

Summary
The ppk gene encodes polyphosphate kinase (Ppk), an enzyme that catalyses the polymerization of inorganic phosphate into long chains of polyphosphate (polyP). An insertion mutation in ppk causes a decrease in adaptive mutation in Escherichia coli strain FC40. Adaptive mutation in FC40 mostly results from error-prone DNA polymerase IV (Pol IV), encoded by dinB; most of the antimutagenic phenotype of the ppk mutant disappears in a dinB mutant strain. In addition, the ppk mutant causes a decrease in growth-dependent mutations produced by overexpressing Pol IV. However, the amount of Pol IV protein is unchanged in the ppk mutant strain, indicating that the activity or fidelity of Pol IV is altered. Adaptive mutation is inhibited both by the absence of Ppk, which results in low amounts of polyP, and by overproduction of Ppk, which results in high amounts of polyP, suggesting that an optimal level of polyP is necessary. Taken together, these results suggest a novel mechanism involving polyP that directly or indirectly regulates DNA polymerase activity or fidelity.
Introduction
Escherichia coli DNA polymerase IV (Pol IV) is a member of the Y-family of DNA polymerases that is characterized by their ability to synthesize past DNA lesions. However, Y-family DNA polymerases insert incorrect nucleotides at higher frequencies than normal replicative polymerases. Therefore, when DNA lesions are present (i.e. after UV irradiation) Y-family polymerases cause increased mutation rates (reviewed by Goodman, 2002). Error-prone polymerases, as they have been called, are found in all three domains of life. A deficiency in the human homologue, Pol η, causes a variant of the skin disease Xero-derma pigmentosum that results in increased sensitivity to UV light and a predisposition to skin cancer (Yamada et al., 2000).
Both of E. coli’s Y-family polymerases, Pol IV and Pol V, are induced as part of the LexA-controlled SOS regulon that responds to DNA damage (Kenyon and Walker, 1980; Bagg et al., 1981; Courcelle et al., 2001). A current model for trans-lesion DNA synthesis is as follows (Goodman, 2002). The normal replicative polymerase, Pol III, stalls at a DNA lesion, which results in induction of the SOS response; one of the Y-family polymerases (Pol IV or Pol V) is recruited to the replication fork, synthesizes past the lesion and continues replicating for a short period. The Y-family polymerase then is replaced by Pol III, allowing normal DNA replication to continue. Because of the high rate at which incorrect nucleotides are incorporated, DNA synthesis by Pol IV or Pol V results in mutations.
When bacteria are held under non-lethal selection, the non-growing cells accumulate mutations that relieve the selective pressure and allow the bacteria to grow. This phenomenon is known as adaptive mutation (Cairns and Foster, 1991). Pol IV is responsible for 50–80% of the adaptive mutations produced in the highly studied E. coli strain FC40 (Foster, 2000; McKenzie et al., 2001). FC40 is deleted for lac on the chromosome and the lac region is carried on the episome, F′128. lacI and lacZ are fused, eliminating the last four residues of lacI, all of lacP and lacO, and the first 23 residues of lacZ (Mueller-Hill et al., 1964). The lacI–Z gene fusion has a +1 frameshift in lacI, adding a G:C base pair to a run of three G:C base pairs, resulting in a Lac− phenotype (Calos and Miller, 1981).
During non-selective growth, Lac+ mutants of FC40 arise at a rate of about 10−9 per cell per generation. When incubating on minimal lactose media, Lac+ mutants arise for about a week at a rate of 10−7 per cell per day (Cairns and Foster, 1991). Adaptive mutation in FC40 differs from growth-dependent mutation in several ways. Adaptive Lac+ mutations result almost exclusively from −1 frame-shifts in runs of consecutive bases (Foster and Trimarchi, 1994; Rosenberg et al., 1994). In contrast, the mutations that occur during growth include deletions and duplications as well as simple frameshifts. The rate of adaptive mutation in FC40 is 100 times higher when the lac allele is on the episome than when it is on the chromosome (Foster and Trimarchi, 1995; Radicella et al., 1995). Furthermore, unlike growth-dependent mutation, adaptive mutation depends on the double-strand break repair activities of RecABCD (Cairns and Foster, 1991; Foster, 1993; Harris et al., 1994) and is enhanced by the expression of conjugal functions (Foster and Trimarchi, 1995; Galitski and Roth, 1995; Radicella et al., 1995). Evidence suggests that the conjugal function that is required for adaptive mutation is nicking the DNA at the conjugal origin (Rodriguez et al., 2002). Although the mechanism by which adaptive mutation occurs is controversial (Foster, 2004; Rosenberg and Hastings, 2004; Roth and Andersson, 2004), it is widely accepted that Pol IV causes most of the frameshifts that result in adaptive mutations.
Adaptive mutation is increased 100-fold in a strain defective for the RecG helicase (Foster et al., 1996; Harris et al., 1996). This increase is eliminated when Pol IV is also defective, indicating that the high rate of adaptive mutation in recG mutants is almost exclusively due to Pol IV (Layton and Foster, 2003). Because this 100-fold increase in adaptive mutation is easy to assay, we used a recG mutant strain to screen for mutations that decrease the amount or activity of Pol IV. A transposon was used to generate random insertions, and the resulting mutants were screened for low levels of adaptive mutation. One gene found during this screen was rpoS, which encodes the stationary-phase sigma factor, σ38, that is required for transcription of genes induced upon entry into stationary phase (reviewed by Hengge-Aronis, 2002). Further analysis showed that Pol IV is induced about threefold during late stationary phase, and this induction is RpoS dependent (Layton and Foster, 2003).
Another gene that was found in the screen was ppk, which encodes polyphosphate kinase (Ppk). Ppk polymerizes the gamma phosphate of ATP into long chains of inorganic polyphosphate (polyP) (reviewed by Kornberg et al., 1999). ppk is in an operon with ppx that encodes an exopolyphosphatase (Ppx) responsible for degrading polyP (Akiyama et al., 1993). PolyP is found in all studied organisms in all domains of life (reviewed by Kornberg et al., 1999), and many functions have been proposed for it. PolyP activates Lon protease by binding to its DNA-binding domain; this binding allows Lon to degrade ribo-somal proteins in response to amino acid starvation (Kuroda et al., 2001; Nomura et al., 2004). recA and rpoS mRNA levels are decreased when polyP levels are undetectable (Shiba et al., 1997; Tsutsumi et al., 2000). Ppk is one of several proteins that form the RNA degradosome that regulates RNA turnover (Blum et al., 1997; Carpousis, 2002; Bernstein et al., 2004). PolyP that was purified with DNA from the fungus Colletotrichum inhibited DNA replication by Taq polymerase in vitro (Rodriguez, 1993). Finally, polyP forms a complex with RNA polymerase; in vitro studies have shown that polyP inhibits transcription from σ70 promoters but allows transcription from RpoS-dependent promoters at high salt concentrations (Kusano and Ishihama, 1997).
Here we report that Ppk is necessary for normal levels of adaptive mutation. Furthermore, Ppk is required for the increased rate of growth-dependent mutations produced by Pol IV overexpression. Our results suggest that Ppk is affecting the activity of Pol IV in a manner that is independent of any previously known factors that regulate this polymerase.
Results
Mutations in Ppk reduce adaptive mutation in FC40
In E. coli strain FC40 adaptive mutation is increased up to 100-fold by mutations in recG (Foster et al., 1996; Harris et al., 1996), and this increase is dependent on Pol IV (encoded by dinB) (Layton and Foster, 2003). To search for genetic factors that control Pol IV, we made random insertions of a transposon encoding chloramphenicol resistance (CmR), miniTn10Cm (Kleckner et al., 1991), into a recG mutant strain, and then screened the CmR isolates for reduced levels of adaptive mutation. One mutant found in this screen has an insertion in the ppk gene at nucleotide 345 in its coding sequence. The ppk::miniTn10Cm (hereafter referred to as ppk::Cm) mutation decreased adaptive mutation by approximately 80% in the recG mutant strain (Fig. 1) and up to 65% in the wild-type strain (see Fig. 2A). To determine whether another ppk mutant allele has a similar phenotype, we tested an allele that has the C-terminal end of ppk and the N-terminal end of ppx (the second gene in the operon) replaced by a kanamycin-resistance (KnR) cassette (Δppk::Kn); a strain carrying this allele was shown to have 1.5% of the Ppk and 22% of the Ppx activities of the wild-type strain (Kuroda et al., 1997). When transduced into FC40, the Δppk::Kn allele conferred a phenotype similar to that conferred by ppk::Cm (Fig. 1). To test whether the phenotype conferred by ppk::Cm is due solely to loss of ppk instead of to a polar effect on ppx, we deleted ppx from codon 94 to the end of its coding sequence and inserted a KnR gene to create Δppx::Kn. The Δppx::Kn allele did not affect adaptive mutation (Fig. 1). Thus, wild-type ppk, but not ppx, is required for normal levels of adaptive mutation.
Fig. 1.

Disrupting the ppk gene reduces Lac+ adaptive mutation. Approximately 107 cells from four independent cultures were spread on each quadrant of a lactose minimal plate in a small-scale experiment (see Experimental procedures). The data are the mean of the total number (±SEM) of Lac+ colonies appearing on days 3–5. All strains are recG mutants. ppk+ppx+ = FC526, ppk::Cm = PFG39, Δppk-ppx::Kn = PFG203, Δppx::Kn = PFG162.
Fig. 2.

Most of the effect of ppk on adaptive mutation results from decreased Pol IV activity.
A. The cumulative number of Lac+ revertants per 108 Lac− cells. About 108 cells from six independent cultures of FC40 and PFG74 (ppk), and 109 cells from six independent cultures of PFB243 (dinB) and PFG274 (ppk dinB) were plated on lactose minimal plates. Data points are the means (±SEM). ♦, wild type = FC40; ▪, ppk::Cm = PFG74; ▴, ΔdinB::Zeo = PFB243; •, ppk::Cm ΔdinB::Zeo = PFG274.
B. The viability of the strains during incubation on lactose plates. Each day, three plates per strain were used to determine the number of viable Lac− cells (see Experimental procedures). Data points are the means ± SEM. Symbols are the same as in (A).
To determine whether ppk and dinB are in the same pathway for adaptive mutation, we performed an epistasis test comparing adaptive mutation in the ppk, dinB and ppk dinB double mutant strains. Figure 2A shows that the double mutant strain had approximately half of the level of adaptive mutation as did the dinB mutant strain, indicating that most but not all of the Ppk antimutator effect can be attributed to decreased Pol IV activity. None of these mutant backgrounds affected cell viability on lactose plates (Fig. 2B).
Intermediate levels of polyP appear to be required for optimal levels of adaptive mutation
Polyphosphate kinase produces long chains of inorganic polyP and other studies have shown that ppk mutant strains have decreased amounts of polyP (Shiba et al., 1997). To test whether the observed decrease in adaptive mutation resulted from a lack of polyP, we attempted to complement the ppk::Cm defect with a multicopy plasmid (pBR322) carrying ppk under control of its own promoter. Presumably expressing ppk on a multicopy plasmid would increase polyP levels, especially in a strain without functional ppx. Surprisingly, the presence of this plasmid did not complement ppk::Cm for adaptive mutation (Fig. 3). When the same plasmid was transformed into the wild-type strain, the adaptive mutation levels were not affected (Fig. 3). Cell viability did not change over the course of the experiment (data not shown), indicating that overexpressing ppk is not toxic. In addition, the amount of Pol IV measured by Western blot analysis was not affected by overexpression of ppk (data not shown). As the ppk::Cm mutation is polar on ppx, these results suggest that when ppk is overexpressed, Ppx activity is required for normal levels of adaptive mutation. This conclusion was further supported by the fact that overexpressing ppk in the Δppx::Kn mutant strain also inhibited adaptive mutation (Fig. 3). As Ppx degrades polyP, overexpressing ppk in the Δppx::Kn mutant background should result in relatively high levels of polyP. These results suggest that adaptive mutation is inhibited both when polyP levels are high and when they are low.
Fig. 3.
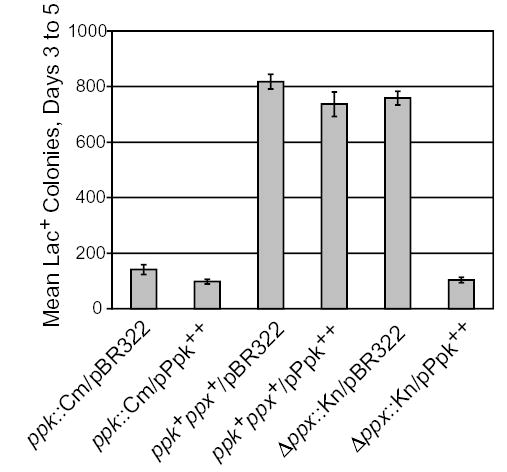
Overexpressing Ppk is antimutagenic in ppx mutant cells. Approximately 107 cells from four independent cultures were spread on each quadrant of a lactose minimal plate in a small-scale adaptive mutation experiment (see Experimental procedures). The data are the means of the total number (±SEM) of Lac+ colonies appearing on days 3–5. All strains are recG mutants. ppk::Cm/pBR322 = PFG87; ppk::Cm/pPpk++ = PFG176; ppk+ppx+/pBR322 = PFG89; ppk+ppx+/pPpk++ = PFG175; Δppx::Kn/pBR322 = PFG323; Δppx::Kn/pPpk++ = PFG322.
Polyphosphate kinase does not affect the amount of Pol IV protein
Adaptive mutation could be low in ppk mutant strains because either the amount or the activity of Pol IV is decreased. For example, polyP levels have been shown to be important in transcription of rpoS, which encodes the RNA polymerase sigma factor that regulates the stationary-phase response (Shiba et al., 1997). Pol IV protein is induced about threefold during stationary phase, and this induction is dependent on rpoS (Layton and Foster, 2003). Therefore, it was possible that in ppk mutant strains, RpoS transcription is low and the RpoS-dependent induction of dinB is attenuated. To test this hypothesis, we incubated ΔrecG, ΔrecG ppk::Cm and ΔrecG rpoS::Cm strains in minimal glycerol medium for 3 days. Samples were taken on days 2 and 3, and the amounts of Pol IV measured by Western blot analysis. Unlike rpoS::Cm mutant cells, ppk::Cm mutant cells had the same amount of Pol IV as wild-type cells after 3 days of continuous incubation (Fig. 4). This result indicates that the ppk antimutator phenotype is not due to a reduction in the amount of Pol IV or an increase in its degradation. Furthermore, the ppk antimutator phenotype is not exerted via rpoS.
Fig. 4.

The amount of Pol IV declines in an rpoS mutant strain but not in wild type or a ppk mutant strain during prolonged incubation in minimal medium. Samples were taken 2 and 3 days after inoculation and equal amounts of total protein from each strain were loaded in each lane. Pol IV was detected by immunoblotting. ΔrecG::Kn = FC526; ΔrecG::Kn rpoS::Cm = PFG36; ΔrecG::Kn ppk::Cm = PFG39.
Loss of Ppk inhibits growth-dependent mutations due to overexpression of Pol IV
Because there are other factors in addition to Pol IV that affect adaptive mutation (see Introduction), we used another assay that is independent of adaptive mutation to test the effect of Ppk on Pol IV activity. The rate of mutation in growing cells can increase up to 800-fold when Pol IV is overexpressed (Kim et al., 1997). Growth-dependent mutations giving resistance to tetracycline (TcR) can be measured easily in strain FC722, which has a +1 frameshift mutation in the tetA gene carried on its episome (Foster, 1997). Overexpression of Pol IV under control of an exogenous promoter on a multicopy plasmid resulted in an eightfold increase in the mutation rate to TcR (Fig. 5). The same plasmid carried by the ppk::Cm mutant strain resulted in only a threefold increase in the mutation rate to TcR (Fig. 5). The amount of Pol IV in these strains as determined by Western blot analysis was not affected by the ppk::Cm mutation (data not shown). These results indicate that Ppk is necessary for optimally high mutation rates due to Pol IV whether these mutations occur during growth or during stationary phase. Furthermore, as the ppk::Cam mutation affects Pol IV activity even when dinB is being transcribed from an exogenous promoter, Ppk is not exerting its effects by regulating dinB transcription.
Fig. 5.
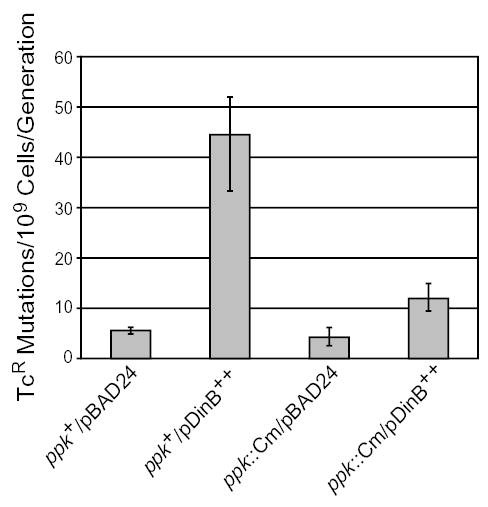
The ppk mutant allele reduces the growth-dependent mutation rate due to overexpression of Pol IV. Values are mutation rates to tetracycline resistance; error bars are 95% confidence levels. ppk+/pBAD24 = PFG265; ppk+/pDinB++ = PFG266; ppk::Cm/pBAD24 = PFG267; ppk::Cm/pDinB++ = PFG268.
Polyphosphate kinase does not affect the β-galactosidase levels of Lac+ revertants
Both adaptive mutation to Lac+ and growth-dependent mutation to TcR are dependent on the cell’s ability to replicate the episome, transcribe the genes encoding the mutational targets and translate the corresponding proteins. To test the possibility that ppk mutants are defective in one of these processes, we mated a Lac+ episome into the ppk::Cm mutant and wild-type strains and compared the β-galactosidase activities. The small (approximately 10%) difference in β-galactosidase activity between these strains is not statistically significant (t = 1.49, P = 0.20) (Table 1). As the amount of β-galactosidase (which is expressed solely from the episome) is nearly the same regardless of ppk activity, we conclude that ppk does not affect the copy number of the lacZ gene or the episome.
Table 1.
β-Galactosidase activity of wild-type and ppk mutant strains.
| Straina | Relevant genotype | β-Galactosidase activity (Miller units)b | SEM |
|---|---|---|---|
| FC420 | ppk+ | 341 | 19.8 |
| PFG282 | ppk::Cm | 306 | 11.7 |
Lac+ derivatives of the wild-type and ppk mutant strains.
Four independent cultures for each strain were assayed. Data are the mean and SEM.
Overexpressing RecA does not suppress the ppk antimutator phenotype
Adaptive mutation is dependent on the recombination functions of RecA. Previous studies have shown that strains with no detectable polyP have decreased levels of the recA transcript (Tsutsumi et al., 2000). As expected, the levels of RecA protein were slightly lower in ppk::Cm mutant cells than in wild-type cells (Fig. 6A). To test whether this reduction was responsible for the ppk antimutator phenotype, a multicopy plasmid encoding RecA was transformed into FC40 and its ppk::Cm derivative; the resulting strains were tested for adaptive mutation. As shown in Fig. 6B, overexpressing RecA did not suppress the ppk::Cm antimutator phenotype for adaptive mutation, indicating that the antimutator phenotype does not result from low levels of RecA.
Fig. 6.

Overexpression of RecA does not suppress the ppk antimutator phenotype.
A. Western blot showing that a ppk mutant strain has less RecA than a ppk+ strain, but not if RecA is overexpressed from a plasmid. Equal amounts of total protein from each strain were loaded in each lane; 10 μg of purified RecA protein (New England Biolabs) was loaded in the right lane. (The lower band is probably a degradation product).
B. Overexpression of RecA does not suppress the ppk mutant phenotype for adaptive mutation. Approximately 107 cells were spread on each quadrant of a lactose minimal plate. The data are the means of the total number (±SEM) of Lac+ colonies appearing on days 3–5. ΔrecA = FC348; ppk::Cm/pBR322 = PFG88; ppk+/pBR322 = PFG90; ppk::Cm/pRecA++ = PFG92; ppk+/pRecA++ = PFG94.
Polyphosphate kinase also affects Pol V-dependent mutagenesis
In E. coli, Pol V (encoded by the umuDC genes) is necessary for mutagenesis by DNA-damaging agents such as UV light (Tang et al., 2000). To test whether Ppk affects Pol V activity, we assayed UV-induced mutagenesis to nalidixic acid resistance (NalR), which occurs by base substitutions in the genes that encode DNA gyrase (Yoshida et al., 1988). The ppk::Cm mutant strain had a 40% decrease in UV mutagenesis to NalR compared with the wild-type strain, but the ppk::Cm mutation had no effect in a umuDC mutant strain, which is missing Pol V (Fig. 7). These results indicate that Ppk not only affects Pol IV, but also affects the mutagenic activity of Pol V.
Fig. 7.

The ppk mutant allele reduces UV mutagenesis when Pol V is present. Data points are the mean (±SEM) of NalR mutants per 108 cells. Wild type = FC40; ppk::Cm = PFG74; ΔumuDC::Erm = PFB286; ΔumuDC::Erm ppk::Cm = PFG414.
Discussion
Escherichia coli DNA Pol IV belongs to a family of error-prone polymerases some of whose members perform translesion DNA synthesis. A second important function for these polymerases may be to restart stalled replication forks (Goodman, 2002). It is hypothesized that error-prone polymerases gain access to replication forks by interacting with the β-clamp of DNA polymerase III (Wagner et al., 2000). Purified Pol IV can replicate only a few nucleotides by itself, but when interacting with the β-clamp it can replicate as many as 400 nucleotides (Tang et al., 2000; Wagner et al., 2000). Because many proteins (polymerase and others) bind to the β-clamp (Lopez de Saro et al., 2003), the polymerase that is most available may be the one targeted to the replication fork when normal replication stalls (Goodman, 2002). Polymerases II, IV and V are all induced in response to DNA damage (Goodman, 2002). Pol IV is also induced late in stationary phase independently of the SOS response (Layton and Foster, 2003). As there are about 250–1000 molecules of Pol IV in a non-induced cell and at least 2500 molecules in SOS-induced cells (Kim et al., 2001), there would appear to be enough molecules of Pol IV under most conditions to compete with other polymerases for the replication fork. The results presented here suggest that the mutagenic activity of Pol IV is modulated by Ppk and the amount of cellular polyP.
The ppk mutant derivative of the wild-type strain, FC40, exhibited a threefold decrease in adaptive mutation with no loss in cell viability (Fig. 2). Pol IV is responsible for up to 80% of the adaptive mutations in the wild-type strain (Foster, 2000; McKenzie et al., 2001); in recG mutants, adaptive mutation is increased 10- to 100-fold (Foster et al., 1996; Harris et al., 1996), and all these extra mutations result from Pol IV (Layton and Foster, 2003). As expected, defects in ppk had a relatively larger effect (fivefold decrease) in recG mutant strains because more of the mutations result from Pol IV. An epistasis test in recG+ strains showed that the ppk dinB double mutant had a greater reduction in adaptive mutation than the dinB single mutant (Fig. 2A), suggesting that Ppk has yet an additional effect on adaptive mutation. However, the increase in growth-dependent mutations caused by overexpression of Pol IV was also reduced by the ppk mutation, independently indicating that Ppk affects Pol IV mutagenic activity. Finally, we demonstrated that the activity of β-galactosidase expressed from the episome is not affected by loss of Ppk, eliminating the trivial possibility that Ppk affects the number or expression of the mutational targets (episomal lacZ or tetA). All of these results indicate that Ppk and/or its product, polyP, directly or indirectly affects the ability of Pol IV to cause mutations. Ppk also affects Pol V-dependent UV mutagenesis, suggesting that mutagenesis of all Y-family polymerases may be regulated by Ppk. However, we do not know as yet whether it is the polymerase activity or the fidelity of polymerization that is affected. In addition, it is possible that loss of Ppk reduces the amount of Pol V after UV damage.
The fact that loss of Ppk inhibited Pol IV-dependent mutation during growth is especially intriguing. Ppk is known to affect cells under stress (UV irradiation, nutritional downshift, amino acid starvation), but our results were obtained while the cells were growing in rich medium. The target for mutations during growth was reversion of a frameshift mutation in the tetA gene carried on the episome. Preliminary results show that when the same target for mutations is on the chromosome, the ppk mutant allele did not reduce growth-dependent mutations caused by overexpression of Pol IV (data not shown). One explanation for this difference is that Pol IV is more likely to be involved in DNA synthesis on the episome than on the chromosome. For example, Pol IV may participate in recombination-dependent DNA synthesis, which may occur more often on the episome because of the episome’s high frequency of recombination (Seifert and Porter, 1984; Carter et al., 1992). PolyP could facilitate the recruitment of Pol IV for DNA synthesis primed by recombination intermediates. If this hypothesis is true, ppk and recA should be epistatic for reduction of growth-dependent mutations produced by Pol IV overexpression, a hypothesis which we are currently testing.
The only known function for Ppk is the production of chains of inorganic polyP. In ppk mutants, the cellular levels of long-chained polyP molecules (about 600 inorganic phosphate residues) are reduced, but detectable levels of short-chained polyP molecules (about 60–70 inorganic phosphate residues) remain (Castuma et al., 1995). The source of these short-chained polyP molecules in ppk mutant strains is unknown (Castuma et al., 1995). When Ppx is overexpressed, all classes of polyP are undetectable (Shiba et al., 1997). Strains that overexpress Ppx have lower levels of RecA during SOS induction than do wild-type cells (Tsutsumi et al., 2000). We found that ppk mutant cells also have less RecA protein even when not induced for SOS (Fig. 6A). Adaptive mutation does not occur in the absence of RecA (Foster, 1993); however, as overexpressing RecA from a multicopy plasmid did not suppress the ppk mutant phenotype (Fig. 6B), we conclude that the ppk antimutator effect does not result from low levels of RecA. Thus, although RecA levels are decreased in a ppk mutant strain, they appear to be sufficient for adaptive mutation. Note that having more RecA did not increase mutation in the wild-type strain, suggesting that RecA is normally not limiting for adaptive mutation.
Bacterial strains that overproduce Ppx also have decreased levels of RpoS (Shiba et al., 1997). As the induction of Pol IV in late stationary phase is dependent on RpoS (Layton and Foster, 2003), it was possible that RpoS levels were low in ppk mutants, resulting in less Pol IV. However, the results shown in Fig. 4 indicate that in late stationary phase ppk mutant cells have the same amount of Pol IV protein as do wild-type cells. Thus, the amount of RpoS in ppk mutant strains is sufficient to keep the levels of Pol IV normal. RpoS also affects adaptive mutation independently of Pol IV (Layton and Foster, 2003, Lombardo et al., 2004); therefore, it is possible that Ppk affects these other RpoS-dependent functions in adaptive mutation, which would explain why the ppk::Cm mutant has an antimutator phenotype even in the absence of Pol IV (Fig. 2A). However, it is clear that the majority of the effect of Ppk on adaptive mutation is exerted on Pol IV independently of RpoS.
The simplest explanation for our results is that polyP modulates the mutagenic activity of Pol IV by affecting its ability to synthesize DNA, the accuracy at which it does so, or both. While overexpressing Ppk had no effect on adaptive mutation in a strain with functional Ppx, it strongly inhibited adaptive mutation in a strain with no Ppx (Fig. 3). As Ppx degrades polyP, these results, together with the antimutagenic phenotype of ppk mutants, suggest that there is an optimal level of polyP for maximum expression of Pol IV’s mutagenic activity. At this optimal level, polyP may be itself a positive activator of Pol IV’s mutagenic activity, or it may act indirectly by activating an activator or inhibiting an inhibitor of Pol IV’s mutagenic activity. It is also possible that the activity of Pol IV is affected by the loss of Ppk protein itself (although the antimutagenic effect of overproducing Ppk must have some other cause). For example, Ppk is one of several proteins that bind to the C-terminus of RNase E to form the RNA degradosome that regulates mRNA turnover (Blum et al., 1997; Carpousis, 2002; Bernstein et al., 2004). Thus, Ppk could indirectly regulate Pol IV by changing the mRNA levels of a direct regulator of Pol IV. However, the function of Ppk in the RNA degradosome has not been clearly defined (Blum et al., 1997). In addition, a C-terminus deletion of rne, the gene that encodes RNase E, does not inhibit adaptive mutation (J.D. Stumpf and P.L. Foster, unpubl. data).
PolyP has been shown to inhibit Thermus aquaticus DNA polymerase activity in vitro (Rodriguez, 1993), so another possibility is that polyP directly interacts with Pol IV and either inhibits its ability to synthesize DNA or reduces the accuracy at which it does so. As polyP is a long, negatively charged molecule like DNA, it may interact with many DNA-binding proteins. As mentioned above, polyP affects RpoS, which binds to DNA, and the RNA degradosome, which binds to RNA. Recently, polyP and DNA have been shown to compete for the same binding region of the Lon protease; polyP binding is hypothesized to activate Lon by freeing it from the DNA (Nomura et al., 2004). If polyP competes with DNA or RNA for binding sites to other proteins, this may explain how polyP levels influence many different cellular processes, including DNA replication. Future experiments are needed to determine whether polyP binds to DNA polymerases, or to factors that affect them, and whether this binding regulates the amount or accuracy of DNA synthesis.
Experimental procedures
Bacterial strains
All bacterial strains are E. coli K12 derivatives and are listed in Table 2. Genetic manipulations were as described previously (Miller, 1992). The ppk::Cm insertion was isolated in a screen for antimutators that was previously described (Layton and Foster, 2003). The Δppx::Kn allele was created using the techniques described by Datsenko and Wanner (2001). Mutant alleles were transferred by P1 transduction, selecting for appropriate markers. PFG282 was constructed by mating a Lac+ episome into PFG281 (FC36 ppk::Cm) and selecting for Pro+.
Table 2.
Bacterial strains and plasmids used in this study.
| Strain/plasmid | Relevant genotype | Source/reference |
|---|---|---|
| E. coli strains | ||
| FC29 | ara Δ(lac-proB)XIII thi/F′ Δ (lacIZ) Pro+ | Cairns and Foster (1991) |
| FC36 | F− ara Δ (lac-proB)XIII thi RifR | Cairns and Foster (1991) |
| FC40 | FC36/F′ Φ(lacI33-lacZ) Pro+ | Cairns and Foster (1991) |
| FC348 | FC40 ΔrecA srl::Tn10 | Berardini et al. (1999) |
| FC420 | FC36/F′ Lac+ Pro+ | This study |
| FC438 | FC40 recG162 zib636::Tn10 | Foster et al. (1996) |
| FC526 | FC40 ΔrecG263::Kn | Foster et al. (1996) |
| FC722 | FC40 with a Tcs allele on the episome | Foster (1997) |
| PFB243 | FC40 dinB::Zeo on chromosome and episome | Layton and Foster (2003) |
| PFB286 | FC40 ΔumuDC::Erm | Frank et al. (1996) |
| PFG36 | FC526 rpoS::miniTn10 Cm | Layton and Foster (2003) |
| PFG39 | FC526 ppk::miniTn10 Cm | This study |
| PFG74 | FC40 ppk::miniTn10 Cm | This study |
| PFG87 | PFG39/pBR322 | This study |
| PFG88 | PFG74/pBR322 | This study |
| PFG89 | FC526/pBR322 | This study |
| PFG90 | FC40/pBR322 | This study |
| PFG92 | PFG74/pPF2041 | This study |
| PFG94 | FC40/pPF2041 | This study |
| PFG152 | FC40 Δppk::Kn | This study |
| PFG162 | FC438 Δppx::Kn | This study |
| PFG175 | FC526/pPFG95 | This study |
| PFG176 | PFG39/pPFG95 | This study |
| PFG202 | FC722 ppk::miniTn10 Cm | This study |
| PFG203 | FC438 Δppk-ppx35::Kn | Kuroda et al. (1997); this study |
| PFG265 | FC722/pBAD24 | This study |
| PFG266 | FC722/pPFG96 | This study |
| PFG267 | PFG202/pBAD24 | This study |
| PFG268 | PFG202/pPFG96 | This study |
| PFG274 | PFB243 ppk::miniTn10 Cm | This study |
| PFG281 | FC36 ppk::miniTn10 Cm | This study |
| PFG282 | PFG281/F′ Lac+ Pro+ | This study |
| PFG322 | PFG162/pPFG95 | This study |
| PFG323 | PFG162/pBR322 | This study |
| PFG414 | PFB286 ppk::miniTn10 Cm | This study |
| Plasmids | ||
| pBR322 | None | Bolivar et al. (1977) |
| pPFG95 | ppk+ on pBR322 | This study |
| pPF2041 | recA+ on pBR322 | R. Brent (pers. comm.) |
| pBAD24 | None | Guzman et al. (1995) |
| pPFG96 | dinB + on pBAD24 | This study |
Plasmids
The plasmids used are listed in Table 2. Standard molecular biology techniques were as described (Ausubel et al., 1988). To make the ppk-overexpressing plasmid, ppk was amplified with primers whose sequences were CAGCCGGATCCCTG TAAATCGCAAGCTCC and GCCGAAAGCTTTTTGAAC CAAGATCGACC; the resulting polymerase chain reaction (PCR) fragment contained the whole ppk gene plus 121 nucleotides before the start codon and 69 nucleotides after the stop codon. The fragment was digested and ligated into BamHI- and HindIII-digested pBR322. To make pPFG96, dinB was amplified with primers whose sequences were GCCGATATAGAATTCATGCGTAAAATCATTCATGTGGATA and GCAGCCAAGCTTTCATCATAATCCCAGCACCAGTT GTC; the resulting PCR fragment contained the entire dinB gene and two nucleotides past the stop codon. The fragment was digested and ligated into BamHI- and EcoRI-digested pBAD24. That dinB was under control of the araBAD promoter was confirmed by Western blot analysis (data not shown), which showed that Pol IV was expressed from the plasmid at much higher levels than from the exogenous genes even in the absence of arabinose.
β-Galactosidase assays
Four independent cultures of each strain were grown to saturation in M9 glycerol minimal medium at 37°C. β-Galactosi-dase assays were performed as described (Miller, 1992).
Mutation experiments
Media and protocols were as described previously (Cairns and Foster, 1991; Miller, 1992; Foster, 1994; Foster et al., 1996). When required, antibiotics were added to rich media at the following concentrations: carbinicillin, 100 μg ml−1; kanamycin, 30 μg ml−1; chloramphenicol, 10 μg ml−1; tetracycline, 20 μg ml−1; nalidixic acid, 40 μg ml−1; rifampicin, 100 μg ml−1; when added to minimal media, antibiotics were added at half of the above concentrations except for chloramphenicol. For small-scale adaptive mutation experiments (Figs 1, 3 and 5), approximately 107 cells of each strain were spread on each quadrant of a lactose minimal plate, which was then incubated for 5 days at 37°C (Foster et al., 1996). Lac+ colonies that arose each day were counted, and the results are given as the total number of Lac+ colonies appearing from days 3–5. For the large-scale adaptive mutation experiment (Fig. 2A), lactose minimal plates were spread with about 108 cells of strains FC40 and PFG74 plus 109 cells of FC29 scavenger cells or 109 cells of strains PFB243 and PFG274 and no scavenger cells. Lac+ colonies that arose each day were counted. To determine the viability of the Lac− cells while incubating on lactose, plugs were removed from between Lac+ colonies and the number of viable cells was determined by spreading appropriate dilutions on LB plus rifampicin plates (which selects against FC29). To calculate the number of Lac+ revertants per cell, the number of Lac+ colonies was divided by the mean number of Lac− cells plated (Cairns and Foster, 1991). Statistical calculations were as given in Zar (1984).
To determine growth-dependent mutation rates (Fig. 5), overnight cultures of each strain were diluted 105-fold into LB broth plus appropriate antibiotics and 20 independent 1 ml aliquots were grown at 37°C overnight. A sample of 0.1 ml (approximately 108 cells) of each culture was spread on an LB plus tetracycline (Tc) plate and incubated at 37°C; the colonies arising after 24 h were counted. Mutation rates and confidence limits were calculated using the Lea–Coulson method of the median as described in Rosche and Foster (2000).
For UV mutagenesis (Fig. 7), a saturated culture of each strain was diluted 105-fold into LB broth plus appropriate drugs, and eight independent 1 ml aliquots were distributed and grown overnight at 37°C. An aliquot of 0.5 ml of each culture was added to 4.5 ml of 0.1 M MgCl2 in a glass Petri plate and irradiated with agitation for 25 s with UV light at 1.6 J m−2 s−1. Samples of the cultures were taken before and after irradiation, diluted, and plated on LB plates to determine the fraction of survivors, which ranged from 54% to 68% (data not shown). Before and after irradiation, 0.5 ml aliquots of each culture were diluted into 4.5 ml of LB broth and incubated at 37°C with aeration for at least 4 h. After this outgrowth, a sample of 0.1 ml (approximately 108 cells) was plated on LB plus nalidixic acid plate and incubated at 37°C for 24 h. Samples of the outgrown cultures were also diluted and plated on LB medium to determine total cell numbers. Results are given as the mean number of NalR mutants per 108 cells.
Molecular biology techniques
Molecular biology techniques were used as previously described (Ausubel et al., 1988). For immunoblots, cultures were grown in minimal glycerol medium plus appropriate drugs at 37°C and samples were taken 2 and 3 days after inoculation. Samples were centrifuged, and the cells were resuspended in SDS-PAGE sample loading buffer without dye and boiled for 15 min. Total protein was determined in each sample by Bradford assays (Bio-Rad Laboratories). Aliquots containing 40 μg of protein were diluted into sample loading buffer with dye and loaded onto an SDS 12% polyacrylamide gel. Proteins were separated by electrophoresis and then electrotransferred to Immobilon-P membranes (pore size, 0.45 μm; Millipore). The membranes were probed with rabbit anti-Pol IV polyclonal antiserum (obtained from H. Ohmori) or rabbit anti-RecA polyclonal antiserum (obtained from R. Woodgate). The reactions were visualized using alkaline phosphatase-conjugated goat anti-rabbit secondary antibody and developed using the Western-light chemiluminescence reagent (Applied Biosystems).
Acknowledgments
We thank the following people for bacterial strains, plasmids and reagents: R. Brent, L.M. Guzman, A.R. Fernandez de Henestrosa, K. Iams, A. Kuroda, J. Layton, R. Lloyd, J.H. Miller, H. Ohmori and R. Woodgate. We are grateful to the other members of our laboratory for useful comments and suggestions. This work was supported by USPHS Grant NIH-NIGMS GM651575 and NIH Training Grant T32 G07757.
References
- Akiyama M, Crooke E, Kornberg A. An exopolyphosphatase of Escherichia coli: the enzyme and its ppx gene in a polyphosphate operon. J Biol Chem. 1993;268:633–639. [PubMed] [Google Scholar]
- Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., and Struhl, K. (1988) Current Protocols in Molecular Biology New York: John Wiley & Sons.
- Bagg A, Kenyon CJ, Walker GC. Inducibility of a gene product required for UV and chemical mutagenesis in Escherichia coli. Proc Natl Acad Sci USA. 1981;78:5749–5753. doi: 10.1073/pnas.78.9.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini M, Foster PL, Loechler EL. DNA polymerase II (polB) is involved in a new DNA repair pathway for DNA interstrand cross-links in Escherichia coli. J Bacteriol. 1999;181:2878–2882. doi: 10.1093/gao/9781884446054.article.t031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein JA, Lin PH, Cohen SN, Lin-Chao S. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc Natl Acad Sci USA. 2004;101:2758–2763. doi: 10.1073/pnas.0308747101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum E, Py B, Carpousis AJ, Higgins CF. Polyphosphate kinase is a component of the Escherichia coli RNA degradosome. Mol Microbiol. 1997;26:387–398. doi: 10.1046/j.1365-2958.1997.5901947.x. [DOI] [PubMed] [Google Scholar]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calos MP, Miller JH. Genetic and sequence analysis of frameshift mutations induced by ICR-191. J Mol Biol. 1981;153:39–66. doi: 10.1016/0022-2836(81)90525-8. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ. The Escherichia coli RNA degradosome: structure, function and relationship in other ribonucleolytic multienzyme complexes. Biochem Soc Trans. 2002;30:150–155. [PubMed] [Google Scholar]
- Carter JR, Patel DR, Porter RD. The role of oriT in tra-dependent enhanced recombination between mini-F-lacoriT and lambda plac5. Genet Res. 1992;59:157–165. doi: 10.1017/s0016672300030433. [DOI] [PubMed] [Google Scholar]
- Castuma CE, Huang R, Kornberg A, Reusch RN. Inorganic polyphosphates in the acquisition of competence in Escherichia coli. J Biol Chem. 1995;270:12980–12983. doi: 10.1074/jbc.270.22.12980. [DOI] [PubMed] [Google Scholar]
- Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression pro-files following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2001;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Adaptive mutation: the uses of adversity. Annu Rev Microbiol. 1993;47:467–504. doi: 10.1146/annurev.mi.47.100193.002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Population dynamics of a Lac− strain of Escherichia coli during selection for lactose utilization. Genetics. 1994;138:253–261. doi: 10.1093/genetics/138.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Nonadaptive mutations occur on the F′ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Adaptive mutation in Escherichia coli. Cold Spring Harbor Symp Quant Biol. 2000;65:21–29. doi: 10.1101/sqb.2000.65.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Adaptive mutation in Escherichia coli. J Bacteriol. 2004;186:4846–4852. doi: 10.1128/JB.186.15.4846-4852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. Adaptive reversion of a frameshift mutation in Escherichia coli by simple base deletions in homopolymeric runs. Science. 1994;265:407–409. doi: 10.1126/science.8023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. Adaptive reversion of an episomal frameshift mutation in Escherichia coli requires conjugal functions but not actual conjugation. Proc Natl Acad Sci USA. 1995;92:5487–5490. doi: 10.1073/pnas.92.12.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM, Maurer RA. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank EG, Gonzalez M, Ennis DG, Levine AS, Woodgate R. In vivo stability of the Umu mutagenesis proteins: a major role for RecA. J Bacteriol. 1996;178:3550–3556. doi: 10.1128/jb.178.12.3550-3556.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Roth JR. Evidence that F plasmid transfer replication underlies apparent adaptive mutation. Science. 1995;268:421–423. doi: 10.1126/science.7716546. [DOI] [PubMed] [Google Scholar]
- Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson M, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Longerich S, Rosenberg SM. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- Harris RS, Ross KJ, Rosenberg SM. Opposing roles of the Holliday junction processing systems of Escherichia coli recombination-dependent adaptive mutation. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev. 2002;66:373–395. doi: 10.1128/MMBR.66.3.373-395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ, Walker GC. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc Natl Acad Sci USA. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, et al. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc Natl Acad Sci USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. Roles of chromosomal and episomal dinB genes encoding DNA Pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics. 2001;266:207–215. doi: 10.1007/s004380100541. [DOI] [PubMed] [Google Scholar]
- Kleckner, N., Bender, J., and Gottesman, S. (1991) Uses of transposons with emphasis on Tn10 In Bacterial Genetic Systems: Methods in Enzymology, Vol. 204. Miller, J.H. (ed.). San Diego, CA: Academic Press, pp. 139–180. [DOI] [PubMed]
- Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- Kuroda A, Murphy H, Cashel M, Kornberg A. Guanosine tetra- and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J Biol Chem. 1997;272:21240–21243. doi: 10.1074/jbc.272.34.21240. [DOI] [PubMed] [Google Scholar]
- Kuroda A, Nomura K, Ohtomo R, Kato J, Ikeda T, Takiguchi N, et al. Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science. 2001;293:705–708. doi: 10.1126/science.1061315. [DOI] [PubMed] [Google Scholar]
- Kusano D, Ishihama A. Functional interaction of Escherichia coli RNA polymerase with inorganic poly-phosphate. Genes Cells. 1997;2:433–441. doi: 10.1046/j.1365-2443.1997.13203301320330.x. [DOI] [PubMed] [Google Scholar]
- Layton JC, Foster PL. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol. 2003;50:549–561. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MJ, Aponyi I, Rosenberg SM. General stress response regulator RpoS in adaptive mutation and adaptive amplification in Escherichia coli. Genetics. 2004;166:669–680. doi: 10.1093/genetics/166.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Saro FJ, Georgescu RE, Goodman MF, O’Donnell M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003;22:6408–6418. doi: 10.1093/emboj/cdg603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell. 2001;7:571–579. doi: 10.1016/s1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- Miller, J.H. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Mueller-Hill B, Rickenberg HV, Wallenfels K. Specificity of the induction of the enzymes of the lac operon in Escherichia coli. J Mol Biol. 1964;10:303–318. doi: 10.1016/s0022-2836(64)80049-8. [DOI] [PubMed] [Google Scholar]
- Nomura K, Kato J, Takiguchi N, Ohtake H, Kuroda A. Effects of inorganic polyphosphate on the proteolytic and DNA-binding activities of Lon in Escherichia coli. J Biol Chem. 2004;279:34406–34410. doi: 10.1074/jbc.M404725200. [DOI] [PubMed] [Google Scholar]
- Radicella JP, Park PU, Fox MS. Adaptive mutation in Escherichia coli: a role for conjugation. Science. 1995;268:418–420. doi: 10.1126/science.7716545. [DOI] [PubMed] [Google Scholar]
- Rodriguez RJ. Polyphosphate present in DNA preparations from filamentous fungal species of Colletotrichum inhibits restriction endonucleases and other enzymes. Anal Biochem. 1993;209:291–297. doi: 10.1006/abio.1993.1122. [DOI] [PubMed] [Google Scholar]
- Rodriguez C, Tompkin J, Hazel J, Foster PL. Induction of a DNA nickase in the presence of its target site stimulates adaptive mutation in Escherichia coli. J Bacteriol. 2002;184:5599–5608. doi: 10.1128/JB.184.20.5599-5608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosche WA, Foster PL. Determining mutation rates in bacterial populations. Methods. 2000;20:4–17. doi: 10.1006/meth.1999.0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM, Hastings PJ. Adaptive point mutation and adaptive amplification pathways in the Escherichia coli Lac system: stress responses producing genetic change. J Bacteriol. 2004;186:4838–4843. doi: 10.1128/JB.186.15.4838-4843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM, Longerich S, Gee P, Harris RS. Adaptive mutation by deletions in small mononucleotide repeats. Science. 1994;265:405–407. doi: 10.1126/science.8023163. [DOI] [PubMed] [Google Scholar]
- Roth JR, Andersson DI. Adaptive mutation: how growth under selection stimulates Lac(+) reversion by increasing target copy number. J Bacteriol. 2004;186:4855–4860. doi: 10.1128/JB.186.15.4855-4860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert HS, Porter RD. Enhanced recombination between lambda plac5 and F42lac: identification of cis-and trans-acting factors. Proc Natl Acad Sci USA. 1984;81:7500–7504. doi: 10.1073/pnas.81.23.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba T, Tsutsumi K, Yano H, Ihara Y, Kameda A, Tanaka K, et al. Inorganic polyphosphate and the induction of rpoS expression. Proc Natl Acad Sci USA. 1997;94:11210–11215. doi: 10.1073/pnas.94.21.11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Pham P, Shen X, Taylor JS, O’Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- Tsutsumi K, Munekata M, Shiba T. Involvement of inorganic polyphosphate in expression of SOS genes. Biochim Biophys Acta. 2000;1493:73–81. doi: 10.1016/s0167-4781(00)00165-2. [DOI] [PubMed] [Google Scholar]
- Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2000;1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Madutani C, Iwai S, Hanaoka F. Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase eta. Nucleic Acids Res. 2000;28:2473–2480. doi: 10.1093/nar/28.13.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kojima T, Yamagishi J, Nakamura S. Quinilone resistant mutations of the gyrA gene in Escherichia coli. Mol Gen Genet. 1988;211:1–7. doi: 10.1007/BF00338386. [DOI] [PubMed] [Google Scholar]
- Zar, J.H. (1984) Biostatistical Analysis Englewood Cliffs, NJ: Prentice Hall.