

Abstract
Acetaldehyde is fibrogenic and induces the expression of type I collagen genes in hepatic stellate cells. Some of these acetaldehyde-dependent events are mediated by H2O2 and thus establish a direct connection between oxidative stress and collagen upregulation. We localized to the −378 to −183 region of the α2(I) collagen (COL1A2) promoter an acetaldehyde-responsive element (AcRE) functional in human hepatic stellate cells (HHSCs) and investigated molecular mechanisms whereby acetaldehyde stimulates and modulates its transcriptional activity. Because the AcRE co-localized with a previously described transforming growth factor β(TGF-β)1–responsive element, and both acetal-dehyde and this cytokine induce their effects through H2O2, we investigated whether all fibrogenic actions of acetaldehyde were mediated by this cytokine. Here we show that acetaldehyde-induced COL1A2 upregulation in HHSCs recognizes two distinct but overlapping early and late stages that last from 1 to 6 hours and from 6 to 24 hours, respectively. We present several lines of evidence to show that early acetaldehyde-mediated events are independent of TGF-β1. These include significant time-course differences in the expression of COL1A2 and TGF-β1 mRNAs and inability of neutralizing antibodies to TGF-β1 to inhibit acetaldehyde-dependent collagen gene transcription and Smad 3 phosphorylation. We also show that although acetaldehyde-dependent upregulation of collagen was PI3K dependent, that of TGF-β1 was PI3K independent. In conclusion, acetaldehyde-dependent mechanisms involved in COL1A2 upregulation are similar, but not identical, to those of TGF-β1. We suggest that early acetaldehyde-dependent events induce the late expression of TGF-β1 and create an H2O2-dependent autocrine loop that may sustain and amplify the fibrogenic response of this alcohol metabolite.
Abbreviations: HSC, hepatic stellate cell; Col1a1, α1(I) collagen gene; ROS, reactive oxygen species; COL1A2, α2(I) collagen; HHSC, human HSC; TGF, transforming growth factor; AcRE, acetaldehyde-responsive element; TbRE, TGF-β1—responsive element
A lcohol-induced liver fibrosis is multifactorial and involves local and systemic events. At the systemic level, multiple cytokines, including those involved in the acute phase response, play a role in the pathophysiology of the disease and contribute to the fibrogenic process inducing apoptosis and upregulating the expression of type I collagen and tissue inhibitor of met-alloproteinase-1 mRNAs.1–6 At the local level, fibrogenic mechanisms are quite complex and involve various cell types, including hepatocytes, Kupffer cells, and hepatic stellate cells (HSCs).7–14 Whereas hepatocytes are the main cells involved in ethanol metabolism, Kupffer cells contribute with the production of cytokines, chemokines, and growth factors that play key roles in attracting inflammatory cells to injured sites and in activating and trans-differentiating HSCs to myofibroblasts, the main type I collagen–producing cells in the liver.11,12 Some of the molecular mechanisms whereby the various hepatic cell types contribute to liver fibrogenesis have been unraveled.11–14 However, important key events by which ethanol metabolized to acetaldehyde by hepatocytes activates production of type I collagen in HSCs remain to be fully elucidated. We have already shown that, in a functional co-culture system containing hepatocytes and HSCs, the former cells metabolize ethanol and the latter produce type I collagen.15 We and others have shown that acetaldehyde is fibrogenic per se and induces the expression of the α1(I) collagen (Col1a1) gene16–18 by a mechanism dependent on the accumulation of H2O2.18 We further established that H2O2 induces the activation, nuclear translocation, and DNA binding of members of the C/EBPβ family of transcription factors to the −370 to −345 region of the Col1a1 gene.18 In addition, it has been shown that metabolites and reactive oxygen species (ROS) produced during ethanol metabolism by HepG2 cells or HSCs overexpressing CYP2E1 are fibrogenic.19–22 Taken together, these studies confirm the role of H2O2 and oxidative stress in liver fibrogenesis.
Although some publications have described general mechanisms of collagen production by HSCs and have unraveled some signal transduction pathways activated by acetaldehyde,23–28 little is known regarding to the molecular mechanisms whereby acetaldehyde upregulates the expression of the human α2(I) collagen gene (COL1A2). Therefore, in this communication we describe molecular events triggered by acetaldehyde that result in COL1A2 transcriptional activation. We show that acetaldehyde induces the rapid phosphorylation and nuclear translocation of Smads 3 to 4 via a TGF-β1-independent pathway and that protein complexes containing Sp1 and Smads 3 and 4 are essential for COL1A2 upregulation by acetaldehyde in HHSCs.
Materials and Methods
Plasmids and Reagents
Transforming growth factor-beta-1 (TGF-β1) was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Catalase, cycloheximide, and mithramycin were purchased from Sigma Chemical Co. (St. Louis, MO). Wortmannin was purchased from Calbiochem (La Jolla, CA). Acetaldehyde was purchased from Fisher Scientific (Springfield, NJ). Details about the construction of the COL1A2/LUC chimeric plasmids, the COL1A2 mutant constructs, MSp1 COL1A2/LUC, MCAGA COL1A2/LUC, and Δ5ACOL1A2/LUC, TKCAT plasmid, (AcRE)3(TK)CAT plasmid and dominant negative Smad 2, Smad 3, and Sp1 plasmids have been published (http://www.jbc.org/cgi/content/full/272/32/-B1829,30). The COL1A2 and TGF-β1 cDNAs have been described previously29,30 (http://www.jbc.org/cgi/content/full/272/32/-B26). The GAPDH and S14 ribosomal protein cDNAs were obtained from the ATCC (Rockville, MD). Oligonucleotides containing consensus recognition sequences for Sp1 and AP-1 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
HHSC Isolation and Culture
HHSCs were isolated from samples of human livers obtained during gastric bypass surgery for morbid obesity as previously described.26 Informed consent in writing was obtained from each patient, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a prior approval by the institutional review committee. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Hy-Clone Inc., Logan, UT) and antibiotics. Experiments were performed in triplicate using cells obtained from at least 3 different patients cultured for 2 to 6 passages.
Northern Blot Hybridizations and Run-on Transcription Assays
Confluent HHSCs were placed in a serum-free medium containing glutamine, nonessential amino acids, and antibiotics. Approximately 14 hours later, acetaldehyde was added at a final concentration of 200 μmol/L. For some experiments, cells were preincu-bated with either mithramycin (100 nmol/L), catalase (1,000 U/mL), wortmannin (100 nmol/L), or cycloheximide (30 μg/mL) for 30 minutes before acetaldehyde administration. Viability of the cells was estimated using the trypan blue exclusion test. In all cases, cellular viability was more than 90%. Total RNA was extracted at the times indicated in the figures and processed for Northern blot hybridizations according to standard protocols.18 Likewise, a standard protocol was used to determine rates of COL1A2 and TGF-β1 transcription in control and acetaldehyde-treated cells using GAPDH, S14, and pBR322 DNA as controls.31 In some run-on transcription experiments, cells were pre-incubated for 2 hours before acetaldehyde administration, with a neutralizing antibody to TGF-β1 (Promega Corp., Madison, WI) or an unrelated immunoglobulin G (Santa Cruz Biotechnology, Inc.), at a final concentration of 5 μg/mL. To determine the effectiveness of the anti–TGF-β1 antibody, some HHSCs were also incubated with 2 ng/mL recombinant TGF-β1 in the presence or absence of the corresponding neutralizing antibody, or an unrelated immunoglobulin G, following the protocol previously described. Three hours later, nuclei were isolated and used in run-on transcription assays as previously described. Relative intensity of the signals was determined by laser densitometric analysis of the radiographic films. Values are means of triplicate experiments ± SD and were corrected for loading differences using S14 as control.
Preparation of Nuclear Extracts, Footprinting Analysis, and Gel Mobility Shift Assays
Nuclear extracts were prepared from control and HHSCs treated with acetaldehyde for the times indicated in the figures as previously reported.29 After incubating nuclear extracts with an end-labeled probe, gel mobility shift assays and footprinting analysis were carried out as previously described.29 Binding conditions, as well as the sequences of the AcRE (−313 to −183) oligonucleotide and the oligonucleotide containing 3 copies in tandem of the COL1A2 Smad binding site (−272 to −249) (COL1A2 [CAGA]3) used as either probes or unlabeled competitors, have been previously described.29,30 Identical amounts of nuclear proteins from each cell source were added to the binding reactions. Complexes formed were identified by autoradiography of the dried gels. For antibody interference assays, antibodies against Sp1 (PEP-2), Sp3 (D-20), Smads 2 (S-20), 3 (I-20) and 4 (C-20), c-fos (K-25), c-jun (D), and p50 (H-119), and p65 (C-20) nuclear factor kappaB (NFκB) were purchased from Santa Cruz Biotechnology, Inc. These antibodies were used at a final concentration of 0.16 μg/mL. For competition studies, unlabeled oligonucleotides were added at a 100-fold molar excess.
Western Blot Analysis
Nuclear extracts (20 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on 10% gels and transferred onto a nitro-cellulose membrane. Membranes were probed with Smad 3 or TGF-β1 goat antibodies at a final dilution of 1:1,000, followed by incubation with horseradish peroxidase– conjugated donkey anti-goat immunoglobulin G (Santa Cruz Biotechnology) diluted 1:5,000. Proteins were detected with an enhanced chemiluminescence system (Renaissance; NEN Life Science Products, Boston, MA), as recommended by the manufacturer. Conditions used to assess the expression of FLAG epitope in cells stably transfected with the different DN proteins were described before using the M2 anti-Flag antibody from Sigma (http://mcb.asm.org/cgi/content/full/20/3/-B16).31 For detection of phosphorylated Smad 3, nuclear extracts were immunoprecipitated with 5 μg anti-phosphoserine antibody (Poly-Z-PS 1), (Zymed Laboratories, San Francisco, CA) and then subjected to Western blot analysis with Smad3 antibodies as described above.
Cell Transfections
Conditions for the preparation and transfection of plasmids into HHSCs by the calcium phosphate procedure have been detailed before (http://www.jbc.org/cgi/content/full/275/50/-B27).30,31 For the functional characterization of the acetaldehyde-responsive elements, HHSCs were treated with 10% glycerol for 90 seconds 16 hours after transfection and then placed in medium containing 0.1% fetal bovine serum. Two hours later, acetaldehyde was added at a 200 μmol/L final concentration. Twenty-four hours later, a second dose of acetaldehyde was added. Cells were procured 48 hours after the addition of the first dose of acetaldehyde, and used to determine luciferase or CAT assays as previously described.29,32 Transcriptional activity of each of the chimeric constructs was normalized against co-transfected pSv2CAT (when using luciferase vectors) or pSV40 LUC (when using CAT vectors). Transfections were performed multiple times and in duplicate. The statistical value of the data was evaluated by the Mann-Whitney U test. Stable transfectants were selected by culturing the cells for approximately 3 weeks in the presence of 400 μg/mL G418.33 For some experiments, HHSCs were co-transfected with 7.0 μg of the −378 COL1A2LUC reporter construct and 2.5 μg of either one of the dominant negative Smad2, Smad3, or Sp1 vectors followed by treatment with acetaldehyde using the conditions described above.
Statistical Analysis
Values were expressed as mean ± SD and are average from 3 to 6 values per experiment; experiments were repeated at least 3 times. Either Student’s t test or the Mann-Whitney U test was used to evaluate the statistical differences between groups: a P value equal to or less than .05 was considered significant.
Results
Acetaldehyde induces COL1A2 expression in HHSCs by a transcriptional mechanism via a de novo protein synthesis-independent mechanism. Northern blot analysis of total RNA extracted from HHSCs treated with acetaldehyde for 6 hours revealed that α2(I) collagen mRNA was induced 3.4 ± 0.4 fold compared with untreated cells (P < .05) (Fig. 1A). Time-course studies indicated that collagen mRNA upregulation is a relatively early event that starts approximately 3 hours after acetaldehyde administration and reaches its maximum approximately 16 to 24 hours later 4.6 ± 0.7 (P < .05) (Fig. 1B). Lack of inhibition by cycloheximide demonstrated that this early acetaldehyde-dependent increase in α2(I) mRNA is independent of de novo protein synthesis (Fig. 1B). Although cycloheximide did not prevent acetaldehyde-mediated 2(I) collagen mRNA upregulation within the first 3 hours, further increase of COL1A2 gene expression is mostly dependent on de novo protein synthesis (Fig. 1B). Run-on transcription assays performed at 3 hours after acetaldehyde administration showed a 4.3 ± 0.3-fold increase in transcription rates, demonstrating that the increase in COL1A2 gene expression in response to acetaldehyde is exerted at the transcriptional level (4.3 ± 0.3 vs. control untreated cells) (P < .05) (Fig. 1C).
Fig. 1.
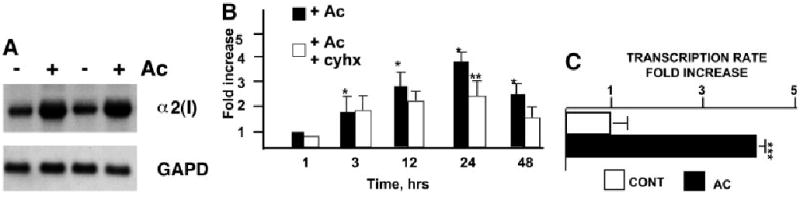
Acetaldehyde stimulates COL1A2 gene expression at the transcriptional level by protein synthesis– dependent and –independent mechanisms. (A) Northern blot analysis of total RNA extracted from HHSCs cultured in the presence or absence of acetaldehyde (AC) for 6 hours; the identity of the probes is indicated on the right side of the autoradiograph. (B) Time-course analysis COL1A2 mRNA expression in AC-treated cells cultured in the absence or presence of cycloheximide (cyhx). (C) Run-on transcription assays of the endogeneous COL1A2 gene in HHSCs treated with AC for 3 hours. *P < .05 when compared with control, untreated HSCs. **P < .05 when compared with AC-treated cells.***P < .05 when compared with control values.
The acetaldehyde-responsive element (AcRE) of the COL1A2 gene is localized to the −378 to −183 promoter region and contains 2 distinct footprints.
To localize the region in the COL1A2 promoter responsible for acetaldehyde-mediated transcriptional activation, transient transfections in HHSCs were performed using luciferase reporter vectors driven by progressive 5′ deletions of the COL1A2 upstream sequence. Cells transfected with a reporter vector driven by the 3,500-bp COL1A2 promoter responded to acetaldehyde by augmenting luciferase activity 2.03 ± 0.1-fold above control values (P < .05) (Fig. 2A). Similarly, cells transfected with a vector driven by the proximal 378-bp promoter responded to acetaldehyde by stimulating luciferase activity 2.47 ± 0.2 fold (P < .05) (Fig. 2A). By contrast, luciferase activity in cells transfected with a reporter vector driven by the 183-bp COL1A2 promoter was not altered after acetaldehyde administration (Fig. 2A). These analyses therefore located the acetaldehyde responsive element (AcRE) somewhere within the −378 to −183 region of the COL1A2 promoter. Footprinting analysis using the −378 to −183 sequence, and nuclear extracts prepared from control and acetaldehyde-treated HHSCs showed 2 distinct sites of DNA–protein interaction that were noticeably wider and more evident in acetaldehyde-treated than untreated cells (Fig. 2B). Interestingly, the 2 footprints are virtually identical to those previously reported in TGF-β–treated fibroblasts.29 Further confirmation of the functional role of the footprinted region was obtained by showing that this sequence was capable of conferring acetaldehyde responsiveness to the otherwise unresponsive TK promoter (Fig. 2C).
Fig. 2.
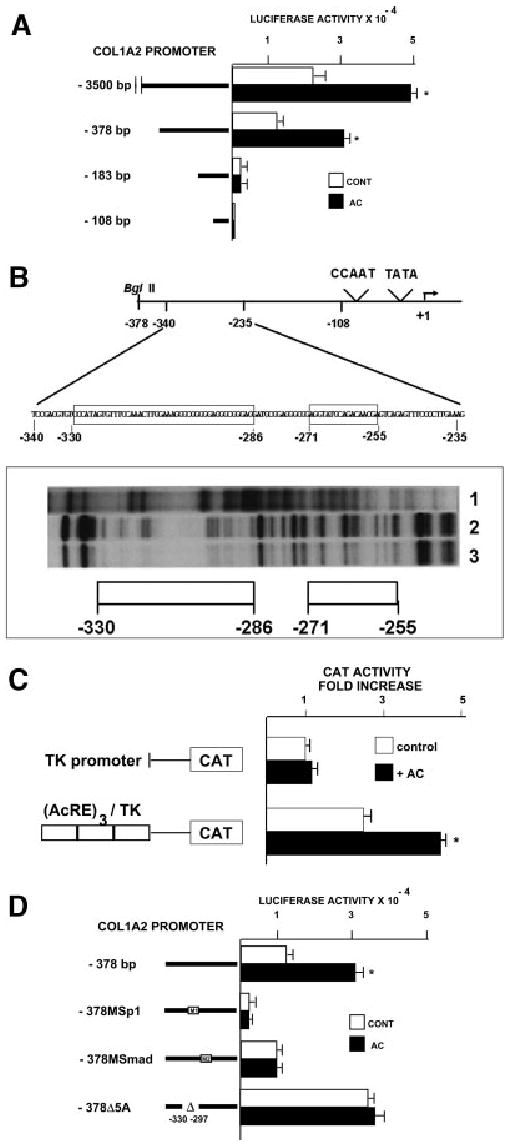
The acetaldehyde-responsive element of the COL1A2 gene in HHSCs is located between nucleotides −378 and −183. (A) Deletion analysis in control and acetaldehyde (AC)-treated HHSCs transfected with luciferase reporter constructs driven by different lengths of the COL1A2 promoter. The length of each of the constructs used is shown on the left side of the figure. *P < .05 vs control without AC. (B) Footprinting analysis of the −378 to the −183 COL1A2 promoter region performed with nuclear extracts from control (lane 2) and acetaldehyde-treated HHSCs (lane 3) for 3 hours. As a negative control, DNA incubated without nuclear extracts was used (lane 1). (C) CAT activity in control and acetaldehyde-treated HHSCs (AC) transfected with a reporter CAT vector driven by the TK promoter alone or with a vector containing 3 copies in tandem of the acetaldehyde-responsive element (AcRE) upstream of the TK promoter. *P < .05 vs. cells transfected with the (AcRE)3 TKCAT vector without AC. (D) Mutation analysis in control and acetaldehyde-treated HHSCs transfected with luciferase reporter constructs driven by mutated versions of the −378 COL1A2LUC vector. For these experiments, the following sequences of the −378COL1A2LUC were mutated: −301 to −302, −292 to −291 and −275 to 274, sequences corresponding to the 3 COL1A2 Sp1 binding sites (MSp1 COL1A2/LUC) or the −272 to −249 sequence corresponding to the SMAD binding site (MCAGA COL1A2/LUC). In addition, a construct in which the −330 to −297 area was deleted (Δ5A) was also used. *P < .05 vs. control cells without AC.
The AcRE co-localizes with the previously described TGF-β1–responsive element (TbRE) of COL1A2.29 Because the TbRE contains Sp1 and Smad-binding sites, we determined whether these transcription factors are involved in the acetaldehyde-dependent upregulation of the COL1A2 gene. HHSCs were therefore transfected with luciferase reporter vectors driven by the wild-type −378 promoter sequence or by −378 promoters containing mutations within the Sp1 (MSp1COL1A2/LUC) or the Smad (MCAGA COL1A2/LUC) binding sites (Fig. 2D). Whereas mutations in the Sp1-binding sites decreased substantially basal expression of the reporter vector and abrogated response to acetaldehyde, mutations of the Smad-binding site had only a minimal effect on basal gene expression but abrogated acetaldehyde responsiveness (Fig. 2D).
Sp1 and Smads 3 and 4 are required for acetaldehyde-mediated upregulation of COL1A2 gene expression. The electrophoretic mobility shift assay was employed to study the possible role of Sp1 and members of the Smad family of transcription factors in acetaldehyde-induced COL1A2 upregulation. To this end, we used nuclear extracts purified from control cells and from HHSCs treated with acetaldehyde from 1 to 24 hours. Consistent with the footprinting data, electrophoretic mobility shift assay showed increased binding to the −313 to −183 sequence after acetaldehyde treatment, whereas antibody interference assays demonstrated that complex 1 contained Sp1 but not AP-1 or NFκB (Fig. 3A-B). The formation of Sp1-containing DNA complexes increased as early as 1 hour and remained elevated thereafter for at least 24 hours after acetaldehyde administration (Fig. 3A). The nature of complex 2 remains to be determined. However, as shown in Fig. 3A, binding of this complex was also altered by acetaldehyde treatment. However, the increment in complex 2 was found to be variable and not always reproducible (compare Figs. 3A and B). Increased Sp1 binding activity to the COL1A2 promoter after acetaldehyde treatment was not unique; binding to an Sp1 high-affinity recognition sequence also increased (Fig. 3C). The essential role of Sp1 in acetaldehyde-mediated upregulating of COL1A2 was further confirmed by experiments of interference with Sp1 DNA-binding activity. First, mitramycin at a concentration known to specifically inhibit Sp1 binding to DNA-abrogated acetaldehyde-stimulated α2(I) collagen mRNA accumulation (P < .05) (Fig. 4A). Second, overexpression of a dominant negative form of Sp1 completely abrogated acetaldehyde-mediated up-regulation of the −378COL1A2 LUC vector (Fig. 4B).
Fig. 3.
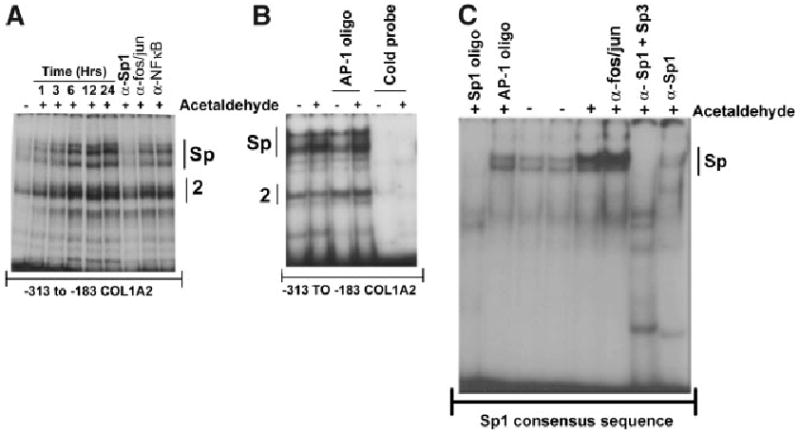
Acetaldehyde enhances binding of a Sp1-containing complex to the −313 to −183 region of the COL1A2 gene in HHSCs and to a Sp1 high-recognition sequence. (A) Electrophoretic mobility shift assays of nuclear extracts from control (−) and acetaldehyde-treated (+) HHSCs for 1, 3, 6, 12, and 24 hours using the −313 to −183 COL1A2 region. As shown in this panel, acetaldehyde significantly enhances protein binding to this region. Antibody interference assays demonstrated that the upper complex contains Sp1, but not AP-1 (fos/jun) or NFκB. The identity of the lower complex (labeled “complex 2” remains unknown). (B) The complexes formed after acetaldehyde treatment (3 hours) are specific, as they can be competed by unlabeled −313 to −183 COL1A2 sequence (“cold probe”) but not by an AP-1 high-affinity recognition oligonucleotide. (C) Increased binding of nuclear extracts of HSCs treated for 24 hours with AC to an Sp1 high-recognition oligonucleotide. The complex is specific, as it can be competed with excess (100-fold molar) unlabeled Sp1 consensus sequence, but not AP-1, and is recognized by antibodies to Sp1 and Sp3 (α-Sp1 + α-Sp3) but not AP-1 (anti-fos/jun). Panel C also shows that whereas the combination of α-Sp1 + α-Sp3 antibodies completely eliminates (or supershifts) the DNA–protein complexes obtained with acetaldehyde, the α-Sp1 antibody was less efficient and did not eliminate the complex completely.
Fig. 4.
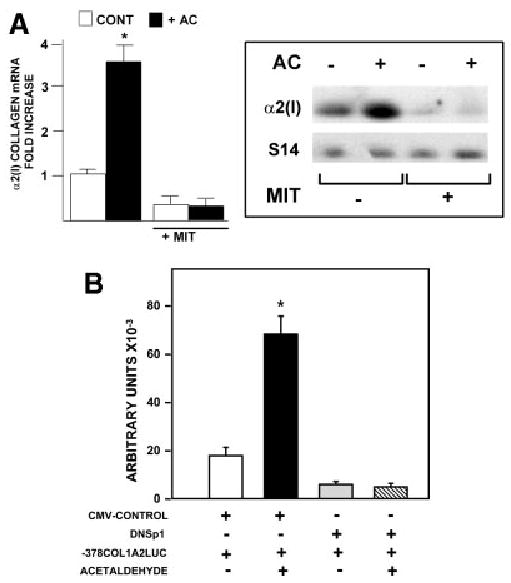
Sp1 is essential for the acetaldehyde-dependent upregulation of the COL1A2 gene in HHSCs. (A) Northern blot analysis of α2(I) collagen mRNA performed with total RNA extracted from control and acetaldehyde (AC)-treated HHSCs cultured in the presence or absence of 100 nmol/L mithramycin (MIT) for 6 hours. The histogram on the left summarizes the results of quadruplicate experiments. The right panel depicts a representative Northern blot. Differences in loading were corrected with the signal generated with an S14 mRNA probe. *P < .05 versus control cells without AC. (B) Effect of acetaldehyde on HHSCs transiently co-transfected with the −378 COL1A2LUC reporter construct and a dominant negative Sp1 expression plasmid (DNSp1). Activity is expressed as arbitrary units of luciferase activity. As a control for these experiments, some HHSCs were co-transfected with the −378 COL1A2LUC and an empty CMV expression vector (CMV-CONTROL). * P < .05 versus control cells without AC.
Similar analyses established the role of Smad proteins in the acetaldehyde response. Electrophoretic mobility shift assays demonstrated binding of Smads 3 and 4 to the cognate site of the COL1A2 promoter, which increased significantly after treatment (Fig. 5A). Time course studies revealed that the Smad 3–4 DNA complexes appear as early as 15 minutes after acetaldehyde administration and remain evident for at least 24 hours thereafter (Fig. 5B). Immunoblots of nuclear proteins from control and acetaldehyde-treated HHSCs demonstrated the presence of Smad 3 in both samples. However, phosphorylated Smad 3 was only present in acetaldehyde-treated cells, and this post-translational modification was not inhibited by cycloheximide (Fig. 5C). Co-transfection of HHSCs with the −378COL1A2LUC construct reporter and vector expressing a dominant negative isoform of Smad 3 documented abrogation of the acetaldehyde response (Fig. 6A). Furthermore, HHSCs stably transfected with the dominant negative Smad 3 expression vector displayed down-regulation of the endogenous COL1A2 gene, in addition to blunting of acetaldehyde responsiveness (Fig. 6B). Similar findings were obtained with HHSCs transfected with a dominant negative Smad 4 construct (data not shown). In marked contrast, a dominant negative isoform of Smad 2, which is not part of the acetaldehyde-induced complex binding to the COL1A2 promoter, had no effect on the activity of the -378 COL1A2LUC plasmid (Fig. 6A). We reasoned from previous study of the col1a1 that accumulation of H2O2 may be similarly responsible for stimulation of COL1A2 expression in HH-SCs. Indeed, Northern blots showed that catalase prevents acetaldehyde-mediated upregulation of α2(I) collagen mRNA accumulation (0.85 ± 0.17 vs. 3.49 ± 0.17; P < .05) (Fig. 7).
Fig. 5.
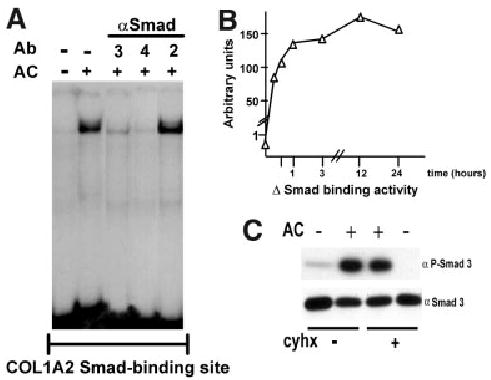
Acetaldehyde enhances phosphorylation of Smad 3 and binding of a Smad3/4-containing complex to the −272 to −249 COL1A2 region by a protein synthesis–independent mechanism. (A) Electrophoretic mobility shift assay performed with nuclear extracts from control and acetaldehyde (AC)-treated HHSCs for 6 hours, using an oligonucleotide containing 3 copies in tandem of the COL1A2 Smad binding site (CAGA)3 as probe. Antibody interference assays with antibodies (Ab) to either Smads 2, 3, or 4 revealed that the complex contained Smads 3/4 but not Smad 2. (B) Time-course analysis of the effect of acetaldehyde on the binding of the Smad 3/4-containing complex to the (CAGA)3 oligonucleotide. The graph was constructed with values obtained after densitometric analysis of bands generated in duplicate electrophoretic mobility shift assays performed at the indicated times and expressed as arbitrary units. (C) Western blot analysis of nuclear extracts obtained from control and acetaldehyde (AC)-treated HHSCs for 6 hours in the presence or absence of cycloheximide (cyhx). Samples were probed with an anti-Smad3 antibodies directly (αSmad3) or after immunoprecipitation with an anti-phosphoserine antibody (αP-Smad3).
Fig. 6.
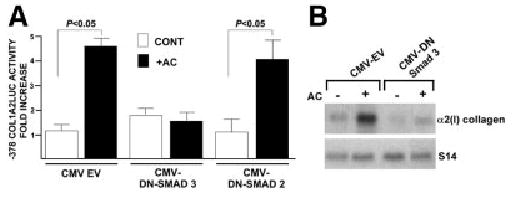
Smad 3 but not Smad 2 is essential for acetaldehyde-dependent COL1A2 gene upregulation in HHSCs. (A) Effect of acetaldehyde (AC) on luciferase activity of HHSCs transiently co-transfected with the −378 COL1A2LUC reporter vector and either a control empty CMV vector, or dominant negative Smad3 (DN-SMAD3) or Smad2 (DN-SMAD2) expression vectors. Values are expressed as fold increase above controls. (B) Northern blot analysis of total RNA extracted from HHSCs harboring stably integrated copies of an empty CMV vector or an expression vector encoding DN-Smad3 cultured in the presence or absence of AC. The identity of the probes is indicated on the right side of the autoradiography.
Fig. 7.
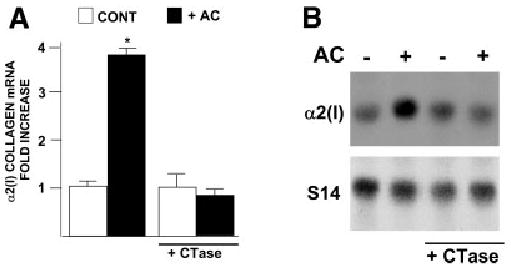
Catalase abrogates acetaldehyde-induced α2(I) collagen mRNA expression in HHSCs. Northern blot analysis of α2(I) collagen mRNA performed with total RNA extracted from control and acetaldehyde (AC)-treated HHSCs cultured in the presence or absence of catalase (1,000 U/mL) for 24 hours. (A) The histogram summarizes the results of quadruplicate experiments. (B) Representative Northern blot. Differences in loading were corrected with the signal generated with an S14 mRNA probe. *P < .05 versus control cells without AC.
Early acetaldehyde-elicited upregulation of COL1A2 is TGF-β1 independent. The finding that the AcRE and the TbRE co-localize to the same promoter element and that Sp1 and Smads 3 and 4 are involved in both transcriptional responses raised the possibility of a functional relationship between these 2 signaling pathways. However, this possibility was not supported by experimental evidence. First, a neutralizing antibody to TGF-β1 failed to prevent acetaldehyde-elicited phosphorylation of Smad 3 in HHSCs or acetaldehyde-dependent COL1A2 upregulation (3.6 ± 0.36 vs. 3.03 ± 0.22) (Fig. 8A-B). Second, acetaldehyde treatment had virtually no effect on the expression of the TGF-β1 gene in HHSCs at the point when COL1A2 expression is already upregulated (Fig. 8C). Last, addition of an acetaldehyde inhibitor, wortmannin, had no effect on TGF-β stimulation of COL1A2 expression in HHSCs (Fig. 8D) but completely abrogated the acetaldehyde-elicited response. Interestingly, and in agreement with data obtained by others, showing that acetaldehyde induced the late expression of TGF-β1 mRNA and its receptor,16,28 we found that acetaldehyde upregulated the expression of this cytokine mRNA by 6 to 8 hours at a point in which COL1A2 mRNA was already upregulated. As shown in Fig. 9A-B, changes in TGF-β1 mRNA were accompanied by similar elevations in TGF-β1 protein as determined by Western blot analysis.
Fig. 8.
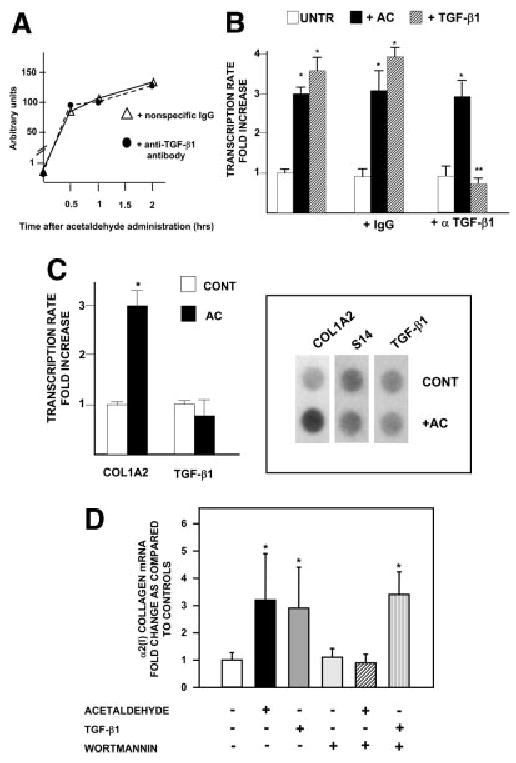
TGF-β1 is not involved in early acetaldehyde-dependent up-regulation of the COL1A2 gene. (A) Effect of neutralizing antibodies to TGF-β1 on acetaldehyde-induced phosphorylation of Smad3. HHSCs were incubated with acetaldehyde in the presence of a nonspecific immunoglobulin G (triangles) or an anti–TGF-β1 neutralizing antibody (circles). At the indicated times, nuclear extracts were obtained for Western blot analysis of phosphorylated Smad 3 as described in Materials and Methods. The graph was constructed with values obtained after densitometric analysis of bands generated in duplicate Western blots performed at the indicated times and expressed as arbitrary units. (B) Run-on transcription assays of control and acetaldehyde-treated HHSCs for 3 hours in the presence or absence of a nonspecific immunoglobulin G or a neutralizing antibody to TGF-β1 (α TGF-β1). As a control for these experiments, some HHSCs were treated with 2 ng/mL of TGF-β1 in the absence or presence of the neutralizing antibody to the cytokine. *P < .05 versus control untreated cells. **P < .05 when compared with TGF-β1–treated cells in the absence of the neutralizing antibody. (C) Run-on transcription assays of COL1A2 and TGF-β1 gene expression performed with nuclear extracts from control and acetaldehyde (AC)–treated HHSCs for 1 hour. The histogram on the left summarizes the results of triplicate experiments. The right panel depicts a representative run-on assay. Differences in loading were corrected with the signal generated with an S14 probe.*P < .05 versus control cells without AC. (D) Northern blot analysis of HHSCs treated with either acetaldehyde (200 μmol/L) or TGF-β1 (2 ng/mL) in the absence or presence of 100 nmol/L wortmannin. For these experiments, HHSCs were pretreated for 3 hours with wortmannin followed by 12 hours with acetaldehyde or TGF-β1. The histogram summarizes results of triplicate experiments, and the results are expressed as fold-increase above control untreated HHSCs. *P < .05 versus control cells without AC, TGF-β1, or wortmannin.
Fig. 9.
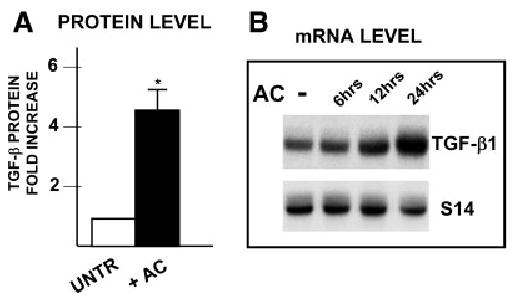
Acetaldehyde induces late expression of α2(I) collagen mRNA by a TGF-β1– dependent mechanism. (A) Western blot analysis of TGF-β1 expression in control and acetaldehyde-treated HHSCs for 24 hours. The histogram summarizes triplicate experiments, and the results are expressed as fold increase above untreated controls. *P < .05 versus control cells without AC. (B) Time-course analysis of TGFβ1 mRNA expression in HHSCs treated with 200 μmol/L acetaldehyde. The identity of the probes is indicated on the right side of the autoradiograph.
Discussion
That acetaldehyde, the first metabolite of ethanol, is a fibrogenic factor in HSCs is well established.16,18,24–26 Thus, irrespective of whether ethanol is metabolized via alcohol dehydrogenase or CYP2E1, generated acetaldehyde triggers a signal transduction cascade that results in increased expression of type I collagen genes.17 Previous studies from our laboratory have localized an acetaldehyde-responsive element in the −370 to −344 of the col1a1 promoter and that this element co-localized with TGF-β1- and TNF-α–responsive elements.18,32,33 Furthermore, we established that acetaldehyde upregulates col1a1 gene expression by a mechanism dependent on the generation of H2O2 and involving nuclear translocation and binding of members of the C/EBPβ family of transcription factors. Type I collagen is a trimeric molecule product of 2 distinct genes that are coordinately expressed through apparently distinct transcriptional mechanisms. This study has further corroborated the diversity of the regulatory mechanisms in type I collagen genes by demonstrating that responsiveness of COL1A2 to acetaldehyde is mediated by Sp1 and Smads 3 and 4 transcription factors.
Like the murine col1a1, the AcRE of the human COL1A2 gene co-localizes with the TbRE.29 However, several lines of evidence exclude TGF-β involvement in acetaldehyde upregulation of COL1A2 at early points after acetaldehyde administration. First, COL1A2 transcription is elevated in HHSCs within 1 hour after acetaldehyde administration, whereas that of TGF-β remains at background levels. Second, acetaldehyde-induced α2(I) collagen mRNA accumulation does not require de novo protein synthesis, whereas that of TGF-β1 does. Third, neutralizing antibodies to TGF-β1 have no effect on acetaldehyde-mediated phosphorylation of Smad 3 and on transcriptional upregulation of the COL1A2 gene. Fourth, wortmannin inhibits early acetaldehyde-induced accumulation of α2(I) collagen mRNA, but has no effect on TGF-β– dependent COL1A2 up-regulation. Although our findings did not address the formal possibility that acetaldehyde may activate pre-existing TGF-β and that this in turn may lead to autocrine stimulation of TGF-β expression, studies performed by others demonstrated that cytokine activation by acetaldehyde is a late event occurring at least 6 to 12 hours after acetaldehyde administration.28 Moreover, his findings also showed that acetaldehyde induces the late expression of TGF-β1 receptor type II (6 hours) and enhances the response of HSCs to TGF-β.28 Altogether, these findings suggest that acetaldehyde exerts its fibrogenic actions by TGF-β1–independent and TGF-β1– dependent mechanisms. Because the late events are cytokine dependent, the findings strongly suggest that acetaldehyde starts the fibrogenic response and primes HSCs to further respond to this cytokine. Moreover, based on our previous findings showing that H2O2 is a mediator of acetaldehyde and TGF-β1– dependent responses, one could speculate that acetaldehyde, through this ROS, activates a TGF-β1–dependent autocrine loop.
Data from several laboratories have shown the critical nature of the proximal regions of the COL1A1 and COL1A2 promoters in regulating collagen gene expression.29–31,34–42 The region encompassing −183 to −378 of the COL1A2 gene harbors TGF-β,29 TNF-α,30 acetaldehyde (this communication), and interferon-responsive elements.36 These responsive elements bind members of the C/EBP family of transcription factors,38 Sp1,29,31,43 (this communication), Smads 3 and 4,31,44,45 (this communication), Ets 1,35 Y box-binding protein YB-1, and the co-activator p300.36 Whereas Sp1, Smads 3 and 4, and p300 form DNA complexes that upregulate the transcription of the COL1A2 gene, the others down-regulate gene expression. Ets 1 is acetylated after TGF-β1 treatment; it associates with CBP/p300 protein complexes and shifts TGF-β response from collagen production to matrix degradation.35 Conversely, YB-1 binds to the interferon responsive element and also interferes with the formation of Smad 3/p300 complexes.36
Our studies demonstrate that acetaldehyde enhances binding of Sp1 and Smads 3 and 4 to the AcRE and that these transcription factors are essential for COL1A2 acetaldehyde responsiveness. These findings are in agreement with previous studies from our and other laboratories, demonstrating that Sp1 is essential for basal as well as TGF-β1–induced type I collagen gene expression.29,31,39,43–46 The molecular mechanisms whereby acetaldehyde activates Sp1 binding activity are yet to be determined, as is the potential role of Sp3. Smads 3 and 4 are essential for the acetaldehyde-dependent upregulation of the COL1A2 gene because overexpression of their respective dominant negative isoforms completely abrogates acetaldehyde responsiveness. As shown in Fig. 8A, HHSC nuclei contain phosphorylated Smad 3 within 30 minutes after acetaldehyde administration.
Studies performed with mouse HSCs have localized an AcRE within a similar region of the col1a2 promoter. However, the transacting factor binding to the AcRE after acetaldehyde was identified as NF-1.47 This nuclear transcription factor has been shown to play an important role in TGF-β1–mediated upregulation of the mouse col1a2.48 However, our studies have failed to demonstrate binding of NF1 to the COL1A2 AcRE, as antibody interference assays with NF1 antibodies had no effect on retarded bands induced by acetaldehyde (data not shown). These findings suggest that in addition to tissue-specific transcription factors there might be species-specific factors as well.
Previous work from our laboratory has established that several kinases, including PI3K and MAPK, are involved in acetaldehyde-dependent upregulation of COL1A2 mRNA.26 However, we further need to establish which of these kinases, if any, are directly involved in phosphorylation of Smads or activation of Sp1 binding to DNA. Such studies will allow us to determine whether activation and phosphorylation of Smads 3–4 in acetaldehyde-treated cells is similar to that induced by TGF-β1 and, therefore recognize a common mechanism.
We have already shown that acetaldehyde and TGF-β1 induce accumulation of H2O2 in rat HSCs.18,32 In this communication, we further showed that H2O2 is involved in acetaldehyde-mediated upregulation of COL1A2 in human HSCs. Therefore, this could be a common link in the fibrogenic cascade.18 Indeed, data in the literature suggest that H2O2 can induce the expression of TGF-β1 in cultured hepatic stellate cells.49 Accordingly, this could establish an important autocrine loop whereby acetaldehyde enhances the accumulation of H2O2 and this ROS upregulates expression of TGF-β1 that will further generate more H2O2. This autocrine loop, which generates a strong oxidative stress response in HHSCs, could play a key role in perpetuation of liver fibrosis on discontinuation of the injurious stimuli. Therefore, agents that interfere with this autocrine loop directly or through modification of the protein kinases that activates it could represent a novel form of therapy for alcohol-induced liver fibrosis.
In summary, our findings demonstrate that acetaldehyde-induced fibrogenesis follows a complex signaling pathway that is independent of TGF-β1 at early points but activates similar mediators used by the cytokine in upregulation collagen gene expression. In addition, this and previous work from our laboratory establishes the importance of oxidative stress in general and of H2O2 in particular in the fibrogenic mechanisms of acetaldehyde. Thus, antioxidants designed for blocking the production of H2O2 by HSCs and Kupffer cells could be of use in the treatment of liver fibrosis.
Footnotes
Supported by NIH grants AA10541 and AA9231 (M.R.); AR38648 and the St. Giles Foundation (F.R.); N. 2003060137_004 from MIUR (G.S.-B. and S.S.); and Fondi Ricerca Scientifica di Ateneo (A.B.).
Potential conflict of interest: Nothing to report.
References
- 1.McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Dis. 1999;19:205–219. doi: 10.1055/s-2007-1007110. [DOI] [PubMed] [Google Scholar]
- 2.McClain CJ, Barve S, Barve S, Deaciuc I, Hill DB. Tumor necrosis factor and alcoholic liver disease. Alcohol Clin Exp Res. 1998;22:248S–252S. doi: 10.1097/00000374-199805001-00006. [DOI] [PubMed] [Google Scholar]
- 3.Hill DB, Marsano L, Cohen D, Allen J, Shedlofsky S, McClain CJ. Increased plasma interleukin-6 concentrations in alcoholic hepatitis. J Lab Clin Med. 1992;119:547–552. [PubMed] [Google Scholar]
- 4.Nieto N, Dominguez-Rosales JA, Fontana L, Salazar A, Armendariz-Borunda J, Greenwel P, et al. Rat hepatic stellate cells contribute to the acute-phase response with increased expression of alpha1(I) and alpha1(IV) collagens, tissue inhibitor of metalloproteinase-1, and matrix-metalloproteinase-2 messenger RNAs. Hepatology. 2001;33:597–607. doi: 10.1053/jhep.2001.22520. [DOI] [PubMed] [Google Scholar]
- 5.Greenwel P, Rojkind M. Accelerated development of liver fibrosis in CCl4-treated rats by the weekly induction of acute phase response episodes: upregulation of alpha1(I) procollagen and tissue inhibitor of metal-loproteinase-1 mRNAs. Biochim Biophys Acta. 1997;1361:177–184. doi: 10.1016/s0925-4439(97)00028-8. [DOI] [PubMed] [Google Scholar]
- 6.Greenwel P, Iraburu MJ, Reyes-Romero M, Meraz-Cruz N, Casado E, Solis-Herruzo JA, et al. Induction of an acute phase response in rats stimulates the expression of alpha 1(I) procollagen messenger ribonucleic acid in their livers: possible role of interleukin-6. Lab Invest. 1995;72:83–91. [PubMed] [Google Scholar]
- 7.Marra F. Chemokines in liver inflammation and fibrosis. Front Biosci. 2002;7:d1899–d1914. doi: 10.2741/A887. [DOI] [PubMed] [Google Scholar]
- 8.Enomoto N, Ikejima K, Bradford BU, Rivera CA, Kono H, Goto M, et al. Role of Kupffer cells and gut-derived endotoxins in alcoholic liver injury. J Gastroenterol Hepatol. 2000;15(Suppl):D20–D25. doi: 10.1046/j.1440-1746.2000.02179.x. [DOI] [PubMed] [Google Scholar]
- 9.Lieber CS. Alcoholic liver disease: new insights in pathogenesis lead to new treatments. J Hepatol. 2000;32:113–128. doi: 10.1016/s0168-8278(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 10.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31:1544–1549. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 11.Safadi R, Friedman SL. Hepatic fibrosis—role of hepatic stellate cell activation. Med Gen Med. 2002;4:27. [PubMed] [Google Scholar]
- 12.Reeves HL, Friedman SL. Activation of hepatic stellate cells: a key issue in liver fibrosis. Front Biosci. 2002;7:d808–d826. doi: 10.2741/reeves. [DOI] [PubMed] [Google Scholar]
- 13.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 14.Reynaert H, Thompson MG, Thomas T, Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut. 2002;50:571–581. doi: 10.1136/gut.50.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontana L, Jerez D, Rojas-Valencia L, Solis-Herruzo JA, Greenwel P, Rojkind M. Ethanol induces the expression of alpha 1(I) procollagen mRNA in a co-culture system containing a liver stellate cell-line and freshly isolated hepatocytes. Biochim Biophys Acta. 1997;1362:135–144. doi: 10.1016/s0925-4439(97)00056-2. [DOI] [PubMed] [Google Scholar]
- 16.Casini A, Cunningham M, Rojkind M, Lieber CS. Acetaldehyde increases procollagen type I and fibronectin gene transcription in cultured rat fat-storing cells through a protein synthesis-dependent mechanism. Hepatology. 1991;13:758–765. [PubMed] [Google Scholar]
- 17.Greenwel P. Acetaldehyde-mediated collagen regulation in hepatic stellate cells. Alcohol Clin Exp Res. 1999;23:930–933. [PubMed] [Google Scholar]
- 18.Greenwel P, Dominguez-Rosales JA, Mavi G, Rivas-Estilla AM, Rojkind M. Hydrogen peroxide: a link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology. 2000;31:109–116. doi: 10.1002/hep.510310118. [DOI] [PubMed] [Google Scholar]
- 19.Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology. 1999;30:987–996. doi: 10.1002/hep.510300433. [DOI] [PubMed] [Google Scholar]
- 20.Nieto N, Friedman SL, Cederbaum AI. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology. 2002;35:62–73. doi: 10.1053/jhep.2002.30362. [DOI] [PubMed] [Google Scholar]
- 21.Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem. 2002;277:9853–9864. doi: 10.1074/jbc.M110506200. [DOI] [PubMed] [Google Scholar]
- 22.Nieto N, Greenwel P, Friedman SL, Zhang F, Dannenberg AJ, Cederbaum AI. Ethanol and arachidonic acid increase alpha 2(I) collagen expression in rat hepatic stellate cells overexpressing cytochrome P450 2E1: role of H2O2 and cyclooxygenase-2. J Biol Chem. 2000;275:20136–20145. doi: 10.1074/jbc.M001422200. [DOI] [PubMed] [Google Scholar]
- 23.Miao K, Potter JJ, Anania FA, Rennie-Tankersley L, Mezey E. Effect of acetaldehyde on Sp1 binding and activation of the mouse alpha 2(I) collagen promoter. Arch Biochem Biophys. 1997;341:140–152. doi: 10.1006/abbi.1997.9948. [DOI] [PubMed] [Google Scholar]
- 24.Anania FA, Womack L, Potter JJ, Mezey E. Acetaldehyde enhances murine alpha2(I) collagen promoter activity by Ca2+-independent protein kinase C activation in cultured rat hepatic stellate cells. Alcohol Clin Exp Res. 1999;23:279–284. [PubMed] [Google Scholar]
- 25.Attard FA, Wang L, Potter JJ, Rennie-Tankersley L, Mezey E. CCAAT/enhancer binding protein beta mediates the activation of the murine alpha1(I) collagen promoter by acetaldehyde. Arch Biochem Biophys. 2000;378:57–64. doi: 10.1006/abbi.2000.1803. [DOI] [PubMed] [Google Scholar]
- 26.Svegliati-Baroni G, Ridolfi F, Di Sario A, Saccomanno S, Bendia E, Benedetti A, et al. Intracellular signaling pathways involved in acetaldehyde-induced collagen and fibronectin gene expression in human hepatic stellate cells. Hepatology. 2001;33:1130–1140. doi: 10.1053/jhep.2001.23788. [DOI] [PubMed] [Google Scholar]
- 27.Attard FA, Wang L, Potter JJ, Rennie-Tankersley L, Mezey E. Identification of new sites of binding and activation of the murine alpha1(I) collagen promoter by CCAAT/enhancer binding protein beta. DNA Cell Biol. 2001;20:455–463. doi: 10.1089/104454901316976082. [DOI] [PubMed] [Google Scholar]
- 28.Chen A. Acetaldehyde stimulates the activation of latent transforming growth factor-beta1 and induces expression of the type II receptor of the cytokine in rat cultured hepatic stellate cells. Biochem J. 2002;368:683–693. doi: 10.1042/BJ20020949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inagaki Y, Truter S, Ramirez F. Transforming growth factor-beta stimulates alpha 2(I) collagen gene expression through a cis-acting element that contains an Sp1-binding site. J Biol Chem. 1994;269:14828–14834. [PubMed] [Google Scholar]
- 30.Inagaki Y, Truter S, Tanaka S, Di Liberto M, Ramirez F. Overlapping pathways mediate the opposing actions of tumor necrosis factor-alpha and transforming growth factor-beta on alpha 2(I) collagen gene transcription. J Biol Chem. 1995;270:3353–3358. doi: 10.1074/jbc.270.7.3353. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W, Ou J, Inagaki Y, Greenwel P, Ramirez F. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor beta1 stimulation of alpha 2(I)-collagen (COL1A2) transcription. J Biol Chem. 2000;275:39237–39245. doi: 10.1074/jbc.M003339200. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A, et al. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 33.Iraburu MJ, Dominguez-Rosales JA, Fontana L, Auster A, Garcia-Trevijano ER, Covarrubias-Pinedo A, et al. Tumor necrosis factor alpha down-regulates expression of the alpha1(I) collagen gene in rat hepatic stellate cells through a p20C/EBPbeta- and C/EBPdelta-dependent mechanism. Hepatology. 2000;31:1086–1093. doi: 10.1053/he.2000.5981. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh AK, Yuan W, Mori Y, Chen Sj, Varga J. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta: integration at the level of p300/CBP transcriptional coactivators. J Biol Chem. 2001;276:11041–11048. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- 35.Czuwara-Ladykowska J, Sementchenko VI, Watson DK, Trojanowska M. Ets1 is an effector of the transforming growth factor beta (TGF-beta) signaling pathway and an antagonist of the profibrotic effects of TGF-beta. J Biol Chem. 2002;277:20399–20408. doi: 10.1074/jbc.M200206200. [DOI] [PubMed] [Google Scholar]
- 36.Higashi K, Inagaki Y, Fujimori K, Nakao A, Kaneko H, Nakatsuka I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with Smad3. J Biol Chem. 2003;278:43470–43479. doi: 10.1074/jbc.M302339200. [DOI] [PubMed] [Google Scholar]
- 37.Inagaki Y, Truter S, Greenwel P, Rojkind M, Unoura M, Kobayashi K, et al. Regulation of the alpha 2(I) collagen gene transcription in fat-storing cells derived from a cirrhotic liver. Hepatology. 1995;22:573–579. [PubMed] [Google Scholar]
- 38.Greenwel P, Tanaka S, Penkov D, Zhang W, Olive M, Moll J, et al. Tumor necrosis factor alpha inhibits type I collagen synthesis through repressive CCAAT/enhancer-binding proteins. Mol Cell Biol. 2000;20:912–918. doi: 10.1128/mcb.20.3.912-918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greenwel P, Inagaki Y, Hu W, Walsh M, Ramirez F. Sp1 is required for the early response of alpha2(I) collagen to transforming growth factor-beta1. J Biol Chem. 1997;272:19738–19745. doi: 10.1074/jbc.272.32.19738. [DOI] [PubMed] [Google Scholar]
- 40.Artlett CM, Chen SJ, Varga J, Jimenez SA. Modulation of basal expression of the human alpha1(I) procollagen gene (COL1A1) by tandem NF-1/Sp1 promoter elements in normal human dermal fibroblasts. Matrix Biol. 1998;17:425–434. doi: 10.1016/s0945-053x(98)90102-0. [DOI] [PubMed] [Google Scholar]
- 41.Chen SJ, Artlett CM, Jimenez SA, Varga J. Modulation of human alpha1(I) procollagen gene activity by interaction with Sp1 and Sp3 transcription factors in vitro. Gene. 1998;215:101–110. doi: 10.1016/s0378-1119(98)00268-6. [DOI] [PubMed] [Google Scholar]
- 42.Li L, Artlett CM, Jimenez SA, Hall DJ, Varga J. Positive regulation of human alpha 1 (I) collagen promoter activity by transcription factor Sp1. Gene. 1995;164:229–234. doi: 10.1016/0378-1119(95)00508-4. [DOI] [PubMed] [Google Scholar]
- 43.Tamaki T, Ohnishi K, Hartl C, LeRoy EC, Trojanowska M. Characterization of a GC-rich region containing Sp1 binding site(s) as a constitutive responsive element of the alpha 2(I) collagen gene in human fibroblasts. J Biol Chem. 1995;270:4299–304. doi: 10.1074/jbc.270.9.4299. [DOI] [PubMed] [Google Scholar]
- 44.Chen SJ, Yuan W, Lo S, Trojanowska M, Varga J. Interaction of smad3 with a proximal smad-binding element of the human alpha2(I) procollagen gene promoter required for transcriptional activation by TGF-beta. J Cell Physiol. 2000;183:381–392. doi: 10.1002/(SICI)1097-4652(200006)183:3<381::AID-JCP11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 45.Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J Invest Dermatol. 1999;112:49–57. doi: 10.1046/j.1523-1747.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 46.Ihn H, LeRoy EC, Trojanowska M. Oncostatin M stimulates transcription of the human alpha2(I) collagen gene via the Sp1/Sp3-binding site. J Biol Chem. 1997;272:24666–24672. doi: 10.1074/jbc.272.39.24666. [DOI] [PubMed] [Google Scholar]
- 47.Anania FA, Potter JJ, Rennie-Tankersley L, Mezey E. Effects of acetaldehyde on nuclear protein binding to the nuclear factor I consensus sequence in the alpha 2(I) collagen promoter. Hepatology. 1995;21:1640–1648. [PubMed] [Google Scholar]
- 48.Rossi P, Karsenty G, Roberts AB, Roche NS, Sporn MB, de Crombrugghe B. A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-beta. Cell. 1988;52:405–414. doi: 10.1016/s0092-8674(88)80033-3. [DOI] [PubMed] [Google Scholar]
- 49.De Bleser PJ, Xu G, Rombouts K, Rogiers V, Geerts A. Glutathione levels discriminate between oxidative stress and transforming growth factor-beta signaling in activated rat hepatic stellate cells. J Biol Chem. 1999;274:33881–33887. doi: 10.1074/jbc.274.48.33881. [DOI] [PubMed] [Google Scholar]