

Summary
Recent evidence has implicated protein phosphatase PP5 in a variety of signaling pathways. While several proteins have been identified that interact with PP5 and regulate its activity, a possibility of its regulation by second messengers remains speculative. Activation of PP5 in vitro by polyunsaturated fatty acids (e.g. arachidonic acid) and fatty acyl-CoA esters (e.g. arachidonoyl-CoA) has been reported. We report here that PP5 is strongly inhibited by micromolar concentrations of a natural polyamine spermine. This inhibition was observed both in assays with a low molecular weight substrate p-nitrophenyl phosphate as well as phospho-casein and apoptosis signal-regulating kinase 1 (ASK1), thought to be a physiological substrate of PP5. Furthermore, a decrease in polyamine levels in COS-7 cells induced by α-difluoromethylornithine (DFMO), an inhibitor of ornithine decarboxylase, led to accelerated dephosphorylation of oxidative stress-activated ASK1. This effect was suppressed by okadaic acid and by siRNA-mediated PP5 depletion, indicating that the effect of polyamine levels on ASK1 dephosphorylation was mediated by PP5. In line with the decreased ASK1 activation, polyamine depletion in COS-7 cells abrogated oxidative stress-induced activation of caspase-3, which executes ASK1-induced apoptosis, as well as caspase-3 activation induced by ASK1 overexpression, but had no effect on basal caspase-3 activity. These results implicate polyamines, emerging intracellular signaling molecules, as potential physiological regulators of PP5. Our findings also suggest a novel mechanism of the anti-apoptotic action of a decrease in polyamine levels via de-inhibition of PP5 and accelerated dephosphorylation and deactivation of ASK1.
Keywords: Protein phosphorylation, protein serine/threonine phosphatase, oxidative stress, apoptosis, intracellular polyamines, spermine
Abbreviations. ASK1, apoptosis signal-regulating kinase 1; DTT, dithiothreitol; HEP, 1-(2-hydroxyethyl)piperazine; IPTG, isopropyl β-D-1-thiogalactopyranoside; Ni-NTA, Ni2+-nitrilotriacetic acid; pNPP, 4-nitrophenyl phosphate; PPP, phosphoprotein phosphatases of P family; TPR, tetratricopeptide repeat
Introduction
Protein Ser/Thr phosphatase PP5 is a member of the PPP family, one of the two known families of Ser/Thr-specific protein phosphatases [1]. PP5 (also termed PPT in fungi) was identified in 1994 by three independent groups [2-4] as a phosphatase with tetratricopeptide repeat (TPR) domain, N-terminally fused to a typical PPP catalytic domain. PP5 appears to be ubiquitous in eukaryotes [5]. The last years have seen a steadily increasing number of publications addressing the ways of PP5 regulation and its roles in intracellular signaling. A variety of PP5-interacting proteins have been reported; interestingly, all known interactions require the TPR domain of PP5.
A significant proportion of PP5 in the cell is bound to a molecular chaperone Hsp90 and is a component of glucocorticoid receptor - Hsp90 heterocomplexes [6,7]. Hsp90-associated PP5 was found to regulate nucleocytoplasmic shuttling of glucocorticoid receptors [8]. Another function of Hsp90-associated PP5 is suggested by the finding that it negatively modulates maturation of the Hsp90-dependent heme-regulated eIF2α kinase [9].
While PP5 was initially reported to be predominantly nuclear, later studies established that it is localized in both the nucleus and the cytoplasm (reviewed in [4]). Recently, the ability of PP5 to translocate to the nucleus during apoptosis has been reported [10]. PP5 has an atypical nuclear localization signal in its C-terminal extension [11]. Cytoplasmic PP5 has been found to interact with the intermediate chain of cytoplasmic dynein and to colocalize with both cytoplasmic dynein and microtubules [12]. Copurification of PP5 with microtubules has also been documented, as well as its ability to dephosphorylate a microtubule-associated protein tau in vitro [13]; physiological relevance of the latter observation has not yet been demonstrated.
PP5 participates in the regulation of the cell cycle in a dual way: (i) it binds to CDC16 and CDC27, two subunits of the anaphase-promoting complex, and is thought to maintain it in an inactive dephosphorylated state [14]; (ii) it regulates transcriptional induction of p21WAF1/Cip1 (a universal inhibitor of cyclin-dependent protein kinases), presumably by directly controlling phosphorylation status of p53 [15]. Furthermore, a recent study [16] has suggested a link between glucocorticoid receptor activation and the phosphorylation status of p53 that regulates expression of p21WAF1/Cip1. Therefore, PP5 appears to control two distinct steps of the signaling pathway that links glucocorticoid receptor activation and cell growth arrest.
A link between PP5 and G protein-coupled signaling has recently been found. PP5 directly interacts with and is stimulated by Gα12/Gα13 [17]. On the other hand, under redox stress PP5 associates with and inhibits apoptosis signal-regulating kinase 1 (ASK1) [18; 19], which in its turn is stimulated by Gα12/Gα13 [20]. Potential interplay between PP5, Gα12/Gα13 and ASK1 is further complicated by the fact that Gα12 [21] and PP5 (see above) both interact with Hsp90.
In addition to the above findings, which already suggest PP5 involvement in elaborate signaling pathways, interaction of this phosphatase with a number of other proteins has been reported, but functional significance of these interactions is less clear. Interaction of PP5 with atrial natriuretic factor receptor (ANP) has been detected using yeast two-hybrid screening [4]. There is an extensive but indirect evidence for the involvement of PP5 in regulation of K+ channels (reviewed in [4]). PP5 has also been reported to associate with both isoforms of chryptochromes (blue light photoreceptors) and to be stimulated by at least one of them [22]. Intriguingly, a distantly related plant PPP phosphatase PP7, which has no TPR domain but shares with PP5 a common atypical nuclear targeting signal [23], has recently been implicated in signaling downstream of cryptochromes [24]. Since animal and plant chryptochromes are thought to have evolved from DNA photolyases independently [25], this may suggest convergent evolution not only of these photoreceptors but also of the signaling mechanisms they trigger. PP5 has also been found to interact with scaffold proteins, such as calcium-dependent phospholipid-binding proteins copines [26] and the A subunit of the protein phosphatase PP2A [27]. Another protein that interacts with both PP5 and PP2A is G5PR, homologous to B″ subunit of PP2A [28]. Functional significance of these interactions remains to be tested.
In contrast to the wealth of data about interactive partners of PP5, relatively little is known about its potential regulation by low molecular weight compounds or second messengers. Arachidonic acid and other polyunsaturated fatty acids, as well as phospholipids, were found to stimulate PP5 via its TPR domain [6,,29,30]. While polyunsaturated fatty acids are known to exert physiologically relevant effects, their concentrations required for PP5 stimulation in vitro are higher than those thought to occur in vivo. Ramsey and Chinkers [31] have recently found that long-chain fatty acid-CoA esters (with acyl chain length of 12 – 20 carbon atoms, saturated or not), are more potent stimulators of PP5 than polyunsaturated fatty acids, and suggested that these compounds may be physiological stimulators of this phosphatase.
Natural polyamines (putrescine, spermidine and spermine) are ubiquitous cations found in almost any living organism, able to bind to nucleic acids and to acidic phospholipids in the membranes [32]. Polyamines are involved in diverse functions such as control of cell cycle and apoptosis, DNA conformation and stability, regulation of transcription, ion channel regulation and protein phosphorylation [32–35]. Due to the role of polyamines in regulation of cell proliferation, polyamine analogs have been envisaged as possible anticancer drugs [36,37]. Although only a limited number of proteins are known that are specifically regulated by polyamines, at least on one occasion polyamines have been shown to act as signaling molecules [38].
We report here that PP5 is strongly inhibited by micromolar concentrations of a polyamine spermine. We demonstrate that polyamine levels modulate activation of ASK1, thought to be a physiological substrate of PP5, and affect ASK1-induced apoptotic signaling in vivo. This implicates polyamines, emerging intracellular signaling molecules, as potential physiological regulators of PP5. Our findings also suggest a novel mechanism of the anti-apoptotic action of a decrease in polyamine levels via de-inhibition of PP5 and accelerated dephosphorylation and deactivation of ASK1.
Materials and Methods
Expression in E.coli and purification of recombinant PP5.
pET15b-PP5 plasmid was kindly provided by Dr M. Chinkers (University of South Alabama). PP5 was expressed in BL21(DE3)pLysS E. coli cells (Stratagene) according to the manufacturer’s instructions, except that expression was induced with 0.2 mM IPTG at 13°C overnight. Cells from 1 l culture were harvested by centrifugation and resuspended in a lysis buffer, containing 20 mM Tris-HCl (pH 7.5), 5 mM imidazole, 0.3 M NaCl, and a cocktail of protease inhibitors (Sigma). After sonication, lysate was centrifuged in an SS-34 rotor (Sorvall) at 15000 rpm for 15 min, supernatant mixed with a suspension of 5 ml of Ni-NTA agarose (Qiagen) and incubated on a shaker for 1 h at 4°C. Suspension was then poured into a column, gel washed with the lysis buffer, and proteins eluted by a stepwise gradient of imidazole up to 250 mM. Fractions were analysed by SDS-PAGE, and if necessary diluted with lysis buffer without imidazole and rechromatographed on Ni-NTA. For final purification, fractions eluted from Ni-NTA agarose were diluted to decrease NaCl concentration to 30 mM, and applied on a 3 ml DEAE-Sepharose (Sigma) column. Proteins were eluted by a stepwise NaCl gradient in 20 mM Tris-HCl (pH 7.5), 1 mM DTT. Fractions containing PP5 were pooled and concentrated by centrifugation in Centricon units (Millipore) to yield 10 mg/ml protein concentration. Purified PP5 was stored in aliquots at −80 °C.
PP5 and ASK1 immunoprecipitation from COS-7 cells.
Confluent COS-7 cells on a 10 cm plate were transfected with 2 μg pCMV6-FLAG-PP5 (kindly provided by Dr M. Chinkers) or pcDNA-3-HA-ASK1 [20]. For immunoprecipitation of activated ASK1, 1 mM H2O2 was added to the growth medium 15 min prior to cell collection. Cells were solubilized in 2 ml 20 mM Hepes (pH 7.5), 5 mM MgCl2, 0.5% lubrol, 1:200 dilution of proteinase inhibitor cocktail (Sigma) by passing through a 25 gauge needle. Debris was pelleted, supernatants supplemented with 5 μg M2 FLAG monoclonal antibody (Sigma) or F-7 HA antibody (Santa Cruz Biotechnology) and 50 μl Protein A/G Agarose suspension (Santa Cruz Biotechnology), and rotated for 5 h at 4°C. Protein A/G Agarose beads were pelleted and washed 3 times with 1 ml lysis buffer and twice with the same buffer without lubrol. PP5 was eluted with 0.1 mg/ml FLAG peptide (Sigma) in lysis buffer wihout lubrol for 15 min at room temperature. ASK1 and PP5 preparations were stored at −20 °C in a 1:1 mixture of glycerol and lysis buffer wihout lubrol.
Phosphatase assays.
Phosphatase assays with p-nitrophenyl phosphate (pNPP) were performed as follows. PP5 was diluted to 50–100 nM (final concentrations) with 40 mM Hepes (pH 7.5), 100 nM BSA and varying concentrations of spermine. Reactions were started by addtion of pNPP to 1.25 mM (final concentration) from a stock solution (25 mM) adjusted with NaOH to pH 7.5. Reaction was continuously monitored by measuring absorption at 410 nm. Reaction rates were calculated from the linear parts of the plots using a molecular absorption coefficient of 15 x 103 mol L−1 cm−1.
Colorimetric phosphatase assays with phospho-casein (Sigma C5890) were performed using procedure of Ekman and Jäger [39].
For In vitro ASK1 dephosphorylation assays, ASK1 isolated from COS-7 cells as described above was autophosphorylated in a buffer containing 20 mM Hepes (pH 7.5), 5 mM MgCl2, and 8 μM [γ-32P] ATP (5 μCi) for 15 min at room temperature, beads were washed 3 times in the same buffer without ATP and stored at −20 °C in 50 % glycerol. Autophosphorylated ASK1 was incubated at room temperature with PP5 isolated from COS-7 cells under conditions indicated in the figure legends. Dephosphorylation reaction was started by addition of [γ- 32P] ASK1 and stopped by addition of trichloroacetic acid and BSA to 15% and 0.2 mg/ml, respectively. Proteins were precipitated by centrifugation, and phosphate released into supernatants was quantitated by scintillation counting.
PP5 suppression using PP5 siRNA.
siRNA-mediated suppression of PP5 was performed essentially according to [19]. siRNA for PP5 suppression (PP5TG2) and mismatched control (PP5TG2MM) were synthesized by Invitrogen.
In vivo ASK1 phosphorylation assay.
Cells were grown in 24 well plates and subjected to oxidative stress by addition of 1 mM H2O2 (final concentration). After various incubation times, cells were washed twice with ice-cold PBS, resuspended in 20 mM Na-phosphate (pH 7.5), 25 mM NaF, 1 mM orthovanadate, 5 mM EDTA, lysed by addition of a standard Laemmli SDS buffer and boiled for 10 min. Cell lysates were separated on 5–20% gradient SDS-polyacrylamide gels and proteins transferred onto a PVDF membrane (Osmonics). Membranes were probed with phospho-ASK1(Thr845) polyclonal antibody (Cell Signaling Technology) and developed using ECL Plus reagents (Amersham Biosciences). Densitometry of ASK1 bands was performed on scanned images using NIH Image 1.63 software. Data were normalized to total ASK1 content in respective samples. For data normalization, membranes were reprobed with ASK1 monoclonal antibody (Cell Signaling Technology), recognizing ASK1 independently of its phosphorylation state.
Difluoromethylornithine (DFMO)- induced ASK1 dephosphorylation was calculated for each time point as the difference between the amounts of ASK1(pThr845) in DFMO-treated and untreated samples, normalized to untreated samples. To assess the effects of PP5 inhibition on DFMO-induced ASK1 dephosphorylation, time points were chosen where the effect of DFMO was maximal without PP5 inhibition. DFMO-induced ASK1 dephosphorylation was then calculated at corresponding (or adjacent if greater, see figure legends) time points in the cells treated with okadaic acid or subjected to siRNA-mediated PP5 depletion. If DFMO-induced dephosphorylation in the PP5-inhibited samples was negative (i.e. the amount of ASK1(pThr845) was greater in the DFMO-treated than iun untreated cells) it was taken as 0. The values were expressed as percentage of those in the respective control cells. A paired two-tailed Student’s t test was used to assess statistical significance.
In the experiments involving PP5 depletion by siRNA, the extent of depletion was tested using PP5 monoclonal antibody (Cell Signaling Technology) and equal loading was checked with α-tubulin and β-actin monoclonal antibodies (Sigma).
Since the film response may not be linear with ECL signal and with the amount of antigen, quantitation was performed from more than one exposure for each experiment to ensure the consistency of the results.
Caspase-3 activation was assessed in a dual way: using colorimetric assay and by detection of cleaved caspase-3 by immunoblotting. Caspase-3 substrate N-Acetyl- Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) was purchased from Sigma. Colorimetric assay was performed according to manufacturer’s instructions. Activated caspase-3 was detected by immunoblotting using an antibody specifically recognizing cleaved form of the enzyme (Cell Signaling Technology).
Results
1. PP5 is highly sensitive to inhibition by spermine.
A substantial body of evidence points to the ability of several phosphatases of the PPP family to be regulated by polycations. PP1 is thought to be regulated by polyamine levels in vivo [40–43]. PP2A regulation by polyamines in vitro was reported [40], although in this case effective polyamine concentrations may be above the physiological range. Plant PP7, although not tested with polyamines, was found to be regulated by micromolar concentrations of hexalysine and was suggested to possess a polycation-binding site [44].
Therefore, we examined the effects of polyamines on PP5 activity. Purified recombinant PP5, assayed with p -nitrophenyl phosphate pNPP, was found to be highly sensitive to inhibition by spermine (Fig. 1A). Scatchard analysis (Fig. 1A, insert) gave an estimate for the Kd of ~4 μM. Another naturally occurring polyamine, putrescine, was much less potent, inhibiting PP5 by only ~30 % at 1 mM concentration (data not shown). To ensure that the observed effect was not an artifact of bacterial expression of PP5, we overexpressed FLAG-tagged PP5 in COS-7 cells, isolated it by immunoprecipitation with FLAG antibody and examined the effect of spermine on its activity. Similar to bacterially expressed PP5, strong inhibition by spermine was observed for PP5 expressed in mammalian cells (Fig. 1B).
Figure 1. Effect of spermine on PP5 activity tested with p-nitrophenyl phosphate (p- NPP).

A, Activity of PP5 (50 nM) was measured with 1.25 mM p-NPP in the absence or in the presence of varying concentrations of spermine. Data are calculated from the linear regression on points between 7.5 and 30 min. Insert: Scatchard plot of the same data. PP5 inhibited by saturating concentrations of spermine was postulated to bind 1 mol spermine/mol protein. A linear fit of these data is consistent with the single binding site and gives a value for the apparent Kd of 3.8 μM. B, FLAG-tagged PP5 was expressed in COS-7 cells, immunoprecipitated using FLAG monoclonal antibody and its activity was measured as described in Materials and Methods. Data shown are the means of three replicates, with error bars indicating values from each replicate. Experiment shown is representative of two similar experiments. Insert, a Coomassie stained gel of material immunoprecipitated from cells expressing FLAG-PP5 (+) and control cells transfected with vector alone (−). Identity of PP5 and Hsp90 was verified by immunoblotting (not shown).
2. Dephosphorylation of protein substrates by PP5 is inhibited by spermine.
While pNPP is convenient as it permits to follow the reaction course in real time, observations with this low molecular weight substrate may not adequately reflect regulation of phosphatase activity towards protein substrates [44,45]. Therefore, we tested wether dephosphorylation of a protein substrate by PP5 would also be affected by spermine. PP5 activity tested with phosphocasein was also strongly inhibited by spermine (Fig. 2). Although apparent Kd ( ~30 μM) for spermine with phosphocasein was higher than that observed with pNPP, it was still within the supposed physiological range of spermine concentrations (see Discussion) and is similar to that reported for PP1 (40 μM [40]). These results indicated that PP5 may be regulated by physiological concentrations of intracellular polyamines.
Figure 2. Effect of spermine on PP5 activity tested with phospho-casein.
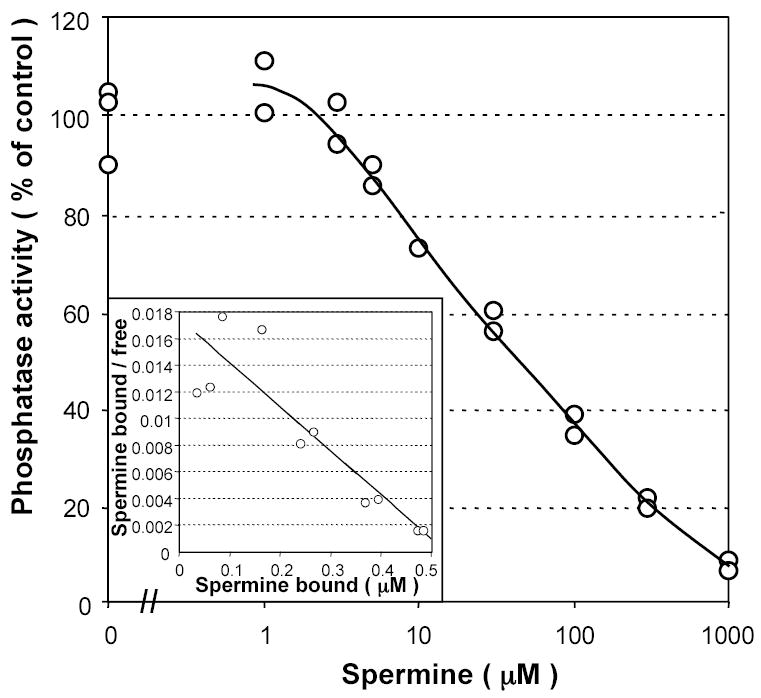
PP5 (600 nM) was incubated for 30 min with 0.2 mg/ml phospho-casein in the absence or in the presence of varying concentrations of spermine. Insert: Scatchard plot of the same data. PP5 inhibited by saturating concentrations of spermine was postulated to bind 1 mol spermine/mol protein. A linear fit of these data gives a value for the apparent Kd of 30 μM. Experiment shown is representative of two different experiments.
3. In vitro dephosphorylation of ASK1, a physiological substrate of PP5, is inhibited by spermine.
As the next step to validate the physiological relevance of PP5 regulation by spermine, we examined its effect on dephosphorylation of ASK1, a MAPKK kinase which is thought to be a physiological substrate of PP5 [18,19]. ASK1 is activated by autophosphorylation on Thr845 and inactivated by dephosphorylation of this residue by PP5 [18].
Recombinant PP5 purified from E. coli was found to be unable to dephosphorylate ASK1, suggesting that either its folding differs from that of the native enzyme, or that an additional factor absent in E. coli is required for efficient dephosphorylation of ASK1. This is in line with observations by Morita and co-workers [18] that GST-PP5 fusion protein expressed in E. coli was unable to dephosphorylate ASK1 unless activated with arachidonic acid. However using polyunsaturated fatty acids or fatty acyl CoA (which are more efficient activators of PP5 [31]) would not be possible in our experiments since spermine would presumably neutralize these exogenously added acidic compounds.
Therefore, we tested whether PP5 expressed in COS-7 cells was able to dephosphorylate ASK1. FLAG-tagged PP5 was purified from COS-7 cells by immunoprecipitation (see above and Fig. 1B) and eluted with FLAG peptide. These PP5 preparations were found to efficiently dephosphorylate [32P]-labeled ASK1, prepared by autophosphorylation in vitro.
In the absence of spermine, the release of phosphate from ASK1 was linear at least up to ~25 % release of total Pi (Fig. 3A). In the presence of spermine (100 μM), an initial stimulation of ASK1 dephosphorylation was observed during the first minutes of incubation, followed by inhibition (Fig. 3B). This biphasic kinetics of ASK1 dephosphorylation was observed with three independent PP5 preparations, although the extent of the initial stimulation varied. The initial stimulation by spermine followed by inhibition was also observed when ASK1 autophosphorylated in vivo upon H2O2 treatment and isolated by immunoprecipitation was dephosphorylated in vitro by purified PP5 or when ASK1 dephosphorylation was assessed in a mixture of crude lysates of COS-7 cells expressing ASK1 and PP5 (data not shown). Since in these experiments ASK1 dephosphorylation was followed using phospho-Thr845 specific antibody rather than release of radioactive Pi, the initial stimulation by spermine cannot be attributed to ASK1 dephosphorylation at a site distinct from Thr845.
Figure 3. Dephosphorylation of ASK1 by PP5 in vitro is inhibited by spermine.

HA-ASK1 and FLAG-PP5 were purified from COS-7 cells by immunoprecipitation and PP5 eluted with FLAG peptide. ASK1 was autophosphorylated in the presence of [γ32P] ATP. A, Time course of ASK1 dephosphorylation by PP5 in the absence (empty circles) or in the presence (filled circles) of spermine (100 μM). B, Dephosphorylation of ASK1 by PP5 as a function of varying concentrations of spermine. Dephosphorylation was allowed to proceed for 30 min. Phosphatase activity was normalized to the activity in the absence of spermine. The experiment shown is representative of four different experiments performed with two independent preparations of ASK1 and three preparations of PP5.
ASK1 dephosphorylation by PP5 was half inhibited at 20–30 μM spermine (Fig. 3B). Although non-linearity of dephosphorylation in the presence of spermine does not allow an accurate Kd estimate, inhibition of ASK1 dephosphorylation clearly occurs within the presumed physiological range of spermine concentrations.
4. ASK1 activation by oxidative stress is decreased by polyamine depletion in vivo.
Observations described above led us to a prediction that if regulation of PP5 by polyamines takes place in vivo, polyamines might regulate the extent of activation of ASK1 by affecting the rate of its dephosphorylation by PP5. Therefore, we examined the time course of ASK1 activation and deactivation in COS-7 cells subjected to oxidative stress in the absence or in the presence of α-difluoromethylornithine (DFMO), an irreversible inhibitor of ornithine decarboxylase, the first enzyme in polyamine synthesis.
In control cells, ASK1 was maximally activated in ~5 min, followed by a partial dephosphorylation and the second activation wave at 20–30 min (Fig. 4A,B Fig. 5A,B). While the peak time of the first and the second phosphorylation waves and the extent of their separation by a dephosphorylation phase varied between the experiments (compare Fig. 4B and Fig. 5B), the two wave-like profile of ASK1(Thr845) phosphorylation was reproducible.
Figure 4. Dephosphorylation of ASK1 in COS-7 cells is enhanced by polyamine depletion in an okadaic acid-sensitive manner.

COS-7 cells growing in a 24 well plate were transfected with 20 ng/well pcDNA3-HA-ASK1. 18 h after transfection, OPTI-MEM medium was changed for DMEM containing 10 % calf serum with or without 5 mM DFMO. Cells were grown for further 30 h and and ASK1 autophosphorylation on Thr845 was induced by adding H2O2 (1 mM). 1.5 h prior to H2O2 addition, cells were pretreated with okadaic acid (150 nM; OkA) or vehicle (DMSO) as indicated. Cell lysates were analyzed by immunoblotting with phospho-specific ASK1(pThr845) antibody or ASK1-specific antibody (A). Data were normalized by dividing the extent of ASK1 phosphorylation at different time points by that in the DFMO- and okadaic acid-untreated cells without H2O2 addition, and plotted as a function of time after stimulation (B, C). Empty and filled circles, cells grown in the absence or in the presence of DFMO, respectively. Arrows in B and C indicate the time points used to calculate the extent of suppression of DFMO-induced ASK1 dephosphorylation in okadaic acid-treated cells (D) as described in Materials and Methods. Data shown in D are derived from the experiment shown in A–C (150 nM okadaic acid) and from a similar experiment using a 40 min pretreatment with 100 nM okadaic acid. Filled bars are the means of three time points from each experiment ± S.D. *, P < 0.05 in a paired two-tailed Student’s t test.
Figure 5. DFMO-induced ASK1 dephosphorylation is decreased by siRNA-mediated PP5 depletion.
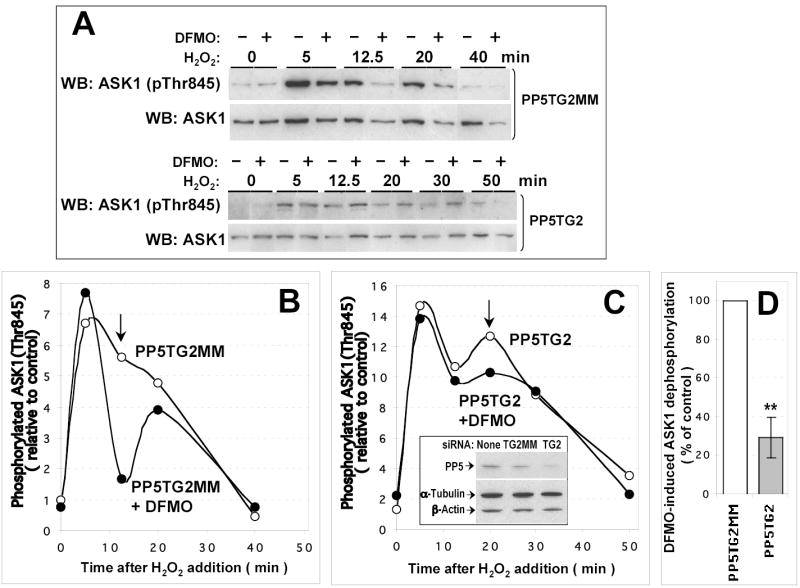
COS-7 cells growing in a 24 well plate were transfected with 20 ng/well pcDNA3-HA-ASK1 and 10 pmoles/well siRNA duplexes (PP5TG2, PP5-targeting siRNA; PP5G2MM, mismatched control). 6 h after transfection, OPTI-MEM medium was changed for DMEM containing 10 % calf serum with or without 5 mM DFMO. Cells were grown for further 20 h and H2O2 (1 mM) was added at indicated time intervals before cell collection. The extent of ASK1(Thr845) phosphorylation (A) was quantified and data plotted (B, C) as in Fig. 4. Empty and filled circles, cells grown in the absence or in the presence of DFMO, respectively. Insert in C shows typical extent of PP5 depletion; actin and tubulin content is shown as control of equal loading. Arrows in B and C indicate the time points used to calculate the extent of suppression of DFMO-induced ASK1 dephosphorylation in PP5-depleted cells (D) as described in Materials and Methods. Filled bar in (D) represents a mean (± S.D.) of the data from the experiment shown in A–C and two similar experiments. *, P < 0.01 in a paired two-tailed Student’s t test.
In polyamine-depleted cells, the initial ASK1 activation was not affected (Fig. 4A, Fig. 5A), however ASK1 dephosphorylation was accelerated, which affected the second phosphorylation wave (Fig. 5A and Fig. 4A, respectively; note the difference between the control and DFMO-treated cells indicated by arrows). The effect of DFMO was specific, since it could be reversed by addition of exogenous putrescine (precursor of other polyamines), which permits cells to bypass the block of polyamine biosynthesis (data not shown). These results indicated that either a kinase reponsible f o r the second phosphorylation wave was inhibited by polyamine depletion, or ASK1 phosphatase activated.
To determine whether the effect of polyamine depletion requires PP5, we examined whether DFMO would have the same effect when PP5 activity is downregulated. While no specific inhibitors of PP5 are available, it is potently inhibited by okadaic acid, a membrane-permeable compound that also inhibits some other PPP phosphatases (PP1, PP2A), but neither PPM nor dual specificity phosphatases. Pretreatment of the cells with okadaic acid led to a severalfold increase in the basal level of ASK1(Thr845) phosphorylation and also enhanced its maximal activation (Fig. 4). In the presence of okadaic acid, DFMO did not accelerate ASK1 dephosphorylation (compare Fig. 4C and Fig. 4B). Quantitation of the effect of okadaic acid in two experiments using different concentrations of okadaic acid showed a robust suppression of DFMO-induced ASK1 dephosphorylation (Fig. 4D).
These findings demonstrate that the effect of DFMO is mediated by an okadaic acid-sensitive protein phosphatase, as it would be expected for PP5. Notably, despite the presence of okadaic acid, an efficient dephosphorylation of ASK1 after the first phosphorylation wave was observed, suggesting a compensatory effect of an okadaic acid-insensitive phosphatase. This phosphatase activity was unlikely to be due to Cdc25 (which has been identified as an ASK1-interacting protein [46]) since it was not suppressed by NSC95397, a selective cell-permeable inhibitor of Cdc25 (data not shown). We made no further attempts to identify the okadaic acid-resistant ASK1 phosphatase.
To further confirm that the effect of polyamine depletion specifically requires PP5, we downregulated endogenous PP5 by siRNA and checked whether this would affect DFMO-induced ASK1 dephosphorylation (Fig. 5). Transfection of COS-7 cells with PP5 siRNA resulted in a 50–60 % decrease in the levels of PP5 compared to mismatched control, as determined by immunoblotting (Fig. 5C, insert). Like treatment with okadaic acid, depletion of PP5 led to an increased basal level of ASK1 phosphorylation and higher maximal activation (Fig. 5). Under these conditions, the effect of DFMO treatment was still observable but was much weaker and slightly delayed (Fig. 5C; compare the difference between DFMO-treated and untreated cells indicated by arrows in Fig. 5C and in Fig. 5B). Quantitation of the data from three experiments showed a statistically significant decrease in DFMO-induced ASK1 dephosphorylation in PP5- depleted cells as compared to the cells transfected with mismatched control siRNA (Fig. 5D).
Note that since PP5 was only partially depleted in these experiments one would expect the effect of DFMO to be decreased but not completely abolished.
These observations are consistent with polyamine depletion leading to de-inhibition of PP5, which in its turn results in a more efficient dephosphoryaltion of ASK1.
5. Polyamine depletion suppresses pro-apoptotic signaling in COS-7 cells.
ASK1 plays a pivotal role in the pro-apoptotic signaling induced by oxidative stress [47]. At the same time, polyamines have been implicated in apoptosis, acting both as apoptosis promoting or protective agents depending on a stress type and cellular context [48]. Since polyamines appear to be required for full ASK1 activation in COS-7 cells, the decrease in polyamine levels would be expected to protect COS-7 cells from oxidative stress-induced apoptosis.
ASK1 signaling is known to induce apoptosis via activation of caspase-3 [49]. Therefore, we examined oxidative stress-induced activation of this caspase in COS-7 cells grown in the absence or in the presence of DFMO. As expected, polyamine depletion inhibited caspase-3 activation upon H2O2 treatment, as evidenced both by caspase-3 activity assay and immunoblotting with an antibody specifically recognizing cleaved form of caspase-3 (Fig. 6A).
Figure 6. Polyamine depletion inhibits H2O2-induced and ASK1-induced caspase-3 activation in COS-7 cells.
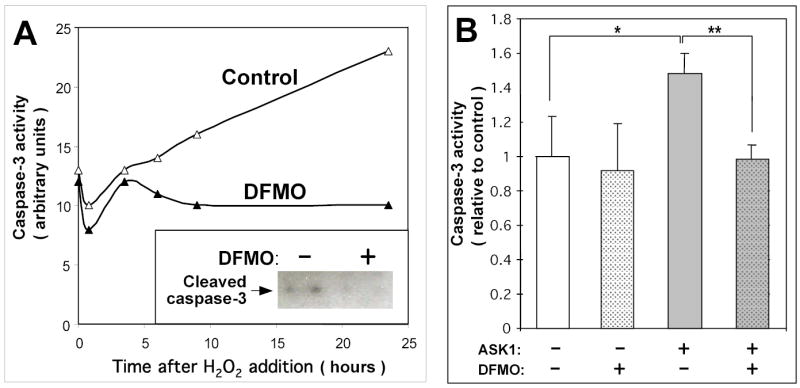
(A) COS-7 cells were grown in DMEM medium containing 10 % calf serum with or without 5 mM DFMO for 48h, then medium was changed to DMEM without serum ± 2 mM DFMO, and 1 mM H2O2 was added at indicated time intervals before cell collection. Cell lysates were assayed for caspase-3 activity (see Materials and Methods for details) and analyzed by immunoblotting with cleaved caspase-3-specific antibody (insert; cells were collected 6 h after stimulation). (B) Cells were grown in DMEM medium containing 10 % calf serum with or without 5 mM DFMO for 48h and then transfected with ASK1 or vector as indicated (DFMO pretreatment did not affect transfection efficiency, data not shown). Cells were grown in OPTI-MEM with or without 2 mM DFMO for further 24 h, lysed and assayed for caspase-3 activity. Data shown are the means of three replicates ± S.D. *, P < 0.05; **, P < 0.01 in a paired two-tailed Student’s t test.
To further confirm that polyamine depletion inhibits ASK1-mediated apoptotic signaling, we examined the ability of ASK1 overexpression to induce caspase-3 activation in the absence or in the presence of DFMO. In the absence of DFMO, ASK1 overexpression induced an increase in caspase-3 activity, while in the DFMO-pretreated cells the level of caspase-3 activity was similar to that in control cells, independently of ASK1 overexpression (Fig. 6B). Note that DFMO had no significant effect on basal caspase-3 activity (compare DFMO-untreated or treated samples at zero time in Fig. 6A and sampes without ASK1 overexpression in Fig. 6B).
These results indicate that a decrease of polyamine levels in vivo specifically inhibits ASK1-induced caspase-3 activation.
Discussion
Recent evidence has implicated protein phosphatase PP5 as a component of several signaling pathways, and a number of potential regulatory and effector proteins have been shown to interact with PP5 (reviewed in [50,51]). A possibility of PP5 regulation by lipid second messengers (arachidonic acid and coenzyme A fatty acid esters) has been invoked [6,29,31,52], although it is based so far solely on in vitro evidence.
In this report, we have documented the evidence for in vitro regulation of PP5 by spermine, one of natural polyamines. Moreover, we have demonstrated that depletion of polyamine levels in vivo leads to a more efficient inactivation of apoptosis signal-regulating kinase (ASK1), a supposed physiological substrate of PP5, and that this effect is PP5-dependent. These findings further expand existing knowledge about biochemical properties of PP5 and suggest a novel way of modulation of its activity via cellular polyamine levels, as well as a novel mechanism how polyamines affect apoptotic signaling.
Natural polyamines (putrescine, spermidine and spermine) are involved in diverse functions such as control of cell cycle and apoptosis, DNA conformation and stability, regulation of transcription, ion channel regulation and protein phosphorylation [30–32]. Due to the role of polyamines in regulation of cell proliferation, polyamine analogs have been envisaged as possible anticancer drugs [36,37].
Although average concentration of polyamines in the cell is in high submillimolar or low millimolar range, they are mainly bound to RNA, DNA, ATP and membranes, so that free polyamine concentrations are in the micromolar range [53,54]. Evidence for proteins directly regulated by polyamines is scarce. In vitro regulation by polyamines has been reported for a PPP phosphatase PP2A [40,55] and for phosphatidylinositol kinases [56], however polyamine concentrations used in these studies are above presumed physiological range.
In several other instances reported effective polyamine concentrations fall within the physiological range. An early work reported inhibition of tyrosine hydroxylase by spermine with a Ki 68 μ M [57]. PP1, another protein phosphatase of the PPP family, is half inhibited in vitro by 40 μM spermine [40], and physiological relevance of this regulation has been supported by further studies [41–43]. Other identified polyamine-regulated signaling proteins include casein kinase 2 [35,58] and several ion channels [33,54,59,60]. Reported apparent Kd for regulation by spermine in these cases were 1.4 μM [58] and 1.5 – 5.5 μM [54], respectively. Modulation of Ca2+-permeable AMPA-type glutamate receptors via stimulus-dependent regulation of spermine synthesis has been reported in sensory neurons [38], demonstrating that polyamines may act as bona fide signaling molecules in vivo. Polyamine levels are subject to regulation during the cell cycle, as well as by trophic stimuli [32]
Spermine concentrations found in this work to regulate PP5 (apparent Kd for spermine of ~4 μM or ~30 μM for pNPP and protein substrates, respectively) clearly fall within the physiological range. The apparent half-inhibitory concentrations of spermine obtained with protein substrates are very close to that reported for PP1 (40 μM [40]). Thus, our results suggest that PP5 may be regulated by polyamines under physiological conditions.
High-affinity inhibition of PP5 by spermine suggests that PP5 may have a polycation-binding site, as previously documented for PPP protein phosphatases PP1 and plant PP7 (discussed in [44]). Another possibility is that polyamines may interfere with PP5 interaction with its substrates, for example via ionic interactions with the phosphate group. Alternatively, polyamines might counteract PP5 stimulation by some low molecular weight activators (such as arachidonic acid and coenzyme A fatty acid esters ), which may be present in purified PP5 preparations and also be associated with a proportion of PP5 in the cell. Further work is required to distinguish between these possibilities.
Polyamines are known to exert both protective and pro-apoptotic effects depending on the cellular context and the pro-apoptotic stimuli [48], however the precise mechanisms of these effects are unknown. A series of recent publications ([61] and references therein) has reported a protective effect of polyamine depletion in IEC-6 cells against TNF-α- induced apoptosis, which is mediated through the activation of ERK1/2. TNF- α is known to induce production of reactive oxygen species, which leads to ASK1 activation [Takeda et al., 2003]. Our finding that polyamine depletion counteracts ASK1 activation probably via de-inhibition of its negative regulator PP5 provides another mechanism (Fig. 7) for the protective effect of polyamine depletion against stress-induced apoptosis, and for the first time defines a precise molecular basis for such a protective effect.
Figure 7. Suggested mechanism of modulation of ASK1 activity by polyamine levels.

ASK1 is activated by redox stress-induced dissociation of its inhibitor thioredoxin, followed by autophosphorylation on Thr845 [18,62]. Dissociation of thioredoxin allows PP5 interaction with ASK1, resulting in ASK1 dephosphorylation and deactivation [18]. High polyamine levels result in a suppressed activity of PP5 and prolonged ASK1 activity. Decrease in polyamine levels would result in de-inhibition of PP5 and accelerated deactivation of ASK1.
Acknowledgments
We are grateful to Dr M. Chinkers (University of South Alabama) for the pET15b-PP5 and pCMV6-FLAG-PP5 plasmids. This study is supported by National Institutes of Health grants GM56159, GM65160, and HL06078 and American Heart Association (AHA) grant to T.V.Y. who is an Established Investigator of the American Heart Association.
References
- 1.Cohen PTW. Trends Biochem Sci. 1997;22:245–251. doi: 10.1016/s0968-0004(97)01060-8. [DOI] [PubMed] [Google Scholar]
- 2.Becker W, Kentrup H, Klumpp S, Schultz JE, Joost HG. J Biol Chem. 1994;269:22586–22592. [PubMed] [Google Scholar]
- 3.Chen MX, McPartlin AE, Brown L, Chen YH, Barker HM, Cohen PTW. EMBO J. 1994;13:4278–4290. doi: 10.1002/j.1460-2075.1994.tb06748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinkers M. Proc Natl Acad Sci U S A. 1994;91:11075–11079. doi: 10.1073/pnas.91.23.11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreeva AV, Kutuzov MA. Mol Biol Evol. 2001;18:448–452. doi: 10.1093/oxfordjournals.molbev.a003823. [DOI] [PubMed] [Google Scholar]
- 6.Chen MX, Cohen PTW. FEBS Lett. 1997;400:136–140. doi: 10.1016/s0014-5793(96)01427-5. [DOI] [PubMed] [Google Scholar]
- 7.Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, Pratt WB. J Biol Chem. 1999;272:16224–16230. doi: 10.1074/jbc.272.26.16224. [DOI] [PubMed] [Google Scholar]
- 8.Dean DA, Urban G, Aragon IV, Swingle M, Miller B, Rusconi S, Bueno M, Dean NM, Honkanen RE. BMC Cell Biol. 2001;2:6. doi: 10.1186/1471-2121-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao J, Hartson SD, Matts RL. Biochemistry. 2002;41:6770–6779. doi: 10.1021/bi025737a. [DOI] [PubMed] [Google Scholar]
- 10.Shinoda S, Skradski SL, Araki T, Schindler CK, Meller R, Lan JQ, Taki W, Simon RP, Henshall DC. Eur J Neurosci. 2003;17:2065–2076. doi: 10.1046/j.1460-9568.2003.02655.x. [DOI] [PubMed] [Google Scholar]
- 11.Borthwick EB, Zeke T, Prescott AR, Cohen PTW. FEBS Lett. 2001;491:279–284. doi: 10.1016/s0014-5793(01)02177-9. [DOI] [PubMed] [Google Scholar]
- 12.Galigniana MD, Harrell JM, Murphy PJ, Chinkers M, Radanyi C, Renoir JM, Zhang M, Pratt WB. Biochemistry. 2002;41:13602–13610. doi: 10.1021/bi020399z. [DOI] [PubMed] [Google Scholar]
- 13.Gong CX, Liu F, Wu G, Rossie S, Wegiel J, Li L, Grundke-Iqbal I, Iqbal K. J Neurochem. 2004;88:298–310. doi: 10.1111/j.1471-4159.2004.02147.x. [DOI] [PubMed] [Google Scholar]
- 14.Ollendorff V, Donoghue DJ. J Biol Chem. 1997;272:32011–32018. doi: 10.1074/jbc.272.51.32011. [DOI] [PubMed] [Google Scholar]
- 15.Zuo Z, Dean NM, Honkanen RE. J Biol Chem. 1998;273:12250–12258. doi: 10.1074/jbc.273.20.12250. [DOI] [PubMed] [Google Scholar]
- 16.Urban G, Golden T, Aragon IV, Cowsert L, Cooper SR, Dean NM, Honkanen RE. J Biol Chem. 2003;278:9747–9753. doi: 10.1074/jbc.M210993200. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi Y, Katoh H, Mori K, Negishi M. Curr Biol. 2002;12:1353–1358. doi: 10.1016/s0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]
- 18.Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. EMBO J. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou G, Golden T, Aragon IV, Honkanen RE. J Biol Chem. 2004;279:46595–46605. doi: 10.1074/jbc.M408320200. [DOI] [PubMed] [Google Scholar]
- 20.Berestetskaya Yu. V, Faure MP, chijo H, Voyno-Yasenetskaya TA. J Biol Chem. 1998;273:27816–27823. doi: 10.1074/jbc.273.43.27816. [DOI] [PubMed] [Google Scholar]
- 21.Vaiskunaite R, Kozasa T, Voyno-Yasenetskaya TA. J Biol Chem. 2001;276:46088–46093. doi: 10.1074/jbc.M108711200. [DOI] [PubMed] [Google Scholar]
- 22.Zhao S, Sancar A. Photochem Photobiol. 1997;66:727–731. doi: 10.1111/j.1751-1097.1997.tb03214.x. [DOI] [PubMed] [Google Scholar]
- 23.Andreeva AV, Kutuzov MA. Mol Cell Biol Res Commun. 2001;4:345– 352. doi: 10.1006/mcbr.2001.0302. [DOI] [PubMed] [Google Scholar]
- 24.Møller SG, Kim YS, Kunkel T, Chua NH. Plant Cell. 2003;15:1111–1119. doi: 10.1105/tpc.008649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cashmore AR, Jarillo JA, Wu YJ, Liu DM. Science. 1999;284:760–765. doi: 10.1126/science.284.5415.760. [DOI] [PubMed] [Google Scholar]
- 26.Tomsig JL, Snyder SL, Creutz CE. J Biol Chem. 2003;278:10048–10054. doi: 10.1074/jbc.M212632200. [DOI] [PubMed] [Google Scholar]
- 27.Lubert E. J. Hong Y, Sarge KD. J Biol Chem. 2001;276:38582–38587. doi: 10.1074/jbc.M106906200. [DOI] [PubMed] [Google Scholar]
- 28.Kono Y, Maeda K, Kuwahara K, Yamamoto H, Miyamoto E, Yonezawa K, Takagi K, Sakaguchi N. Genes Cells. 2002;7:821–834. doi: 10.1046/j.1365-2443.2002.00562.x. [DOI] [PubMed] [Google Scholar]
- 29.Skinner J, Sinclair C, Romeo C, Armstrong D, Charbonneau H, Rossie S. J Biol Chem. 1997;272:22464–22471. doi: 10.1074/jbc.272.36.22464. [DOI] [PubMed] [Google Scholar]
- 30.Sinclair C, Borchers C, Parker C, Tomer K, Charbonneau H, Rossie S. J Biol Chem. 1999;274:23666–23672. doi: 10.1074/jbc.274.33.23666. [DOI] [PubMed] [Google Scholar]
- 31.Ramsey AJ, Chinkers M. Biochemistry. 2002;41:5625–5632. doi: 10.1021/bi016090h. [DOI] [PubMed] [Google Scholar]
- 32.Wallace HM, Fraser AV, Hughes A. Biochem J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams K. Biochem J. 1997;325:289–297. doi: 10.1042/bj3250289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas T, Thomas TJ. Cell Mol Life Sci. 2001;8:244–258. doi: 10.1007/PL00000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Childs AC, Mehta DJ, Gerner EW. Cell Mol Life Sci. 2003;60:1394–1406. doi: 10.1007/s00018-003-2332-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas T, Balabhadrapathruni S, Gallo MA, Thomas TJ. Oncol Res. 2002;13:123–135. [PubMed] [Google Scholar]
- 37.Seiler N. Curr Drug Targets. 2003;4:565–585. doi: 10.2174/1389450033490876. [DOI] [PubMed] [Google Scholar]
- 38.Aizenman CD, Muñoz-Elias G, Cline HT. Neuron. 2002;16:623–634. doi: 10.1016/s0896-6273(02)00674-8. [DOI] [PubMed] [Google Scholar]
- 39.Ekman P, Jäger O. Anal Biochem. 1993;214:138–141. doi: 10.1006/abio.1993.1468. [DOI] [PubMed] [Google Scholar]
- 40.Tung HY, Pelech S, Fisher MJ, Pogson CI, Cohen P. Eur J Biochem. 1985;149:305–313. doi: 10.1111/j.1432-1033.1985.tb08927.x. [DOI] [PubMed] [Google Scholar]
- 41.Swärd K, Pato MD, Nilsson B-O, Nordström I, Hellstrand P. Am J Physiol. 1995;269:C563–C571. doi: 10.1152/ajpcell.1995.269.3.C563. [DOI] [PubMed] [Google Scholar]
- 42.Sjöholm Å, Honkanen RE. Pancreas. 2000;20:32–37. doi: 10.1097/00006676-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Nilsson BO, Gomez MF, Sward K, Hellstrand P. Acta Physiol Scand. 2002;176:33–41. doi: 10.1046/j.1365-201X.2002.01013.x. [DOI] [PubMed] [Google Scholar]
- 44.Kutuzov MA, Evans DE, Andreeva AV. FEBS Lett. 1998;440:147–152. doi: 10.1016/s0014-5793(98)01428-8. [DOI] [PubMed] [Google Scholar]
- 45.Rusnak F, Mertz P. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 46.Zou X, Tsutsui T, Ray D, Blomquist JF, chijo H, Ucker DS, Kiyokawa H. Mol Cell Biol. 2001;21:4818–4828. doi: 10.1128/MCB.21.14.4818-4828.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeda K. Matsuzawa A, Nishitoh H, Ichijo, H Cell Struct Function. 2003;28:23–29. doi: 10.1247/csf.28.23. [DOI] [PubMed] [Google Scholar]
- 48.Schipper RG, Penning LC, Verhofstad AAJ. Seminars Cancer Biol. 2000;10:55–68. doi: 10.1006/scbi.2000.0308. [DOI] [PubMed] [Google Scholar]
- 49.Hatai T, Matsuzawa A, noshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, chijo H, Takeda K. J Biol Chem. 2000;275:26576–26581. doi: 10.1074/jbc.M003412200. [DOI] [PubMed] [Google Scholar]
- 50.Andreeva AV, Kutuzov MA. Cell Signal. 1999;11:555–562. doi: 10.1016/s0898-6568(99)00032-7. [DOI] [PubMed] [Google Scholar]
- 51.Chinkers M. Trends Endocrinol Metab. 2001;12:28–32. doi: 10.1016/s1043-2760(00)00335-0. [DOI] [PubMed] [Google Scholar]
- 52.Kang H, Sayner SL, Gross KL, Russell LC, Chinkers M. Biochemistry. 2001;40:10485–10490. doi: 10.1021/bi010999i. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe S, Kusama-Eguchi K, Kobayashi H, Igarashi K. J Biol Chem. 1991;266:20803–20809. [PubMed] [Google Scholar]
- 54.Bowie D, Mayer ML. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 55.Cornwell T, Mehta P, Shenolikar S. J Cyclic Nucleotide Protein Phosphor Res. 1986;11:373–382. [PubMed] [Google Scholar]
- 56.Singh SS, Chauhan A, Brockerhoff H, Chauhan VP. Life Sci. 1995;57:685–694. doi: 10.1016/0024-3205(95)00320-6. [DOI] [PubMed] [Google Scholar]
- 57.Kiuchi K, Kiuchi K, Togari A, Nagatsu T. Biochem Biophys Res Commun. 1987;148:1460–1467. doi: 10.1016/s0006-291x(87)80296-6. [DOI] [PubMed] [Google Scholar]
- 58.Leroy D, Heriché JK, Filhol O, Chambaz EM, Cochet C. J Biol Chem. 1997;272:20820–20827. doi: 10.1074/jbc.272.33.20820. [DOI] [PubMed] [Google Scholar]
- 59.Johnson TD. Trends Pharmacol Sci. 1996;17:22–27. doi: 10.1016/0165-6147(96)81566-5. [DOI] [PubMed] [Google Scholar]
- 60.Guo D, Lu Z. J Gen Physiol. 2003;122:485–500. doi: 10.1085/jgp.200308890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharya S, Ray RM, Johnson LR. Am J Physiol - Gastrointest Liver Physiol. 2004;286:G479–G490. doi: 10.1152/ajpgi.00342.2003. [DOI] [PubMed] [Google Scholar]
- 62.Liu H, Nishitoh H, chijo H, Kyriakis JM. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]