

Abstract
Aberrant accumulation of the Myc oncoprotein propels proliferation and induces carcinogenesis. In normal cells, however, an abundance of Myc protein represses transcription at the c-myc locus. Cancer cells often lose this autorepression. We examined the control of myc in Drosophila and show here that the Drosophila ortholog, dmyc, also undergoes autorepression. We find that the developmental repressor Polycomb (Pc) is required for dmyc autorepression, and that this Pc-dMyc-mediated repression spreads across an 875-kb region encompassing the dmyc gene. To further investigate the relationship between Myc and Polycomb, we used microarrays to identify genes regulated by each, and identify a striking relationship between the two: A large set of dMyc activation targets is normally repressed by Pc, and 73% of dMyc repression targets require Pc for this repression. Chromatin immunoprecipitation confirmed that many dMyc-Pc-repressed loci have an epigenetic mark recognized by Pc. Our results suggest a novel relationship between Myc and Polycomb, wherein Myc enhances Polycomb repression in order to repress targets, and Myc suppresses Polycomb repression in order to activate targets.
Keywords: Myc, Pc, transcription, repression, autorepression, activation
The biological activity of the c-Myc protein is required for growth and proliferation, and its overexpression interferes with differentiation. Myc is a transcription factor with a C-terminal basic helix-loop-helix zipper (bHLHZ), and together with its dimerization partner Max, binds to E-boxes and activates transcription of many target genes (Oster et al. 2002). Myc also acts as a repressor by a mechanism that is less well understood than its mechanism of activation. An interesting aspect of Myc biology is that Myc represses the c-myc locus itself. Autorepression occurs at the level of c-myc transcription initiation and requires cellular factors in addition to Myc protein (Penn et al. 1990). In order to determine the mechanism of myc autorepression, we have studied the myc ortholog in Drosophila melanogaster (dmyc), which shows many structural and functional similarities to its vertebrate counterpart (Gallant et al. 1996; Schreiber-Agus et al. 1997). In addition, dmyc behaves similarly to c-myc in cell transformation assays by collaborating with RAS to generate transformed foci in rat embryonic fibroblasts (Schreiber-Agus et al. 1997). Infection of c-myc-null mouse cells with a dmyc-expressing retrovirus rescues their growth defect (Trumpp et al. 2001). In Drosophila, strong mutant alleles of dmyc cause defects in cell growth and endoreplication in the ovary and larva, and are lethal (Maines et al. 2004; Pierce et al. 2004).
Results and Discussion
To test the ability of ectopic dmyc expression to cause repression of normal dmyc expression, we used the Gal4 UAS system to express ectopic dmyc in embryos. We used RT-PCR to amplify endogenous dmyc and exogenous dmyc, using dRas64B as a control for RNA levels. We observed transcription of UAS dmyc by 6 h at 18°C, and at this stage a twofold reduction of the endogenous dmyc transcript was evident, followed by a threefold reduction by 12-18 h (Fig. 1A). We examined endogenous dmyc transcripts in situ for any tissue specificity of dmyc autorepression in the embryo (Fig. 1B, top panel). Ectopic dMyc reduces endogenous dmyc transcripts overall and throughout embryogenesis, with no apparent tissue specificity (Fig. 1B, bottom panel). We conclude that dmyc undergoes autorepression as does mammalian c-myc and that all cells of the embryo respond similarly.
Figure 1.

Ectopic dMyc induces autorepression of dmyc. (A) Endogenous dmyc is repressed upon ectopic dmyc expression. RT-PCR amplification products are shown of RNA from wild-type (lanes 1,3,5,7) or ectopic dmyc (lanes 2,4,6,8) embryos at 0-6 h (lanes 1,2), 6-12 h (lanes 3,4), 12-18 h (lanes 5,6), and 18-24 h (lanes 7,8) after egg laying, at 18°C. (Top) Ectopic dmyc was amplified using a forward primer specific for an epitope tag present in the transgene. (Middle) Endogenous dmyc was amplified using a forward primer specific for the dmyc 5′-UTR. (Bottom) dRas64B was amplified with dRas64B-specific oligonucleotides as a control for RNA levels. (B) Autorepression in situ: stage 10 embryos stained with a riboprobe specific for the dmyc 5′-UTR in wild-type (top) and ectopic dmyc (bottom) embryos. The region complementary to the probe is not included in the UAS dmyc transgene.
In a large-scale screen for modifiers of dmyc expression using dmycBG02383 (FlyBase, unpubl.), we found that the deficiency Df(3L)fz-CAL5 behaved as a repressor of dmyc expression. Landecker and colleagues found that this deficiency enhances the heterozygous phenotype of strong mutations in Polycomb group genes, and has homeotic phenotypes of its own (Landecker et al. 1994). We found similar effects on dmyc expression with alleles of Pc and Psc (data not shown). In parallel with further genetic investigations into these effects, we initiated a more direct analysis, using RNA interference (RNAi) to reduce Pc levels and test whether the repressive effects of PcG genes were relevant to the mechanisms responsible for dmyc autorepression. Injection of Pc dsRNA produced a 2.3-fold reduction of Pc transcript levels in the embryo (Fig. 2A). This reduction in Pc levels had no effect on dmyc expression in embryos that did not express ectopic dmyc. In control buffer-injected embryos, ectopic dmyc expression led to a 45% decrease in endogenous dmyc levels in the particular experiment shown (Fig. 2B, left panel). Injection of Pc dsRNA blocked this repression, and endogenous dmyc levels were indistinguishable from those in embryos without ectopic Myc (Fig. 2B, right panel). We repeated the experiment, obtaining the same results with RNA from a total of >1400 injected embryos.
Figure 2.

Pc is required for dmyc autorepression. (A) Pc depletion in Pc RNAi embryos, as shown by RT-PCR amplification of Pc (top) and dRas64B (bottom), in embryos injected with buffer (bands on left) and Pc dsRNA (bands on right). These embryos all have ectopic dmyc. (B) Pc RNAi eliminates dmyc autorepression, shown by RT-PCR amplification of endogenous dmyc in wild-type embryos (bands on the left for each panel) or ectopic dmyc embryos (bands on the right for each panel), injected with buffer (left panel) or Pc dsRNA (right panel). Ras64B is shown as a control for RNA levels (lower bands in both panels).
We were curious whether dMyc-induced, Pc-mediated repression shares known characteristics of Pc repression. Specifically, we were interested in the possibility of long-range effects surrounding the dmyc locus, possibly mediated by chromatin reorganization or compaction in the region (Gerasimova and Corces 1998; Francis et al. 2004). We used quantitative RT-PCR to test 14 genes in this region, and of those, 11 were repressed by dMyc, including dmyc itself (Fig. 3A). This repression of the region depends on wild-type levels of Pc, because embryos with ectopic dMyc and Pc RNAi failed to exhibit repression of dmyc and its 10 neighbors (Fig. 3B). The regulation of the region is more complicated than repression by global compaction, however, because we found two genes that are dMyc induced (CG3588 and dnc), and one gene whose expression levels remained unchanged (CG33221). All three genes remained unaffected by the presence or absence of Pc.
Figure 3.

dMyc-mediated repression of dmyc affects cytological region 3C3-3F2. (A) Buffer-injected embryos of either wild type (all bands on the left) or ectopic dmyc (all bands on the right) provided RNA to amplify the following genes in the region of 3C3-3F2, distal to proximal: CG3588, rst, kirre, N, CG3939, dnc, CG14265, CG10793, dmyc, CG14269, CG16782, Rala, CG33221, and fd3F. (Upper right) Pc transcripts are shown by RT-PCR in the box. The X chromosome is depicted by the thick black line. The numbers above or below each band are the relative band intensities between pairs, normalized to the control reaction (dRas64B). The sequence location of the region along the X chromosome for Release 4 is indicated in kilobases. (B) Repression of 3C3-3F2 by dmyc is mediated by Pc. Pc RNAi embryos of either wild type (all bands on the left) or expressing ectopic dmyc (all bands on the right) provided a template to amplify the genes shown in A. (Upper right) Pc transcripts are shown in the box. The numbers above or below each band are the relative band intensities between pairs, normalized to the control, Ras64B, reaction.
To investigate the general relationship between Polycomb and dMyc, we examined the effect of Pc on a large number of dMyc-responsive genes in the Drosophila genome. First, we used Affymetrix Genome Arrays to examine the effect of dmyc on gene expression by comparing RNAs from Gal4 embryos with and without UAS dmyc. We found that 272 of 7421 detectable genes were up-regulated in response to Myc (1.9-fold or greater) (Materials and Methods; Supplementary Data 1). We then identified Pc embryonic targets on Affymetrix arrays, using cDNA from Gal4 embryos injected with Pc dsRNA to reduce levels of Pc transcripts. Of the 7421 genes detected on our arrays, 214 genes were elevated upon reduction of Pc (Materials and Methods; Supplementary Data 2). Surprisingly, among those 214, almost half (95) were also among those activated by ectopic dMyc (Fig. 4A,B). The probability that 95 genes could randomly belong to these two separate groups (272 dMyc activated and 214 Pc repressed among 7421 genes) is represented by the P-value of 10-83 by the hypergeometric distribution. These data suggest that a normal and substantial role for Pc in the cell is to repress more than a third of potential Myc activation targets, and that overexpression of dmyc may release that repression. The expression of these 95 genes in an embryo having both Pc inactivation and dmyc overexpression is somewhat higher than with either inducer/repressor alone, but not to the degree that would suggest two parallel pathways functioning additively (Fig. 4B).
Figure 4.

Microarray data illustrate that a subset of dMyc activation targets is normally repressed by Pc, and a majority of dMyc repression targets require Pc for repression. (A) A Venn diagram shows that dMyc-repressed genes and Pc-repressed genes overlap by a very small number of genes, within the realm of random chance (P = 0.08). In contrast, dMyc-activated genes and Pc-repressed genes overlap by a highly significant amount (P = 10-83). (B,C) Each line illustrates the log10 of normalized intensity of one gene for eight different chip hybridizations: Gal4, buffer injected (GB) and repeat (GB2); Gal4, Pc RNAi (GP) and repeat (GP2); Gal4, UAS dmyc, buffer injected (GMB) and repeat (GMB2); Gal4, UAS dmyc, Pc RNAi (GMP) and repeat (GMP). Each gene in B is dMyc-activated and also Pc-repressed (95 genes), and each gene shown in C is dMyc-repressed (129 genes).
The Affymetrix arrays also allowed the characterization of genes whose expression is down-regulated by Myc. Among the 7421 genes detected on our arrays, 129 were repressed by a factor of 0.533 or more by the ectopic accumulation of dMyc (Fig. 4C; Materials and Methods; Supplementary Data 3). Since we had evidence that dmyc autorepression was Polycomb-dependent (Fig. 2), we compared expression profiles from UAS dmyc-expressing embryos injected with Pc dsRNA to those from UAS dmyc embryos injected with buffer. Depletion of Pc by RNAi eliminates dMyc repression of 73% (94) of 129 targets (Fig. 4C). These results argue that wild-type Polycomb activity is required for ectopic dMyc to repress target gene expression. However, repression by Pc in the absence of ectopic dMyc does not affect these targets, given that only seven genes that are repressed by ectopic dMyc are also among the 214 Pc repression targets (Fig. 4A).
Pc protein binds to methylated histone H3 at Lys 27 (H3K27), a modification that correlates with its repressive activity (Cao et al. 2002; Czermin et al. 2002; Muller et al. 2002; Fischle et al. 2003; Min et al. 2003; Ringrose et al. 2004). To determine whether the behavior of target genes could be correlated with this chromatin modification, we examined the methylation status of four of the dMyc repression targets, as well as two of the Pc repression targets. We used an antibody raised against trimethyl-H3K27, and immunoprecipitated chromatin from 24-h collections of Gal4 and Gal4; UAS dmyc embryos. Anti-trimethyl H3K27 precipitated chromatin in the region of both Pc-repressed genes, CG2065 and Cyp309a1, as we expected (Fig. 5A). The four dMyc-repressed genes, all of which required Pc for repression by dMyc, were also precipitated by anti-trimethyl H3K27, including dmyc (dmyc, CG5002, CG18108, and Hsp70Ab) (Fig. 5B). As a control, we tested three genes whose levels are unaffected by Pc or dMyc, and found that the antibody fails to bind above background levels to chromatin in the region of these genes (CG12703, hsp68, and Ras64B) (Fig. 5C). Similar results were obtained with chromatin from either Gal4 or Gal4;UAS dmyc embryos. The fact that ectopic Myc itself does not increase histone methylation of its targets suggests that those targets may have been previously marked to allow their repression by Polycomb. In this view, ectopic dMyc would build on or mobilize the Pc-repressive complex to decrease transcription at such loci.
Figure 5.
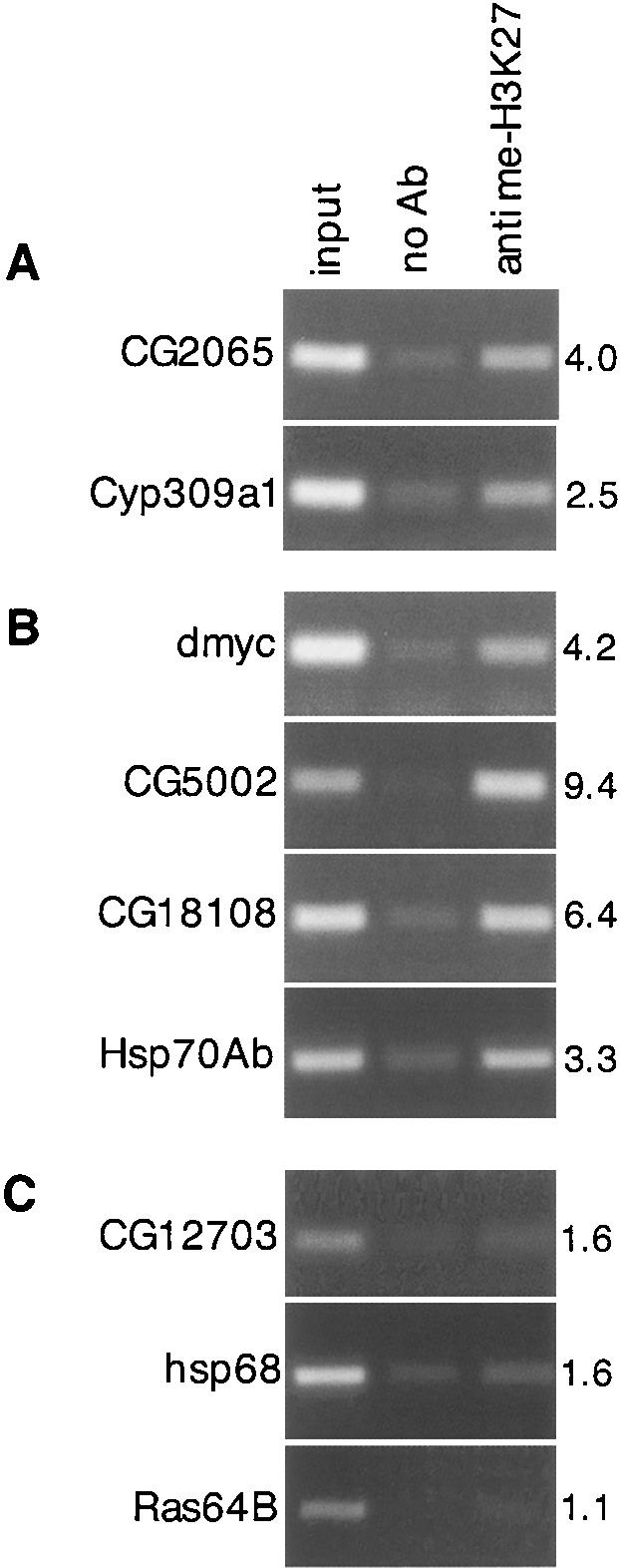
Pc- and dMyc-repressed genes are trimethylated at histone H3 Lys 27. (A-C) Amplified genomic DNA pulled down by ChIP for each of two Pc-repressed (A), four dMyc-repressed (B), and three unaffected (C) genes is shown. Input DNA from sonicated chromatin is shown on the left, ChIP with no antibody is shown in the middle, and ChIP with anti-trimethyl H3K27 is shown on the right. The genotype of the embryos used in this experiment was arm-Gal4. The numbers on the right show the fold changes in band intensities from the second to third lines.
The first Myc-regulated gene ever identified was c-myc itself (Cole 1986). The mechanism of autorepression has remained elusive, and the present study offers new insight into this feedback regulatory loop. We show that myc autorepression is conserved from mammals to flies and that it requires the Pc complex. The myc autoregulation loop is frequently disrupted in cancer cells, and furthermore, it has been suggested that gene repression correlates better with Myc biological activity than does gene activation (Grignani et al. 1990; Penn et al. 1990; Xiao et al. 1998). Our data suggest that autorepression and general repression by Myc are mediated by the same mechanism and that both are dependent on the PcG. Indeed, we show that dMyc repressed genes have the hallmark chromatin modification of Pc-repressed genes. Members of the PcG have previously been implicated in cancer, including Bmi-1 (homologous to Psc), which cooperates with Myc in lymphomagenesis and represses expression of the p16 CDK inhibitor (van Lohuizen et al. 1991; Jacobs et al. 1999). However, no previous connection has been made between general Myc-mediated repression and the PcG. The large chromosomal domain surrounding the dmyc locus that is repressed in concert with dmyc itself is consistent with a PcG-mediated mechanism, since repression by Pc is known to act over long distances (Lewis 1978). Interestingly, repression within this domain is not absolute, since some interspersed genes can resist repression or even be activated. We cannot exclude the possibility that each of the genes in the domain is independently repressed by elevated dMyc expression, but their proximity to dmyc itself seems more consistent with a regional effect.
An unexpected outcome of our studies was the striking observation that one-third of the genes that score as dMyc-activated in early stage embryos were also scored as repressed by Pc, since ablation of Pc by RNAi activated the genes to a similar extent as transgenic dmyc overexpression. Similarly, approximately one-half of the Pc repressed genes were also activated by transgenic dmyc overexpression. The overlap in these two gene sets is statistically highly significant and suggests a mechanistic overlap in the gene response. Since dmyc overexpression was provided via transgene, whereas ablation of Pc was achieved by RNAi, the overlap in gene response is unlikely to be a consequence of experimental manipulation. We have not yet determined if this response is a direct effect of either dMyc or Pc binding to the corresponding genes. Nevertheless, the microarray data suggest that, at the minimum, the two pathways converge on a common cellular network.
For both dMyc-activated and -repressed genes, the Polycomb complex provides an essential context for Myc regulation, but the direction of that regulation depends on Myc itself and the nature of its interaction with a particular target. In the simplest view, Myc repression might work by enhancing Pc's generally negative effects on transcription, whereas it appears to activate other genes by opposing those same effects.
Materials and methods
Fly strains
See FlyBase (http://flybase.bio.indiana.edu) for additional information about the mutants and insertions used. Females with the Gal4 driver matTub-Gal4VP16 67C;15 (homozygous on II and III) were crossed to UAS-dmyc (Johnston et al. 1999) males. Females homozygous for an armadillo-Gal4 driver (Sanson et al. 1996) were crossed to males homozygous for UAS-dmyc for ectopic expression of dmyc in dsRNA-injected embryos. Control embryos were laid from females of either Gal4 driver corresponding to the one used to drive UAS-dmyc in the experiment.
RNA isolation and RT-PCR
We used TRIzol reagent (Invitrogen) to isolate total RNA from dechorionated embryos. RT-PCR reactions were performed using Invitrogen's SuperScript One Step RT-PCR system, according to the manufacturer's instructions. Sense primers were end-labeled using T4 Polynucleotide Kinase (NEB) and [γ-32P]ATP (Perkin Elmer). Primer pairs for all amplification products spanned an intron to control for DNA contamination. The forward primer for endogenous dmyc amplification recognized the 5′-UTR of the dmyc transcript, which is not present in the UAS dmyc transgene. We monitored the ectopic expression of dmyc using a forward primer that would anneal to the c-myc epitope tag (9E10) of the dmyc transgene. We minimized amplification cycles in order to remain in the linear range of amplification for each gene. Please see sequences for oligonucleotides in Supplemental Material.
In situ hybridization
We amplified the dmyc 5′-UTR using the oligonucleotides dmyc.probe.5 (5′-CGACTGGAAAGCAAAGGAAG-3′) and dmyc.probe.3 (5′-GCGATTATGTTGTCTGGGTTT-3′), which is absent in the dmyc transgene. We cloned the product using the pGem T Easy vector (Promega), and synthesized digoxigenin-labeled antisense riboprobes using T7 RNA polymerase and digoxigenin-UTP (Roche). The protocol was performed as previously described (Lehmann and Tautz 1994).
RNAi
We amplified 500 base pairs (bp) of a 5′ region of the Pc cDNA using the oligonucleotides PcS.Xho (5′-CCCTTTCTCGAGATGACTGGTCGAGGCAAGGGG-3′) and PcA.Bcl (5′-GGAGGAGCTGTGATCAATGCG 3′), and cloned the product using the pGem T Easy vector (Promega). We synthesized the sense and antisense transcripts separately using the dual promoters of pGem T Easy, SP6 and T7 polymerases (Ambion), and annealed the transcripts to each other. As previously described (Kennerdell and Carthew 1998), we injecteda5μM solution of Pc dsRNA into embryos with either ectopic Gal4 alone or Gal4 and UAS dmyc, and isolated RNA from the embryos after 20 h at 18°C. Injected embryos were washed off the coverslip with heptane to solubilize the glue and oil.
Microarray
All RNA was prepared from stage 10-11 embryos, having either a Gal4 transgene or having both a Gal4 and UAS dmyc transgene. We generated probes by synthesizing cDNA from 5 μg of total RNA (Invitrogen), followed by biotinylated cRNA synthesis (ENZO). We hybridized Affymetrix Drosophila Genome 1 chips, detected by Affymetrix specifications, and scanned using an Agilent DNA microarray scanner and Affymetrix GeneChip software. We used GeneSpring and Excel for data analysis.
We wanted to eliminate genes with low signal intensities before we conducted our analysis, but we also wanted to retain genes whose levels changed dramatically with changes in dMyc or Pc levels. Therefore, we eliminated genes from our analysis whose signals summed <1500 for eight hybridizations. We chose the average signal per chip per gene of 187.5 (1500/8) because it is more than double the highest intensity of any negative control for the arrays. Summing the intensities of eight hybridizations allowed us to retain genes with dramatic changes in expression with ectopic Myc or depleted Pc. We thus eliminated 6546 genes from our analysis, and we used levels of the remaining 7421 exclusively.
Changes in expression were determined by first averaging the signal intensities for duplicate hybridizations (biological replicates), then calculating ratios of expression levels, such that we had log2 ratios of Gal4 UAS dmyc, buffer injected/Gal4, buffer injected; Gal4 UAS dmyc, Pc RNAi/Gal4, buffer injected; Gal4, Pc RNAi/Gal4, buffer injected for each gene. We computed standard deviations for each of the lists of ratios, and considered genes with ratios 1.5 times the standard deviation in either direction to exhibit significantly different expression. We used two times the standard deviation for our analysis of Pc repression targets. All of our lists of dMyc and Pc targets are available in the Supplemental Material. Quantitative RT-PCR validation of four dMyc-repressed genes, dependent on Pc, is shown in Supplementary Figure 1. The genomic locations of Myc-repressed and Pc-repressed genes are shown in Supplementary Figure 2.
Chromatin immunoprecipitation (ChIP)
We homogenized and sonicated dechorionated embryos in SDS Lysis buffer (Upstate), and proceeded with the protocol as directed, using Upstate's ChIP Assay Kit (catalog #17-295). Anti-trimethyl H3K27 (Upstate) was used at a 1:50 dilution.
Supplementary Material
Acknowledgments
We thank Peter Gallant and Allen Shearn for flies, Mike Beer for assistance in microarray data analysis and comments on the manuscript, Robert Carroll for assistance cloning Pc, Topher Gee for help with statistical analysis, and Anna Sokac for comments on the manuscript. This work was supported by grants from the NCI to M.D.C. and E.W. E.W. is an investigator of the Howard Hughes Medical Institute.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1352305.
References
- Cao R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., Jones, R.S., and Zhang, Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039-1043. [DOI] [PubMed] [Google Scholar]
- Cole M.D. 1986. The myc oncogene: Its role in transformation and differentiation. Annu. Rev. Genet. 20: 361-384. [DOI] [PubMed] [Google Scholar]
- Czermin B., Melfi, R., McCabe, D., Seitz, V., Imhof, A., and Pirrotta, V. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185-196. [DOI] [PubMed] [Google Scholar]
- Fischle W., Wang, Y., Jacobs, S.A., Kim, Y., Allis, C.D., and Khorasanizadeh, S. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes & Dev. 17: 1870-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis N.J., Kingston, R.E., and Woodcock, C.L. 2004. Chromatin compaction by a polycomb group protein complex. Science 306: 1574-1577. [DOI] [PubMed] [Google Scholar]
- Gallant P., Shiio, Y., Cheng, P.F., Parkhurst, S.M., and Eisenman, R.N. 1996. Myc and Max homologs in Drosophila. Science 274: 1523-1527. [DOI] [PubMed] [Google Scholar]
- Gerasimova T.I. and Corces, V.G. 1998. Polycomb and trithorax group proteins mediate the function of a chromatin insulator. Cell 92: 511-521. [DOI] [PubMed] [Google Scholar]
- Grignani F., Lombardi, L., Inghirami, G., Sternas, L., Cechova, K., and Dalla-Favera, R. 1990. Negative autoregulation of c-myc gene expression is inactivated in transformed cells. EMBO J. 9: 3913-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.J., Kieboom, K., Marino, S., DePinho, R.A., and van Lohuizen, M. 1999. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397: 164-168. [DOI] [PubMed] [Google Scholar]
- Johnston L.A., Prober, D.A., Edgar, B.A., Eisenman, R.N., and Gallant, P. 1999. Drosophila myc regulates cellular growth during development. Cell 98: 779-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerdell J.R. and Carthew, R.W. 1998. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell 95: 1017-1026. [DOI] [PubMed] [Google Scholar]
- Landecker H.L., Sinclair, D.A., and Brock, H.W. 1994. Screen for enhancers of Polycomb and Polycomblike in Drosophila melanogaster. Dev. Genet. 15: 425-434. [DOI] [PubMed] [Google Scholar]
- Lehmann R. and Tautz, D. 1994. In situ hybridization to RNA. Methods Cell Biol. 44: 575-598. [DOI] [PubMed] [Google Scholar]
- Lewis E.B. 1978. A gene complex controlling segmentation in Drosophila. Nature 276: 565-570. [DOI] [PubMed] [Google Scholar]
- Maines J.Z., Stevens, L.M., Tong, X., and Stein, D. 2004. Drosophila dMyc is required for ovary cell growth and endoreplication. Development 131: 775-786. [DOI] [PubMed] [Google Scholar]
- Min J., Zhang, Y., and Xu, R.M. 2003. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes & Dev. 17: 1823-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J., Hart, C.M., Francis, N.J., Vargas, M.L., Sengupta, A., Wild, B., Miller, E.L., O'Connor, M.B., Kingston, R.E., and Simon, J.A. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197-208. [DOI] [PubMed] [Google Scholar]
- Oster S.K., Ho, C.S., Soucie, E.L., and Penn, L.Z. 2002. The myc oncogene: MarvelouslY Complex. Adv. Cancer Res. 84: 81-154. [DOI] [PubMed] [Google Scholar]
- Penn L.J., Brooks, M.W., Laufer, E.M., and Land, H. 1990. Negative autoregulation of c-myc transcription. EMBO J. 9: 1113-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce S.B., Yost, C., Britton, J.S., Loo, L.W., Flynn, E.M., Edgar, B.A., and Eisenman, R.N. 2004. dMyc is required for larval growth and endoreplication in Drosophila. Development 131: 2317-2327. [DOI] [PubMed] [Google Scholar]
- Ringrose L., Ehret, H., and Paro, R. 2004. Distinct contributions of histone H3 lysine 9 and 27 methylation to locus-specific stability of polycomb complexes. Mol. Cell 16: 641-653. [DOI] [PubMed] [Google Scholar]
- Sanson B., White, P., and Vincent, J.P. 1996. Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383: 627-630. [DOI] [PubMed] [Google Scholar]
- Schreiber-Agus N., Stein, D., Chen, K., Goltz, J.S., Stevens, L., and DePinho, R.A. 1997. Drosophila Myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc. Natl. Acad. Sci. 94: 1235-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A., Refaeli, Y., Oskarsson, T., Gasser, S., Murphy, M., Martin, G.R., and Bishop, J.M. 2001. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768-773. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M., Verbeek, S., Scheijen, B., Wientjens, E., van der Gulden, H., and Berns, A. 1991. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 65: 737-752. [DOI] [PubMed] [Google Scholar]
- Xiao Q., Claassen, G., Shi, J., Adachi, S., Sedivy, J., and Hann, S.R. 1998. Transactivation-defective c-MycS retains the ability to regulate proliferation and apoptosis. Genes & Dev. 12: 3803-3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.