

Abstract
Carbon monoxide (CO) and nitric oxide (NO) can be involved in regulation of cerebral circulation. Inhibition of production of either one of these gaseous intercellular messengers inhibits newborn pig cerebral arteriolar dilation to the excitatory amino acid glutamate. Glutamate can increase NO production. Therefore, the present study tests the hypothesis that NO, which is increased by glutamate, stimulates the production of CO by cerebral microvessels. Experiments used freshly isolated cerebral microvessels from piglets that express only heme oxygenase-2 (HO-2). CO production was measured by gas chromatography-mass spectrometry. Although inhibition of nitric oxide synthase with L-nitro arginine (LNA) did not alter basal HO-2 catalytic activity or CO production, LNA blocked glutamate stimulation of HO-2 activity and CO production. Further, the NO donor sodium nitroprusside mimicked the actions of glutamate on HO-2 and CO production. The action of NO appears to be via cGMP because 8-br-cGMP mimics and ODQ blocks, glutamate stimulation of CO production and HO-2 catalytic activity. Inhibitors of neither casein kinase nor PI3 kinase altered HO-2 catalytic activity. Conversely, inhibition of calmodulin with calmidazolium chloride blocked glutamate stimulation of CO production and reduced HO-2 catalytic activity. These data suggest that glutamate may activate NOS producing NO that leads to CO synthesis via a cGMP dependent elevation of HO-2 catalytic activity. These results are consistent with the findings in vivo that either HO or NOS inhibition blocks cerebrovascular dilation to glutamate in piglets.
Keywords: heme oxygenase, cyclic GMP, nitric oxide synthase, glutamate
Introduction
Both carbon monoxide (CO) and nitric oxide (NO) are endogenously produced, gaseous, intercellular messengers that can be involved in regulation of cerebral circulation. In neonatal pigs, CO regulation and modulation are involved in cerebrovascular circulatory control in response to neuronal activity, hypoxia, and changing blood pressure (12, 18, 26, 41). While the contributions of NO to cerebral blood flow regulation increase with age (40, 47), NO is important in the newborn as a permissive factor enabling vascular responses to CO (15). In the piglet cerebrovascular circulation, glutamate induced pial arteriolar dilation can be blocked by either inhibiting nitric oxide synthase (NOS) (14, 24), that produces NO, or heme oxygenase (HO)(18, 30), that produces CO. One possible explanation for these apparently conflicting data is that one gaseous messenger is necessary to allow dilation to the other. Indeed, as noted above, such a permissive contribution of NO to CO-induced dilation has been described. Another possibility is that glutamate receptor activation increases the production of one of the two gases and that gas in turn increases the production of the other, which is the final mediator of the dilatory response.
CO has been reported to directly affect NO production. In intestinal smooth muscle, CO increased NO that activated of L-type Ca 2+ channels (19). Conversely, CO dose-dependently inhibited NO synthesis by rat renal arteries, although low concentrations of CO actually increased NO by causing release from a preformed pool (38).
Studies to date regarding the effects of NO on CO production have reported both increases and decreases of CO production caused by NO. Thus, HO-2 expressed in Escherichia coli was inhibited by NO donors via binding of NO to a heme regulatory motif on HO-2 (5). Also, Rodriguez et al (31) found that rats treated for two days with the NOS inhibitor, NG-nitro-L-arginine methyl ester, had increased renal CO production without any change in HO expression. Conversely, in isolated heart (22) and porcine aortic endothelial cells (25) NO increased CO production.
Because both inhibition of NOS and HO block glutamate-induced dilation and glutamate has been reported to increase NO production (6, 8), we wondered if NO could stimulate CO production, adding another form of interaction to the permissive action of NO in CO-mediated dilation to glutamate. Therefore, the present experiments were designed to test the hypothesis that NO stimulates CO production by piglet cerebral microvessels. We also examined potential mechanisms by which NO could stimulate the production of CO.
Materials and Methods
Experiments using animals were reviewed and approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center. Brains were removed from 1–3 day old piglets under ketamine (33 mg/kg) and acepromazine (3.3 mg/kg) anesthesia.
Isolation of cerebral microvessels
Cerebral microvessels were isolated from the brains as described before (15, 16). The isolation was accomplished in cold Krebs solution (mM: 120 NaCl, 5 KCl, 0.62 MgSO4, 1.8 CaCl2, 10 N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), 6 glucose [pH 7.4]). The dura mater and attached vessels were removed from the tissue, and the tissue was washed three times with the isolation solution. The tissue was minced into tiny pieces using two scalpels in isolation solution and then transferred to a 40-ml Dounce homogenizer and homogenized with 10 strokes of a loose-fitting pestle. The homogenate was passed through a 300-μm, nylon-mesh screen. The passage was refiltered over a 60-μm, nylonmesh screen. The screen was removed and placed in a 50-ml centrifuge tube containing Krebs solution. Microvessels that passed through the 300μm but not 60μm mesh screen were washed off by agitation and scraping and then centrifuged at 1200 rpm for 5 min. Experimentation began immediately after vessel isolation (less than 30 min from brain removal) with resuspension of the microvessels in Krebs solution.
Experimental treatments
Treatments were begun by replacement of Krebs in the vial with fresh Krebs containing the experimental treatment. Glutamate (10−4 M), Nω-nitro-L-arginine (LNA)(10−3 M), sodium nitroprusside (SNP) (10−7–10−5 M), 8-bromo-cGMP (10−5 M), TBB (4,5,6,7-tetrabromo-2-azabenzimidazole) (2x10−5 M), LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one) (2.5x10−5 M) and calmidizolium Cl (CzCl) (2x10−5M) were dissolved in Krebs. Heme (5x10−6 M) was prepared as heme-l-lysinate or hemin in basic Krebs and protected from light. Guanylyl cyclase was inhibited with 1H - [1,2,4] oxadiazolo [4,3-a] quinoxaline-l-one (ODQ). ODQ (5x10−4 M) was initially dissolved in DMSO to a concentration of 4x10−2M, and diluted approximately 100 times with Krebs. DMSO at double this concentration did not effect CO production or HO-2 catalytic activity.
The concentrations of treatments used were selected based on one or more of three sources. 1.) Where data were available, inhibitor concentrations were those that had been found to be effective in vivo from topical application to the piglet cerebral cortex using cranial windows (LNA (17) and ODQ (14)). 2.) Other inhibitor concentrations were selected from literature sources of use in vitro (TBB (1), LY294002 (7) CzCl (2)). 3.) Agonist concentrations were in all cases selected from submaximal dilator concentrations in piglet cranial window experiments (heme (18), glutamate (14), SNP (17), 8-br-cGMP (14)).
The apparent catalytic activity of HO-2 in the intact cerebral microvessels was determined by providing exogenous substrate so endogenous substrate availability would not affect CO production. We assume that under the present experimental conditions, treatments used are unlikely to markedly alter O2 partial pressure or cellular reducing equivalents. Thus, it seems reasonable to propose that catalytic activity, defined as CO production per mg protein when substrate concentration is high and constant, includes HO catalytic efficiency and fractional activation by intracellular relocation.
Measurement of CO production
For measurement of CO production, freshly isolated microvessels were placed inside amber vials (2.0 ml) containing Krebs solution. All subsequent assay steps were carried out in the dark to prevent non-enzymatic photo-oxidative production of CO ex vivo due to the photo-degradation of organic compounds. Krebs buffer in each vial was replaced with fresh Krebs or fresh Krebs containing the experimental treatment to begin incubation. The internal standard (see below) was injected into the bottom of the vial and the vial was immediately sealed with a rubberized Teflon lined cap. Cerebral microvessels were incubated for 30 min at 37°C. Incubations were terminated by placing the samples in ice water (0°C) and CO production was determined immediately.
A saturated solution of the isotopically labeled CO (13C16O) (isotopic purity >99%) was used as an internal standard for quantitative measurements by gas chromatography/mass spectrometry (GC/MS) (15, 16).
GC/MS analysis of the headspace gas was performed using a Hewlett-Packard 5970 mass-selective ion detector interfaced to a Hewlett Packard 5890A gas chromatograph. The separation of CO from other gases was carried out on a Varian-5A mole sieve capillary column (30 m; 0.32 mm ID) with a linear temperature gradient from 35°C to 65°C at 5° per minute. Helium was the carrier gas at a column head pressure of 4.0 psi. Aliquots (100μl) of the headspace gas were injected using a gas-tight syringe into the splitless injector having a temperature of 120°C. Ions at m/z 28 and 29 corresponding to 12C16O and 13C16O, respectively, were recorded via selective ion monitoring. The amount of CO in samples was calculated from the ratio of peak areas of m/z 28 and m/z 29. The results are expressed as pmol of CO released into the headspace gas per 100 μg protein in 30 min. Protein was measured by the Bradford method.
Statistical analysis
Values are presented as means ± SEM. The results were subjected to analysis of variance (ANOVA) for repeated measures with Tukey post hoc to isolate differences between groups. A level of P < 0.05 was considered significant.
Results
Acute NOS inhibition did not affect either basal CO production or HO-2 catalytic activity. Thus, 30–60 min following treatment, cerebral microvessels produced 19±3 pmol CO/100 μg.30min without and 24±4 pmol CO/10 μg.30min with LNA treatment (n=4 separate microvessel preparations). Furthermore, HO-2 catalytic activity, detected as heme stimulated CO production, was 51±11 pmol CO/100 μg.30min before and 51±8 pmol CO/100 μg.30min after LNA treatment (n=4).
In contrast, NOS inhibition markedly attenuated glutamate stimulation of CO production (Fig. 1). Furthermore, augmentation of HO-2 catalytic activity by glutamate was attenuated when microvessels were treated with LNA (Fig. 1). Thus, in LNA treated microvessels, exogenous heme-stimulated CO production was no different whether or not glutamate was applied (Fig. 1, last two bars). These data suggest that NO may be involved in the mechanism by which glutamate stimulates CO production.
Figure 1.
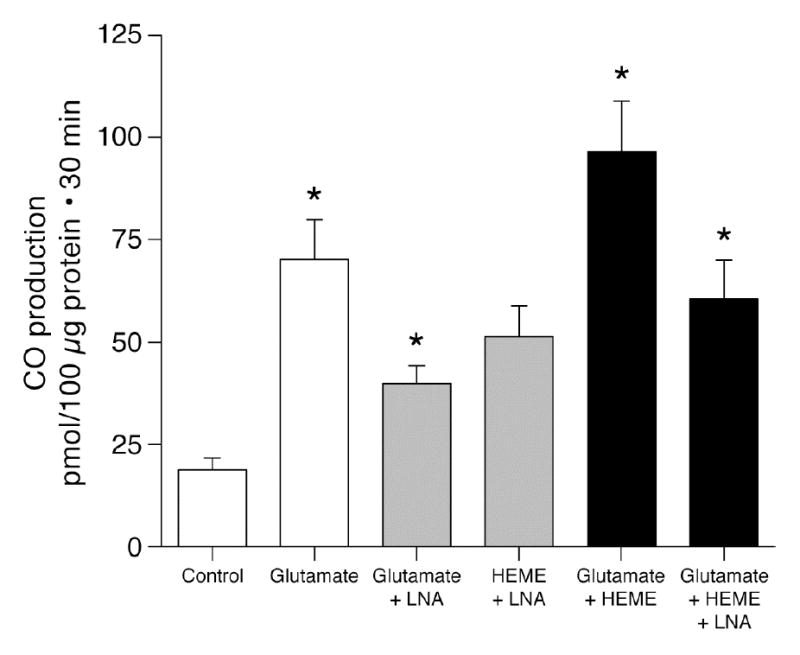
Effect of glutamate (10−4 M) on CO production by piglet cerebral microvessels from endogenous and exogenous heme (5x10−6 M) in the absence and presence of LNA (10−3 M). means±SEM. *P<0.05 compared to preceding bar. N=4.
Because the data above suggest that NO may increase CO production, we addressed this hypothesis directly using the NO donor, SNP. SNP strongly stimulated CO production (Fig. 2). In addition, similarly to glutamate, SNP augmented HO-2 catalytic activity (Fig. 2). These data are consistent with the hypothesis that glutamate increases NO that increases CO production by elevating HO-2 catalytic activity.
Figure 2.
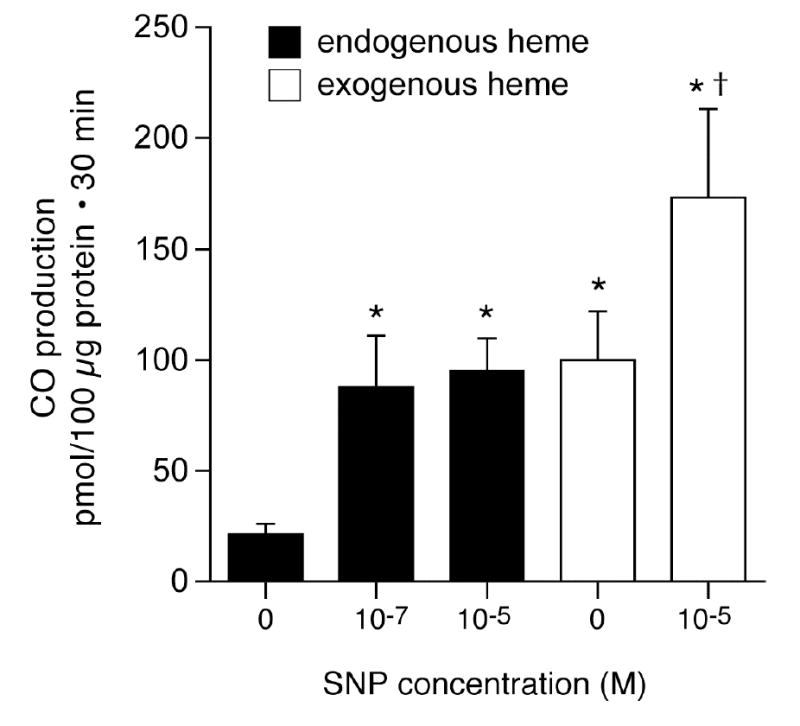
Effect of sodium nitroprusside (SNP) on CO production and HO-2 catalytic activity (exogenous heme, 5x10−6 M) by piglet cerebral microvessels. means±SEM. *P<0.05 compared to no SNP. +P<0.05 compared to exogenous heme with no SNP. N=4.
As NO is a strong activator of guanylyl cyclase, we addressed the hypothesis that the mechanism by which NO increases HO-2 activity and thus CO production is by increasing cGMP. Initially, guanylyl cyclase was blocked with ODQ (14). As anticipated, ODQ decreased CO production by cerebral microvessels and strongly attenuated the increase CO caused by exogenous heme (catalytic activity)(Fig. 3). Furthermore, as would be expected if glutamate increases NO that elevates cGMP that then augments CO production, ODQ blocked glutamate induced CO production (Fig. 4) and glutamate-induced stimulation of HO-2 activity (93 versus 40 pmol CO/10 μg.30min without and with ODQ, respectively). In addition, the stable cGMP analog, 8-br-cGMP, stimulated CO production (Fig. 5). In the presence of sufficient 8-br-cGMP to increase CO production similarly to glutamate, glutamate did not increase and LNA did not decrease CO production (Fig. 5). These data suggest glutamate may increase CO production by stimulating NO production by the microvessels. NO activates guanylyl cyclase producing cGMP that increases the catalytic activity of HO-2. Data in figures 2 and 5 suggest 60–70 pmol CO/100μg protein.30min is approximately maximal CO production without provision of additional substrate (as in Fig. 1,3). The fact that glutamate did not further increase CO production when co-administered with 10−5 M 8-br-cGMP suggests glutamate may not increase cellular heme.
Figure 3.
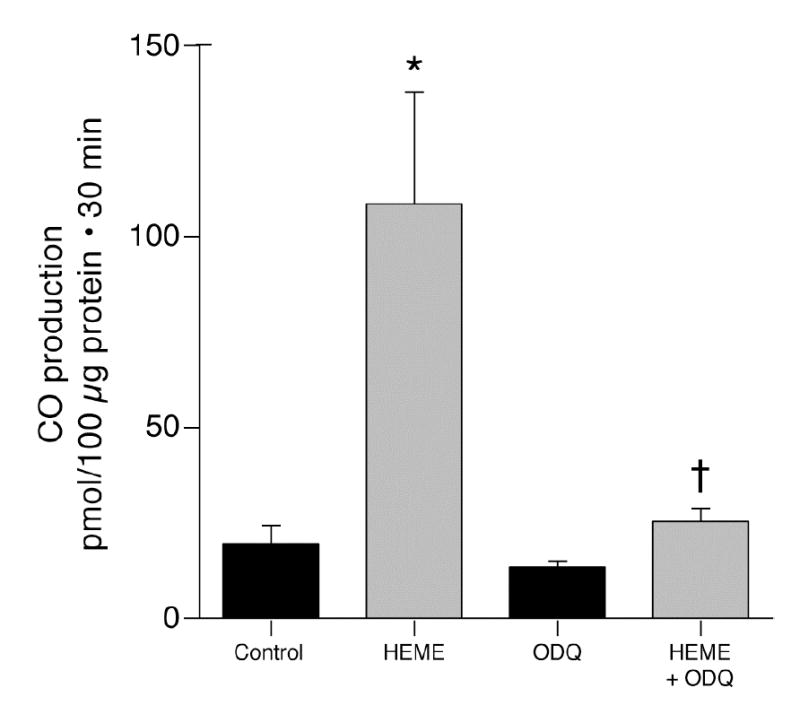
Effect of ODQ (5x10−4M) on CO production from endogenous and exogenous heme (5x10−6 M) by piglet cerebral microvessels. means±SEM. *P<0.05 compared to control. +P<0.05 compared to heme without ODQ. N=6.
Figure 4.
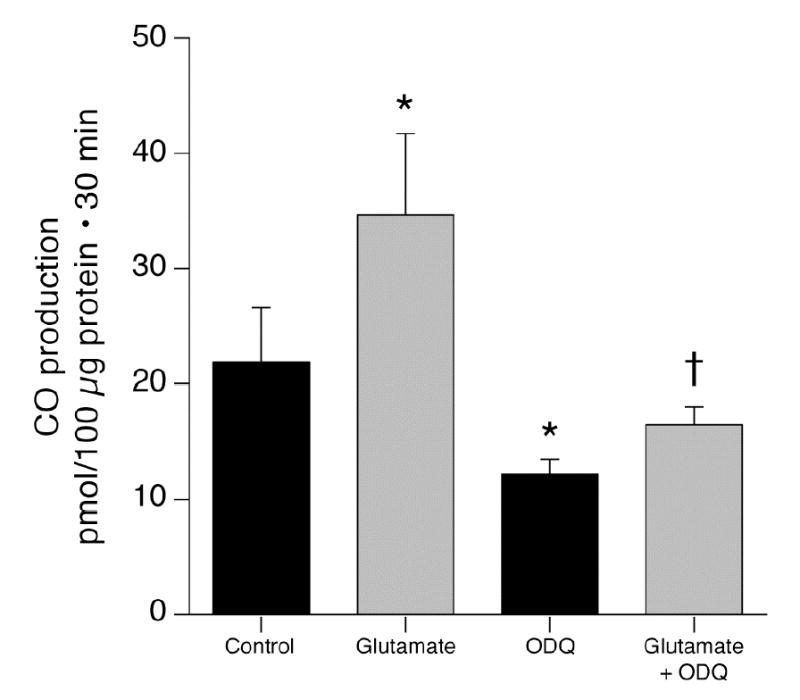
Effect of ODQ (5x10−4M) on glutamate (10−4 M) stimulation of CO production by piglet cerebral microvessels. means±SEM. *P<0.05 compared to control. +P<0.05 compared to ODQ without glutamate. N=6.
Figure 5.
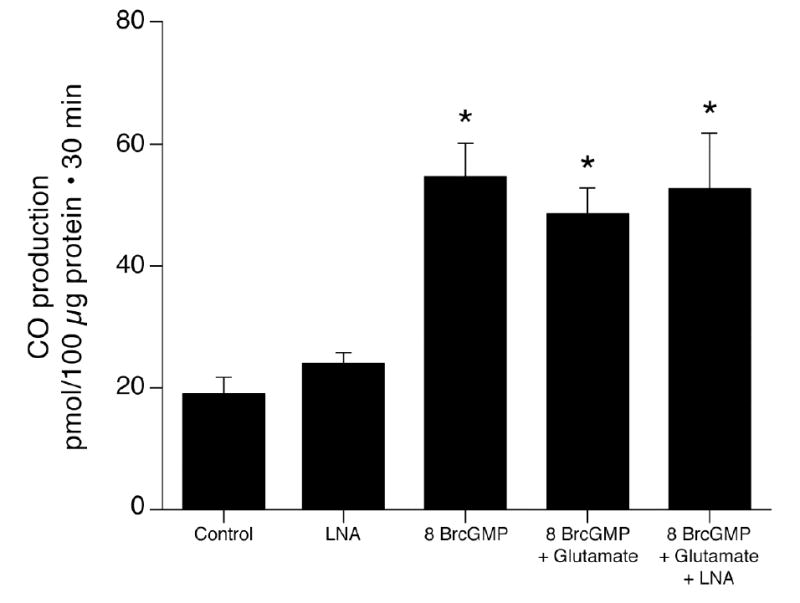
Effect of 8 br-cGMP (10−5 M) on CO production by piglet cerebral microvessels without and with glutamate (10−4 M) and/or LNA (10−3M). means±SEM. *P<0.05 compared to LNA alone. N=8.
The present results and past data from others and us (see discussion) have shown phosphorylation can increase HO-2 catalytic activity. Therefore, we examined the possibility that two kinases that have been shown to alter HO-2 catalytic activity in other tissues of rats might do the same in piglet cerebral microvessels. The casein kinase inhibitor TBB (20μM) did not change HO-2 catalytic activity detected by conversion of exogenous heme (10−6 M) to CO (106±29 and 104±25 pmol/100μg protein.30min without and with TBB, respectively (n=8)) and glutamate (1mM) still increased HO-2 catalytic activity (160±31 and 220±45, without and with TBB, respectively (n=8)). Similarly, the PI3 kinase inhibitor LY294002 (25μM) did not alter catalytic activity in piglet microvessels (at 10−4 M heme: 76±21 and 90±26 pmol/100μg protein.30min without and with LY294002, respectively (n=8)).
Since glutamate may increase cytosolic Ca2+ (37) and Ca2+/calmodulin-dependent eNOS increases NO in endothelial cells in response to increased cytosolic Ca2+ (20, 32), we examined the effect of calmodulin inhibition with calmidazolium chloride (CzCl) on glutamate-induced CO production (Fig. 6). While CzCl had no effect on basal CO production, it completely blocked glutamate stimulation of CO production. Furthermore, CzCl reduced HO-2 catalytic activity (CO production from exogenous heme (10−5 M)) from 53±7 to 36±5 pmol CO/100 μg protein.30min (p<0.05, n=4).
Figure 6.
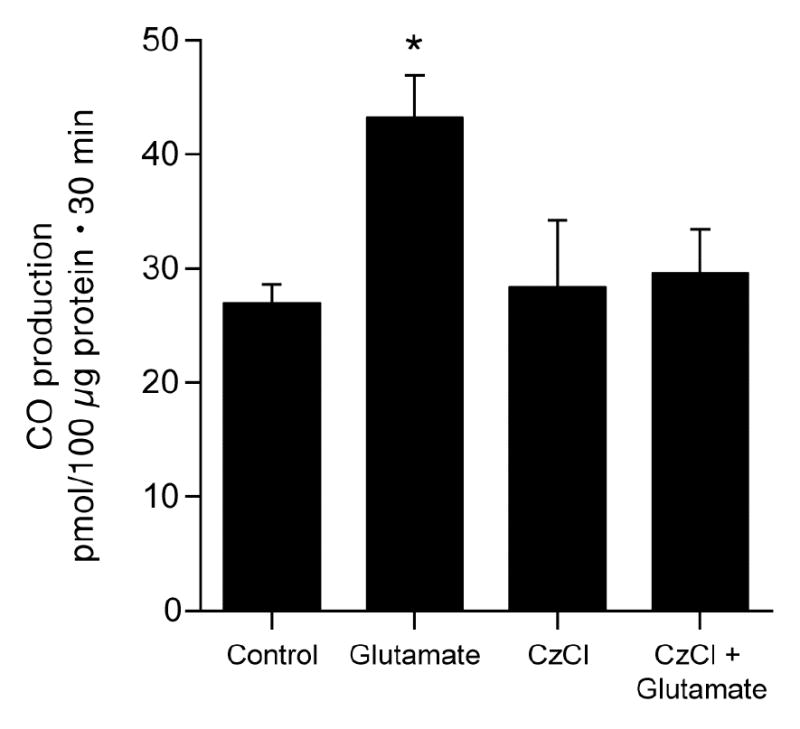
Effect of calmidizolium Cl (CzCl) (2x10−5M) on glutamate (10−4M) stimulation of CO production by piglet cerebral microvessels. means±SEM. *P<0.05 compared to control. N=7.
Discussion
The new findings on newborn pig cerebral microvessels are: 1. Basal NO production does not appear to be involved in either controlling HO-2 catalytic activity or basal CO production. 2. Increasing NO and cGMP elevates HO-2 catalytic activity and stimulates CO production. 3. Glutamate elevates NO and, thereby, cGMP, that enhances HO-2 catalytic activity, causing CO production to increase. 4. Calmodulin is required for glutamate-induced stimulation of HO-2 activity and CO production.
Cellular CO production results from metabolism of heme by HO. In freshly isolated cerebral microvessels from newborn pigs, as in the intact brain, in vivo, of the two known, highly catalytically active isoforms of HO, only HO-2 expression is detectable (28). HO-2 is constitutively expressed and induced by few stimuli (21), so expression of HO in the present experiments can be considered invariant.
Therefore, CO production can be regulated by delivery of substrate (heme) and catalytic activity of HO-2. HO-2 catalytic activity may be altered by co-factor availability, cellular localization, and/or post-translational modifications of the enzyme. Under the experimental conditions used it is unlikely that oxygen, NADPH, or NADPH-cytochrome c reductase would be limiting. The rate-limiting step in heme synthesis is the production in mitochondria of delta-aminolevulinic acid from succinyl CoA and glycine catalyzed by the tightly regulated enzyme delta-aminolevulinic acid synthase (ALAS)(13, 23). However, we did not find evidence of alteration of heme provision contributing to either glutamate- or NO- induced CO production. This conclusion is particularly supported by the finding that glutamate did not increase CO production when catalytic activity of HO-2 was already increased by 8-br-cGMP. If glutamate increased heme delivery, one would expect augmented CO production in the context of elevated HO-2 activity rather than no change. Therefore, it appears that glutamate, NO, and cGMP stimulate CO production by increasing HO-2 catalytic activity. Indeed, glutamate and SNP increased conversion of exogenous heme to CO.
HO-2 catalytic activity control mechanisms may be cell type and tissue specific. In neurons HO-2 activity can be stimulated by CK2 catalyzed phosphorylation of serine 79 (1). Glutamatergic activation of HO-2 results from metabotropic glutamate receptor-induced Ca2+ release, activation of protein kinase C (PKC), and CK2 phosphorylation (3). Conversely, in freshly isolated piglet cerebral microvessels and microvascular endothelial cells in culture, CO production is increased by ionotropic, but not metabotropic, glutamate receptor stimulation (27). In addition, protein tyrosine kinase inhibition decreased and tyrosine phosphatase inhibition increased basal CO production and glutamate stimulated CO production (15). Neither treatment of the cerebral microvessels with phorbol ester to activate PKC, H-7 to inhibit PKC, nor TBB to inhibit CK2 increased HO-2 catalytic activity (16, present study).
In contrast to the present study, in preparations of HO-2 expressed in E. coli, NO donors inhibited HO-2 (5), possibly suggesting that NO can have a direct inhibitory effect on HO-2 that is masked in the intact system by cGMP-induced stimulation. Such a direct inhibitory effect of NO has been reported in HO-1 rich aortic endothelial cell microsomes where nitrosylation of heme prevented catabolism by HO (11). Interestingly, in contrast to the present studies of the effects of acute exposure (minutes) to NO, chronic exposure to NO consistently inhibits HO (11, 31).
CzCl, that inhibits calmodulin, decreased HO-2 catalytic activity and blocked glutamate stimulation of CO production. Also, Boehning (2) showed Ca2+/CaM regulation of HO-2 catalytic activity. Similarly to the present experiments in piglet microvessels, glutamate increased HO-2 activity and that increase was blocked by CzCl in rat cortical neurons. These results were surprising to us because we had found previously that the Ca ionophore, ionomycin, in Ca2+ replete media increased CO production, but did not increase HO-2 catalytic activity (15). Furthermore, ionomycin and Ca2+ free media to deplete cellular Ca2+ did not decrease basal CO production nor prevent glutamate-induced stimulation of CO production. Thus, the results of our earlier study that elevations of cytosolic Ca2+ increased CO production but did not detectably increase HO-2 catalytic activity appear inconsistent with the present findings and those of Boehning et al. However, In our previous study we either flooded the cell with Ca2+ with Ca2+ ionophore or eliminated extracellular Ca2+ and attempted to empty intracellular stores. In the present study, Ca2+ was not manipulated but instead Ca2+/CaM signaling was eliminated by blocking CaM. Interestingly, Ca2+-independent CaM-dependent regulation of enzyme activity has been described (9,35,42), but not for HO-2. However, in rat cortical neurons ionomycin did increase bilirubin production from exogenous heme (2). Explanations for the difference between rat cortical neurons and newborn pig microvascular endothelium in the effects of ionomycin on HO-2 catalytic activity are not immediately apparent.
Although both microvascular smooth muscle and endothelial cells are stimulated to generate CO by glutamate, CO production and the response to glutamate are more pronounced in endothelial cells (15). Piglet cerebrovascular endothelial cells express ionotropic and, to a lesser extent, metabotropic glutamate receptors, as well as glutamate transporters. Ionotropic receptor stimulation causes increased CO production (27). Ionotropic glutamate receptor stimulation can increase reactive oxygen species in endothelial cells (33). However, reactive oxygen species would be expected to decrease NO (10) and thus work against stimulation of CO production. Glutamate increases cytosolic Ca2+ via metabotropic and ionotropic receptor mechanisms in neurons (44) and glia (34,36). Glutamate-induced elevations in Ca2+ also occur in endothelial cells (29). Since eNOS (see below) activity can be regulated by Ca 2+/CaM and elevation of CO production by glutamate was blocked by CzCl, ionotropic glutamate receptor -induced elevation of cytosolic Ca2+ could be involved in glutamate-induced CO production.
The present data suggest NO is an intermediary signal between glutamate receptor stimulation and CO production in cerebral microvessels. The cell types included in the microvessel preparation are primarily endothelial cells and microvascular smooth muscle, with adhering pericytes, astrocytes, and perivascular nerve endings. Because the vessels are used immediately upon collection, iNOS could not be induced. The predominant NOS is probably eNOS, but the potential inclusion of nNOS cannot be excluded. Because eNOS can be activated to produce NO by an elevation of cytosolic Ca2+ (20, 32, 39, 46), glutamate may increase endothelial cell Ca2+ that would activate eNOS. Conversely, eNOS activity can be increased by elevations of eNOS sensitivity to CaM (reviewed in 43) so glutamate could increase NO without increasing cytosolic Ca2+ concentration.
These data on isolated vessels and those from intact cerebrovascular circulation in vivo are not entirely consistent, suggesting potential additional sources of CO in vivo may contribute to pial arteriolar dilation to glutamate. Thus, in the present study LNA totally abolished glutamate –induced CO production. However, in vivo, while LNA blocked glutamate-induced dilation, a constant, background amount of SNP completely restored dilation to glutamate (13). If CO causes dilation to glutamate and NO causes the increase in CO, how could glutamate cause dilation if NO is held constant? In vivo cerebral microvessels are accompanied by astrocytes and neurons, in particular, that also have glutamate receptors, HO-2, and nNOS. In fact, involvement of nNOS in glutamate-induced cerebrovascular dilation in mouse cerebellum has been demonstrated (45). Furthermore, the inclusion of HO-2 and glutamate receptors in astrocytes and neurons provide the possibility of glutamate-induced stimulation of CO independent of NO in either of these cell types. Because the vascular smooth muscle appears to require a permissive level of cGMP that can be provided by eNOS derived NO (15), a constant level of NO, or cGMP, could allow arteriolar dilation to occur in response to increases CO from neurons and/or glia.
cGMP is the predominant mechanism by which NO increases CO production in microvessels. Inhibition of guanylyl cyclase completely blocked the CO increases caused by glutamate and SNP and reduced HO-2 catalytic activity. 8-br-cGMP mimicked the stimulatory effects of glutamate and SNP. Therefore, the present data are consistent with the hypothesis that NO increases HO-2 catalytic activity and glutamate-induced CO production by increasing cGMP.
Acknowledgments
Supported by NHLBI/NIH and NINDS/NIH. We thank G. Short for preparing the figures and M. Lester for clerical assistance.
References
- 1.Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SS. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- 2.Boehning D, Sedaghat L, Sedlak TW, Snyder SS. Heme oxygenase-2 is activated by calcium-calmodulin. J Biol Chem. 2004;279:30927–30930. doi: 10.1074/jbc.C400222200. [DOI] [PubMed] [Google Scholar]
- 3.Boehning D, Snyder SH. Novel neural modulators. Ann Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- 4.Canzoniero LM, Sensi SL, Turetsky DM, Finley MF, Choi DW, Huettner JE. Glutamate receptor-mediated calcium entry in neurons derived from P19 embryonal carcinoma cells. J Neurosci Res. 1996;45:226–36. doi: 10.1002/(SICI)1097-4547(19960801)45:3<226::AID-JNR4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 5.Ding Y, McCoubrey WK, Jr, Maines MD. Interaction of heme oxygenase-2 with nitric oxide donors. Is the oxygenase an intracellular ‘sink’ for NO? Eur J Biochem. 1999;264:854–861. doi: 10.1046/j.1432-1327.1999.00677.x. [DOI] [PubMed] [Google Scholar]
- 6.Domoki F, Perciaccante JV, Shimizu K, Puskar M, Busija DW, Bari F. N -methyl-D-aspartate-induced vasodilation is mediated by endothelium-independent nitric oxide release in piglets. Am J Physiol. 2002;282:H1404–H1409. doi: 10.1152/ajpheart.00523.2001. [DOI] [PubMed] [Google Scholar]
- 7.Duan C, Bauchat JR, Hsieh T. Phosphatidylinositol 3-kinase is required for insulin-like growth factor-I-induced vascular smooth muscle cell proliferation and migration. Circ Res. 2000;86:15–23. doi: 10.1161/01.res.86.1.15. [DOI] [PubMed] [Google Scholar]
- 8.Garthwaite J. Glutamate, nitric oxide, and cell-cell signaling in the nervous system. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- 9.Jenkins CM, Wolf MJ, Mancuso DJ, Gross RW. Identification ot the calmodulin- binding domain of recombinant calcium independent phospholipase A2_. Implications for structure and function. J Biol Chem. 2001;276:7129–7135. doi: 10.1074/jbc.M010439200. [DOI] [PubMed] [Google Scholar]
- 10.Jernigan NL, Resta TC, Walker BR. Contribution of oxygen radicals to altered NO-dependent pulmonary vasodilation in acute and chronic hypoxia. Am J Physiol. 2004;286:L947–55. doi: 10.1152/ajplung.00215.2003. [DOI] [PubMed] [Google Scholar]
- 11.Juckett M, Zheng Y, Yuan H, Pastor T, Antholine W, Wber M, Vercellotti G. Heme and the endothelium, Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J Biol Chem. 1998;273:23388–23397. doi: 10.1074/jbc.273.36.23388. [DOI] [PubMed] [Google Scholar]
- 12.Kanu A, Whitfield J, Leffler CW. Carbon monoxide contributes to cerebrovascular vasodilation in hypotensive piglets. FASEB J. 2005;19:A1249. doi: 10.1152/ajpheart.01368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kikuchi G, Hayashi N. Regulation by heme of synthesis and intracellular translocation of delta-aminolevinulinate synthase in the liver. Mol Cell Biochem. 1981;37:27–41. doi: 10.1007/BF02355885. [DOI] [PubMed] [Google Scholar]
- 14.Koneru P, Leffler CW. Role of cyclic GMP in carbon monoxide induced vasodilation in piglets. Am J Physiol. 2004;286:H304–H309. doi: 10.1152/ajpheart.00810.2003. [DOI] [PubMed] [Google Scholar]
- 15.Leffler CW, Balabanova L, Fedinec AL, Waters CM, Parfenova H. Mechanism of glutamate stimulation of CO production in cerebral microvessels. Am J Physiol. 2003;285:H74–H80. doi: 10.1152/ajpheart.01081.2002. [DOI] [PubMed] [Google Scholar]
- 16.Leffler CW, Balabanova L, Sullivan CD, Wang X, Fedinec AL, Parfenova H. Regulation of CO production in cerebral microvessels of newborn pigs. Am J Physiol. 2003;285:H292–H297. doi: 10.1152/ajpheart.01059.2002. [DOI] [PubMed] [Google Scholar]
- 17.Leffler CW, Nasjletti A, Johnson RA, Fedinec AL. Contributions of prostacyclin and nitric oxide to carbon monoxide induced cerebrovascular dilation in newborn pigs. Am J Physiol. 2001;280:H1490–H1495. doi: 10.1152/ajpheart.2001.280.4.H1490. [DOI] [PubMed] [Google Scholar]
- 18.Leffler CW, Nasjletti A, Yu C, Johnson RA, Fedinec AL, Walker N. Carbon monoxide and cerebral microvascular tone in newborn pigs. Am J Physiol. 1999;276:H1641–H1646. doi: 10.1152/ajpheart.1999.276.5.H1641. [DOI] [PubMed] [Google Scholar]
- 19.Lim I, Gibbons SJ, Lyford GL, Miller SM, Strege PR, Sarr MG, Chatterjee S, Szurszewski, Shah VH, Farrugia G. Carbon monoxide activates human intestinal smooth muscle L-type Ca2+ channels through a nitric oxide dependent mechanism. Am J Physiol. 2005;288:G7–G14. doi: 10.1152/ajpgi.00205.2004. [DOI] [PubMed] [Google Scholar]
- 20.Liou SF, Wu JR, Lai WT, Sheu SH, Chen IJ, Yeh JL. The vasorelaxing action of labedipinedilol_A involves endothelial cell-derived NO and eNOS expression caused by calcium influx. J Cardiovasc Pharmacol. 2005;45:232–240. doi: 10.1097/01.fjc.0000154375.88283.5c. [DOI] [PubMed] [Google Scholar]
- 21.Maines MD. The heme oxygenase system and its functions in the brain. Cell Mol Biol. 2000;46:573–585. [PubMed] [Google Scholar]
- 22.Maulik N, Engelman DT, Watanabe M, Engelman RM, Das DK. Nitric oxide-a retrograde messenger for carbon monoxide signaling in ischemic heart. Mol Cell Biochem. 1996;157:75–86. doi: 10.1007/BF00227883. [DOI] [PubMed] [Google Scholar]
- 23.May BK, Bhasker CR, Bawden MJ, Cox TC. Molecular regulation of 5-aminolevulinate synthase. Diseases related to heme biosynthesis. Mol Biol Med. 1990;7:405–421. [PubMed] [Google Scholar]
- 24.Meng W, Tobin JR, Busija DW. Glutamate-induced cerebral vasodilation is mediated by nitric oxide through N-methyl-D-aspartate receptors. Stroke. 1995;26:857–862. doi: 10.1161/01.str.26.5.857. [DOI] [PubMed] [Google Scholar]
- 25.Motterlini R, Foresti R, Intaglietta M, Winslow RW. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to endothelium. Am J Physiol. 1996;270:H107–H114. doi: 10.1152/ajpheart.1996.270.1.H107. [DOI] [PubMed] [Google Scholar]
- 26.Parfenova H, Daley ML, Carratu P, Leffler CW. Heme oxygenase inhibition reduces neuronal activation evoked by bicuculline in newborn pigs. Brain Res. 2004;1014:87–96. doi: 10.1016/j.brainres.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 27.Parfenova H, Fedinec A, Leffler CW. Ionotropic glutamate receptors in cerebral microvascular endothelium are functionally linked to heme oxygenase. J Cerebral Blood Flow Metab. 2003;23:190–197. doi: 10.1097/01.WCB.000004823561824.C4. [DOI] [PubMed] [Google Scholar]
- 28.Parfenova H, Neff RA, III, Alonso JS, Shlopov BV, Jamal CN, Sarkasova SA, Leffler CW. Cerebrovascular endothelial heme oxygenase: expression, intracellular compartmentalization, and activation by glutamate. Am J Physiol. 2001;281:C1954–C1963. doi: 10.1152/ajpcell.2001.281.6.C1954. [DOI] [PubMed] [Google Scholar]
- 29.Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–7. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- 30.Robinson JC, Fedinec AL, Leffler CW. Role of CO in glutamate receptor-induced dilation of newborn pig pial arterioles. Am J Physiol. 2002;282:H2371–2376. doi: 10.1152/ajpheart.00911.2001. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez F, Lamon BD, Gong W, Kemp R, Nasjletti A. Nitric oxide synthesis inhibition promotes renal production of carbon monoxide. Hypertension. 2004;43:347–351. doi: 10.1161/01.HYP.0000111721.97169.97. [DOI] [PubMed] [Google Scholar]
- 32.Schneider JC, Kebir DE, Chereau C, Lanone S, Huang XL, De Buys Roessingh AS, Mercier JC, Dall’Ava-Santucci J, Dinh-Xuan AT. Involvement of Ca2+/calmodulin-dependent protein kinase II in endothelial NO production and endothelium-dependent relaxation. Am J Physiol. 2003;284:H2311–H2319. doi: 10.1152/ajpheart.00932.2001. [DOI] [PubMed] [Google Scholar]
- 33.Sharp CD, Houghton J, Elrod JW, Warren A, Jackson TH, IV, Jawahar A, Nanda A, Minagar, Alexander JS. N-methyl-D-aspartate receptor activation in human cerebral endothelium promotes intracellular oxidant stress. Am J Physiol. 2005;288:H1893–H1899. doi: 10.1152/ajpheart.01110.2003. [DOI] [PubMed] [Google Scholar]
- 34.Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877–96. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 35.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nature Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 36.Stella N, Tence M, Glowinski J, Premont J. Glutamate-evoked release of arachidonic acid from mouse brain astrocytes. J Neurosci. 1994;14:568–75. doi: 10.1523/JNEUROSCI.14-02-00568.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szabadkai G, Simoni AM, Rizzuto R. Mitochondrial Ca2+ uptake requires sustained Ca2+ release from endoplasmic reticulum. J Biol Chem. 2003;278:15153–15161. doi: 10.1074/jbc.M300180200. [DOI] [PubMed] [Google Scholar]
- 38.Thorup C, Jones CL, Gross SS, Moore LC, Goligorsky MS. Carbon monoxide induces vasodilation and nitric oxide rrelease but suppresses endothelial NOS. Am J Physiol. 1999;277:F882–F889. doi: 10.1152/ajprenal.1999.277.6.F882. [DOI] [PubMed] [Google Scholar]
- 39.Williams JM, Hull AD, Pearce WJ. Maturational modulation of endothelial- dependent vasodilatation in ovine cerebral arteries. Am J Physiol. 2005;288:R149–R157. doi: 10.1152/ajpregu.00427.2004. [DOI] [PubMed] [Google Scholar]
- 40.Willis AP, Leffler CW. Nitric oxide and prostanoids: age-dependence of hypercapnic- and histamine-induced dilations of pig pial arterioles. Am J Physiol. 1999;277:H299–H307. doi: 10.1152/ajpheart.1999.277.1.H299. [DOI] [PubMed] [Google Scholar]
- 41.Winestone JS, Bonner C, Leffler CW. Carbon monoxide as an attenuator of vasoconstriction in piglet cerebral circulation. Experimental Biology and Medicine. 2003;228:46–50. doi: 10.1177/153537020322800106. [DOI] [PubMed] [Google Scholar]
- 42.Wolf MJ, Gross RW. The calcium-dependent association and functional coupling of calmodulin with myocardial phospholipase A2. J Biol Chem. 1996;271:20989–20992. doi: 10.1074/jbc.271.35.20989. [DOI] [PubMed] [Google Scholar]
- 43.Xu HL, Feinstein DL, Santizo RA, Koenig HM, Pelligrino DA. Agonist-specific differences in mechanisms mediating eNOS-dependent pial arteriolar dilation in rats. Am J Physiol. 2002;282:H237–H243. doi: 10.1152/ajpheart.2002.282.1.H237. [DOI] [PubMed] [Google Scholar]
- 44.Yang B, Wang Y, Cynader MS. Synergistic interactions between noradrenaline and glutamate in cytosolic calcium influx in cultured visual cortical neurons. Brain Res. 1996;721:181–90. doi: 10.1016/0006-8993(96)00047-9. [DOI] [PubMed] [Google Scholar]
- 45.Yang G, Zhang Y, Ross ME, Iadecola C. Attenuation of activity –induced increases in cerebellar blood flow in mice lacking neuronal nitric oxide synthase. Am J Physiol. 2003;285:H298–H304. doi: 10.1152/ajpheart.00043.2003. [DOI] [PubMed] [Google Scholar]
- 46.Yi FX, Magness RR, Bird IM. Simultaneous imaging of [Ca 2+] and intracellular NO production in freshly isolated uterine artery endothelial cells: effects of ovarian cycle and pregnancy. Am J Physiol. 2005;288:R140–R148. doi: 10.1152/ajpregu.00302.2004. [DOI] [PubMed] [Google Scholar]
- 47.Zuckerman SL, Armstead WM, Hsu P, Shibata M, Leffler CW. Age dependence of cerebrovascular response mechanisms in the domestic pig. Am J Physiol. 199;271:H535–H540. doi: 10.1152/ajpheart.1996.271.2.H535. [DOI] [PubMed] [Google Scholar]