 monomeric state. An alternative explanation for this observation could be that C34 slows down the formation of the gp41 prebundles in a dose-dependent manner, leading to apparent reduction in NC-1 binding within the first 30 min at 37 °C.
We further studied conformational changes in Env using the 5-helix protein (18, 19), which lacks a third C-helix peptide, and this vacancy is expected to create a high-affinity binding site for the C-helical region of gp41. This protein binds the C-peptide region of gp41 and potently inhibits the fusogenic activity of gp41 at nanomolar concentrations. Although 5-helix binds to native gp41 (45), it binds strongly to gp41 activated by the interaction of the envelope protein with either soluble CD4 or membrane-bound cellular receptors (19). Because 5-helix cannot bind to 6-helix bundles, we would expect 5-helix binding to decrease if there was only a prehairpin–trimer to 6-helix bundle conversion during the fusion reaction. In contrast, we observe a steady increase in the binding of 5-helix to HIV-1 Env-expressing cells (Figures 6 and 7) under conditions where NC-1 MAb binding decreases. This indicates a parallel pathway that involves the generation of gp41 monomers coexisting with the trimers during the fusion reaction (Figure 9). The formation of heterotrimers consisting of gp41 and N36Mut(e,g) results in a shift of the reaction pathway towards gp41 monomers following dissociation of the peptide from the heterotrimer. This pathway may deserve some consideration in the design of entry inhibitors and vaccine candidates.
Acknowledgments
We are very grateful to Shibo Jiang for providing the NC-1 MAb and to David Chan for providing 5-helix and for their insightful comments on the manuscript. We thank Carol Weiss for providing C34 antisera. We are grateful to the NIH AIDS Research and Reference Reagent Program for the supply of Sup-T1 cells, VPE16 recombinant vaccinia, and CHO-WT cells. We thank Satinder Rawat and Mathias Viard for help with the experiments and the members of the Blumenthal lab for their helpful suggestions.
Footnotes
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the AIDS Targeted Antiviral Program of the Office of the Director of the National Institutes of Health.
Abbreviations: HIV, human immunodeficiency virus; DMEM, Dulbecco’s modified Eagle medium; CHO, Chinese hamster ovary; Cy5, Cy5-bis-OSU, N,N′-biscarboxypentyl-5,5′-disulfonatoindodicar-bocyanine; PBS, phosphate-buffered saline; D-PBS, Dulbecco’s PBS; CMAC, 7-amino-4-chloromethylcoumarin; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
For convenience, we use the notation N- and C-helical regions or peptides, although they only become α-helical when associated as homo- or heterotrimers.
References
- 1.Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R. The HIV Env-mediated fusion reaction. Biochim Biophys Acta. 2003;1614:36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 2.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 3.Doms RW, Moore JP. HIV-1 membrane fusion: Targets of opportunity. J Cell Biol. 2000;151:F9–F14. doi: 10.1083/jcb.151.2.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody [see comments] Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 6.Furuta RA, Wild CT, Weng Y, Weiss CD. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol. 1998;5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 7.Weissenhorn W, Dessen A, Calder LJ, Harrison SC, Skehel JJ, Wiley DC. Structural basis for membrane fusion by enveloped viruses. Mol Membr Biol. 1999;16:3–9. doi: 10.1080/096876899294706. [DOI] [PubMed] [Google Scholar]
- 8.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 9.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–428. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 10.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 11.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151:413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: Correlation between solution structure and viral inhibition. Proc Natl Acad Sci USA. 1992;89:10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 15.Louis JM, Bewley CA, Clore GM. Design and properties of N(CCG)-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J Biol Chem. 2001;276:29485–29489. doi: 10.1074/jbc.C100317200. [DOI] [PubMed] [Google Scholar]
- 16.Bewley CA, Louis JM, Ghirlando R, Clore GM. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J Biol Chem. 2002;277:14238–14245. doi: 10.1074/jbc.M201453200. [DOI] [PubMed] [Google Scholar]
- 17.Louis JM, Nesheiwat I, Chang L, Clore GM, Bewley CA. Covalent trimers of the internal N-terminal trimeric coiled-coil of gp41 and antibodies directed against them are potent inhibitors of HIV envelope-mediated cell fusion. J Biol Chem. 2003;278:20278–20285. doi: 10.1074/jbc.M301627200. [DOI] [PubMed] [Google Scholar]
- 18.Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 19.Koshiba T, Chan DC. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J Biol Chem. 2003;278:7573–7579. doi: 10.1074/jbc.M211154200. [DOI] [PubMed] [Google Scholar]
- 20.Gallo SA, Sackett K, Rawat SS, Shai Y, Blumenthal R. The stability of the intact envelope glycoproteins is a major determinant of sensitivity of HIV/SIV to peptidic fusion inhibitors. J Mol Biol. 2004;340:9–14. doi: 10.1016/j.jmb.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 21.Gustchina E, Hummer G, Bewley CA, Clore GM. Differential inhibition of HIV-1 and SIV envelope-mediated cell fusion by C34 peptides derived from the C-terminal heptad repeat of gp41 from diverse strains of HIV-1, HIV-2, and SIV. J Med Chem. 2005;48:3036–3044. doi: 10.1021/jm049026h. [DOI] [PubMed] [Google Scholar]
- 22.Gallo SA, Puri A, Blumenthal R. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry. 2001;40:12231–12236. doi: 10.1021/bi0155596. [DOI] [PubMed] [Google Scholar]
- 23.Gorny MK, Zolla-Pazner S. Recognition by human monoclonal antibodies of free and complexed peptides representing the prefusogenic and fusogenic forms of human immunodeficiency virus type 1 gp41. J Virol. 2000;74:6186–6192. doi: 10.1128/jvi.74.13.6186-6192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sattentau QJ, Zolla-Pazner S, Poignard P. Epitope exposure on functional, oligomeric HIV-1 gp41 molecules. Virology. 1995;206:713–717. doi: 10.1016/s0042-6822(95)80094-8. [DOI] [PubMed] [Google Scholar]
- 25.Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus transmembrane glycoprotein during cell–cell fusion. J Virol. 2002;76:12123–12134. doi: 10.1128/JVI.76.23.12123-12134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallo SA, Clore GM, Louis JM, Bewley CA, Blumenthal R. Temperature-dependent intermediates in HIV-1 envelope glycoprotein-mediated fusion revealed by inhibitors that target N- and C-terminal helical regions of HIV-1 gp41. Biochemistry. 2004;43:8230–8233. doi: 10.1021/bi049957v. [DOI] [PubMed] [Google Scholar]
- 27.De Rosny E, Vassell R, Wingfield PT, Wild CT, Weiss CD. Peptides corresponding to the heptad repeat motifs in the transmembrane protein (gp41) of human immunodeficiency virus type 1 elicit antibodies to receptor-activated conformations of the envelope glycoprotein. J Virol. 2001;75:8859–8863. doi: 10.1128/JVI.75.18.8859-8863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Rosny E, Vassell R, Jiang S, Kunert R, Weiss CD. Binding of the 2F5 monoclonal antibody to native and fusion-intermediate forms of human immunodeficiency virus type 1 gp41: Implications for fusion-inducing conformational changes. J Virol. 2004;78:2627–2631. doi: 10.1128/JVI.78.5.2627-2631.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golding H, Zaitseva M, De Rosny E, King LR, Manischewitz J, Sidorov I, Gorny MK, Zolla-Pazner S, Dimitrov DS, Weiss CD. Dissection of human immunodeficiency virus type 1 entry with neutralizing antibodies to gp41 fusion intermediates. J Virol. 2002;76:6780–6790. doi: 10.1128/JVI.76.13.6780-6790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markosyan RM, Cohen FS, Melikyan GB. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol Biol Cell. 2003;14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang S, Lin K, Lu M. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1998;72:10213–10217. doi: 10.1128/jvi.72.12.10213-10217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PL, Korte T, Blumenthal R. Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and co-receptors. J Biol Chem. 1998;273:404–409. doi: 10.1074/jbc.273.1.404. [DOI] [PubMed] [Google Scholar]
- 33.Earl PL, Koenig S, Moss B. Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: Analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. J Virol. 1991;65:31–41. doi: 10.1128/jvi.65.1.31-41.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss CD, White JM. Characterization of stable Chinese hamster ovary cells expressing wild-type, secreted, and glycosylphosphatidylinositol-anchored human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1993;67:7060–7066. doi: 10.1128/jvi.67.12.7060-7066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caffrey M, Kaufman J, Stahl S, Wingfield P, Gronenborn AM, Clore GM. Monomer–trimer equilibrium of the ectodomain of SIV gp41: Insight into the mechanism of peptide inhibition of HIV infection. Protein Sci. 1999;8:1904–1907. doi: 10.1110/ps.8.9.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S, Boyer-Chatenet L, Lu H, Jiang S. Rapid and automated fluorescence-linked immunosorbent assay for high-throughput screening of HIV-1 fusion inhibitors targeting gp41. J Biomol Screening. 2003;8:685–693. doi: 10.1177/1087057103259155. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Zhao Q, Jiang S. Determination of the HIV-1 gp41 fusogenic core conformation modeled by synthetic peptides: Applicable for identification of HIV-1 fusion inhibitors. Peptides. 2003;24:1303–1313. doi: 10.1016/j.peptides.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Poignard P, Moulard M, Golez E, Vivona V, Franti M, Venturini S, Wang M, Parren PW, Burton DR. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J Virol. 2003;77:353–365. doi: 10.1128/JVI.77.1.353-365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamburger AE, Kim S, Welch BD, Kay MS. Steric accessibility of the HIV-1 gp41 N-trimer region. J Biol Chem. 2005;280:12567–12572. doi: 10.1074/jbc.M412770200. [DOI] [PubMed] [Google Scholar]
- 42.Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, Moore JP, Stiegler G, Katinger H, Burton DR, Parren PW. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol. 2001;75:10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kilgore NR, Salzwedel K, Reddick M, Allaway GP, Wild CT. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J Virol. 2003;77:7669–7672. doi: 10.1128/JVI.77.13.7669-7672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Root MJ, Hamer DH. Targeting therapeutics to an exposed and conserved binding element of the HIV-1 fusion protein. Proc Natl Acad Sci USA. 2003;100:5016–5021. doi: 10.1073/pnas.0936926100. [DOI] [PMC free article] [PubMed] [Google Scholar]