

Abstract
B cells are essential to the immune response in health and disease. Results from knockout (KO) mice for different members of the nuclear factor-κB (NF-κB) family have highlighted the importance of this transcription factor in B cell development and function. The recent generation of additional KO mice for adapters and kinases implicated in NF-κB activation, including several protein kinase C isoforms, has provided new insights into the roles of these proteins in B cell signalling. These studies have also given rise to a number of important questions that must be answered with further experimentation to establish accurately the signalling pathways that regulate B-cell function through NF-κB.
Introduction
It is now widely accepted that nuclear factor-κB (NF-κB) complexes are central to the control of not only the innate but also the acquired immune response. Various NF-κB forms control different aspects of the development and function of the immune system. Of particular relevance to this review is the crucial role that the different subunits of this important transcription factor have in B-cell function, both in vitro and in vivo (reviewed in Gerondakis et al. (1999) and Gugasyan et al. (2000)). NF-κBs are dimers consisting of a variety of combinations of different Rel proteins: RelA (p65), c-Rel, RelB, NF-κB1 (p105) and NF-κB2 (p100). The proteolytic processing of NF-κB1 and NF-κB2 gives rise to p50 and p52, respectively, which lack transactivation domains and act as transcriptional repressors when present in the nucleus as homodimers. In combination with the transactivating Rel proteins, however, these truncated NF-κB proteins produce transcriptionally active complexes targeting genes that are important for the efficient activation of the immune response. Indeed, they are required for robust binding of the Rel proteins to the NF-κB enhancer element (Karin & Ben-Neriah, 2000).
Classic and novel NF-κB pathways in B cell function
Evidence from knockout (KO) mice for different Rel proteins shows that they are required for an adequate humoral response in vivo (Gerondakis et al., 1999). For example, mice in which the p100 gene has been deleted show a cell-autonomous defect in Ig heavy-chain constant region (CH) isotype switching, and mice of the severe-combined immunodeficient (SCID) strain into which fetal liver cells from relA−/− mice have been transplanted have revealed a defect in the ability of B cells to secrete IgA and IgG1. When the c-rel gene is deleted, B cells display defects consistent with a role for this protein in germline transcription of the CH gene. Mice in which deletions of different Rel genes are combined have even more pronounced phenotypes.
RelA- and/or c-Rel-containing NF-κB complexes are retained in the cytosol in a latent inactive form through interaction with the inhibitory protein IκBα (Karin & Ben-Neriah, 2000). These complexes are released only after IκBα has been phosphorylated, and thereby targeted for ubiquitination and proteasome-mediated degradation, by the IκB kinase (IKK) complex. Once released, NF-κB can enter the nucleus, where it can activate transcription provided that it is also phosphorylated (Karin and Ben-Neriah, 2000).
The IKK complex is formed by two catalytic subunits (IKKα and IKKβ) and a regulatory protein named IKKγ, IKKAP or Nemo. The physiological roles of each of these subunits have been revealed by studies of various KO mice (Ghosh & Karin, 2002). Interestingly, from these experiments it is now clear that IKKβ and IKKγ are the main determinants of the ability of the IKK complex to phosphorylate IκBα (see Fig. 1; Israel, 2000). Furthermore, lethally irradiated mice that are reconstituted with ikkβ−/− stem cells are almost completely devoid of both T and B lymphocytes (Horwitz et al., 1997; Senftleben et al., 2001b). The fact that this phenotype is reversed in IKKβ/tumour necrosis factor receptor-1 (TNF-R1) double-KO mice indicates that the absence of B and T cells in the ikkβ−/− reconstituted mice is due to a general role of IKKβ in controlling TNF-α-induced apoptosis (Senftleben et al., 2001b). These studies therefore show that IKKβ has a crucial role in the development of the haematopoietic compartment.
Figure 1.
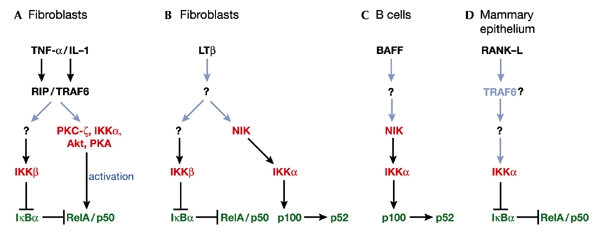
NF-κB signalling pathways in different cell systems. In fibroblasts, the adapters receptor-interacting protein (RIP) and TRAF6 are important intermediaries for the activation of NF-κB complexes (RelA/p50 heterodimers), in response to TNFα and IL-1, respectively (A). Both the inhibition of IκBα by IKKβ and the phosphorylation of RelA by several agents regulate NF-κB activity in this system. In LTβ-activated EFs (B) and in BAFF-stimulated B cells (C), IKKα phosphorylates and triggers the processing of p100, generating a p52-containing complex that might include the transactivating RelB subunit. In mammary epithelial cells (D), it has been proved genetically that IKKα is an IκB kinase in the RANK signalling pathway. It is still unclear whether TRAF6 is upstream of IKKα in this system. Triggers are indicated in black, kinases in red, activities in dark blue, and NF-κB proteins in green; established pathways are indicated by black arrows, and unknown pathways by light blue arrows.
The IKKα subunit KO revealed several surprising results, considering what had been determined for IKKβ. First, the lack of IKKα did not impair the IκB kinase activity of the IKK complex in embryonic fibroblasts (EFs) stimulated by TNF-α or interleukin-1 (IL-1) (Fig. 1A; Hu et al., 2001; Takeda et al., 1999). However, in other systems such as the mammary epithelia (Fig. 1D), IKKα was required for IκB phosphorylation in response to activation of the receptor activator of nuclear factor κ (RANK), a member of the TNF-receptor superfamily (Cao et al., 2001). These results reinforce the notion that different kinase subunits might have distinct roles in different cell systems and organs. Perhaps the most interesting information gathered from the IKKα KO mice relates to the function of B cells. Lethally irradiated mice reconstituted with IKKα−/− stem cells display intact development and proliferation of T cells, but have important defects in B-cell maturation and also in the T-cell-dependent immune response (Kaisho et al., 2001; Senftleben et al., 2001a). Unlike the immunesystem defects observed in the IKKβ-deficient animals, the IKKα-related defects in B-cell maturation cannot be corrected by the inhibition of apoptosis, as was tested by overexpressing the anti-apoptotic protein Bcl-2. More importantly, these defects are not correlated with a general decrease in NF-κB activation. Biochemically, this is consistent with the notion that IKKα does not always contribute significantly to the IκB-directed kinase activity of the IKK complex. IKKα might nevertheless be important in the transcription of subsets of NF-κB-dependent genes in these cells, as suggested by the fact that stimulation of kinase-inactive IKKα knock-in mice (IKKαAA) by lipopolysaccharide fails to induce several typical NF-κB promoter-regulated genes such as bcl-2, inducible nitric oxide synthase (iNOS) and RANK-L in splenic B cells (Senftleben et al., 2001a). One potential explanation for these results is that IKKα could regulate the transcriptional activity of RelA and/or c-Rel, which are the normal activators of these genes. Consistent with this hypothesis are recent results from IKKα−/− EFs indicating that these cells induce the DNA-binding activity of NF-κB perfectly well but are unable to activate a NF-κB-dependent reporter gene (Sizemore et al., 2001). However, the analyses of the IKKα−/− chimaeras and the IKKαAA mice have led to the discovery of an alternative mechanism for the regulation of NF-κB-dependent genes and B-cell function. Both types of mutant fail to develop Peyer's patches, and display marked alterations in the splenic microarchitecture (Kaisho et al., 2001; Senftleben et al., 2001a), a phenotype similar to those exhibited by the KOs of p100, the lymphotoxin-β (LTβ) receptor and NF-κB-inducing kinase (NIK, initially thought to be an IKK kinase) (Caamano et al., 1998; Franzoso et al., 1998; Futterer et al., 1998; Yin et al., 2001).
The biochemical link between NIK, IKKα and p100 has now been established (illustrated in Fig. 1C). For NIK, co-transfection experiments have shown it to act in the processing of p100 to yield p52 (Xiao et al., 2001), and splenic cells isolated from mice carrying the NIK-deficient aly/aly mutation have a defect in p100 processing. Together, these findings led to the hypothesis that the direct phosphorylation of p100 by NIK is the trigger that promotes its proteolytic cleavage to p52 (Fagarasan et al., 2000; Xiao et al., 2001). The fact that splenic B cells from the IKKα−/− reconstituted chimaeras also are defective in p100 processing suggested that NIK and IKKα might be in the same pathway (Senftleben et al., 2001a). Indeed, the ability of NIK to promote p100 processing is inhibited in EFs from IKKα−/− but not from IKKβ−/− mice (Senftleben et al., 2001a). Furthermore, IKKα directly phosphorylates p100 much more efficiently than it does IκBα, whereas IKKβ has the opposite effect (Senftleben et al., 2001a). Therefore, in different cell systems, three functions related to NF-κB signalling have been assigned to IKKα: NF-κB transcriptional activation, p100 phosphorylation and IκBα phosphorylation (Fig. 1). The IKKα-triggered cleavage of p100, which produces a RelB–p52 complex (Solan et al., 2002), is of particular relevance because RelB does not bind any of the IκB subunits but interacts strongly with p100 (Karin, 1998).
The connection between LTβ receptor activation and this non-canonical p100 pathway has also recently been established, with the demonstration that LTβ receptor activation in EFs, via NIK and IKKα, triggers the processing of p100 to generate p52 (Dejardin et al., 2002). The activation of this cascade is slower than that of the classical IκBα phosphorylating pathway when stimulated by TNF-α, or even by LTβ (Dejardin et al., 2002, Fig. 1B). In B cells, the trigger of the IKKα–p100 cascade has recently been identified as the B-cell survival factor BAFF (also known as Blys). BAFF triggers the NIK–IKKα–p100 pathway in B cells independently of IKKγ, through the BAFF receptor (BAFF-R, also called BR3) (Claudio et al., 2002; Kayagaki et al., 2002) (Fig. 1C). Again, the stimulation of this pathway is slow relative to the canonical IκB phosphorylating cascade and, in this case, requires protein synthesis (Claudio et al., 2002; Kayagaki et al., 2002). The identity of the newly synthesized protein(s) required for the activation of the pathway is not known, and is essential for full comprehension of this novel cascade. In any case, these new mechanistic discoveries provide a molecular rationale for the fact that the KO of the BAFF receptor has a phenotype similar to those exhibited by IKKα and p100 KOs (Schiemann et al., 2001; Thompson et al., 2001). However, other ligands would probably activate this non-canonical NF-κB pathway. In this regard, recent evidence suggests that ligand binding to CD40 also activates p100 processing (Coope et al., 2002).
In view of these new developments, the contribution of IKKα to the control of RelA-mediated transcription in EFs (Sizemore et al., 2001) needs to be re-evaluated. The ability of IKKα to regulate RelA transactivation is intriguing because EFs from the NIK KO respond to short-term stimulation of LTβ-receptor signalling with normal DNA-binding activity of NF-κB, but are impaired in NF-κB-dependent transcriptional activation (Yin et al., 2001). It is therefore still possible that NIK and IKKα might be important not only for p100 processing but also for NF-κB-mediated transcription.
Activation of NF-κB signalling by the B-cell receptor
The survival of peripheral B cells depends on the B-cell receptor (BCR), which, when triggered, potently activates NF-κB and induces the expression of anti-apoptotic NF-κB-dependent genes such as bcl-xL and bcl-2. How the activation of the BCR leads to the stimulation of NF-κB is a matter of intense research. Recent genetic evidence from mice deficient in BCR-pathway components has helped greatly in the understanding of this important process (see Gauld et al. (2002) for details of early membrane-proximal events that take place after BCR engagement), while generating new questions. Central to BCR signalling via NF-κB is the complex formed by the Tec family kinase Btk (Bruton's tyrosine kinase), the adapter BLNK (B-cell linker) and PLC-γ2 (phospholipase Cγ2) (Fig. 2). BLNK is phosphorylated on BCR activation and serves to couple the tyrosine kinase Syk to the activation of PLC-γ2, whose complete stimulation requires the action of Btk. Btk also interacts directly with BLNK through its SH2 domain. Natural mutations in the btk gene lead to the X-linked agammaglobulinaemia syndrome in humans (XLA) and a similar, although less penetrant, phenotype in mice (xid) (Rawlings, 1999; Gauld et al., 2002). btk−/−, blnk−/− and PLC-γ2−/− mice have similar phenotypes, which is consistent with the notion that the three proteins, together with Syk, form a functional complex (Gauld et al., 2002). These phenotypes are characterized by a severe inhibition of NF-κB stimulation in response to BCR engagement, but not in response to activation by 12-O-tetradecanoylphorbol-13-acetate plus ionomycin (Petro et al., 2000; Petro & Khan, 2001; Tan et al., 2001), which directly targets the Ca2+- and diacylglycerol (DAG)sensitive protein kinase C (PKC) isoforms (such as PKC-β). Because the activation of PLC-γ2 provokes the release of Ca2+ and DAG, PKC is a strong candidate as a downstream component of the Btk–BLNK–PLC-γ2 pathway. The first evidence that members of the extended family of PKCs might be essential for B-cell function stems from the initial work of Leitges et al. (1996), who showed that PKC-β KO mice have impaired humoral immune responses and defective IgM-triggered B-cell proliferation, despite responding normally in terms of T-cell activation (Leitges et al., 1996). This phenotype is similar to those of btk−/− and PLC-γ2−/− mice, indicating that PKC-β is a genuine downstream target of the BLNK complex.
Figure 2.
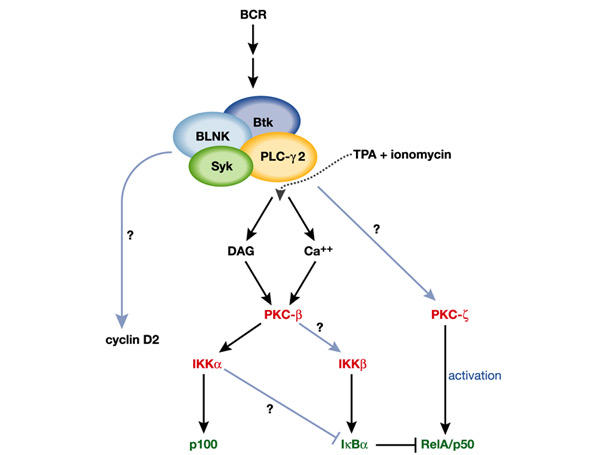
BCR signalling cascades in the activation of NF-κB. Syk, Btk, BLNK, and PLC-γ2 form a functional complex whose signals seem to be mediated by the DAG- and calcium-responsive PKC-β, and whose function can be circumvented through stimulation by 12-O-tetradecanoylphorbol-13-acetate (TPA) plus ionomycin. Some components of this complex also seem to be involved in cell cycle regulation, as demonstrated by their role in cyclin D2 upregulation. Signals from PKC-β are channelled to IKKα or IKKβ either directly or indirectly by controlling the formation of raft complexes, and PKC-ζ most probably controls RelA activity by direct phosphorylation. How PKC-ζ is linked to components of the BCR signalling cascade has not yet been addressed. Triggers are indicated in black, kinases in red, activities in dark blue, and NF-κB proteins in green; established pathways are indicated by black arrows, and unknown pathways by light blue arrows.
PKCs and NF-κB in B cells
Two independent groups have addressed the important question of how the NF-κB pathway is disrupted in PKC-β KO mice. Their main message is that BCR-dependent cell proliferation and survival in these mutants is significantly impaired owing to defective induction of the anti-apoptotic proteins Bcl-xL (Saijo et al., 2002; Su et al., 2002) and Bcl-2 (Saijo et al., 2002). Also noteworthy is the finding that cyclin D2 levels were not affected, indicating that PKC-β is critically involved in B-cell survival but not in cell cycle progression (Su et al., 2002). In marked contrast, blnk−/− B cells show impairment in the induction of both Bcl-xL and cyclin D2 (Tan et al., 2001). This suggests that PKC-β accounts for the control of survival signals emanating from the BLNK complex but not for those that regulate cell growth (Fig. 2).
Interestingly, IκB degradation is impaired in pkcβ−/− B cells activated by IgM crosslinking but not when stimulated through CD40 (Saijo et al., 2002; Su et al., 2002). Su et al. (2002) showed that BCR-induced IKK enzymatic activity in pkcβ−/− B cells is markedly reduced. Together with the fact that IKKα has not been shown to contribute to the IκB kinase activity of the IKK complex (except in mammary epithelial cells; see above), these results suggest that PKC-β directly or indirectly regulates IKKβ activity. However, using anti-phospho-IKK antibodies, Saijo et al. (2002) showed that the pkcβ−/− B cells display only a minor reduction in IKKβ phosphorylation, and a complete inhibition of phospho-IKKα both under basal conditions and when stimulated. The marked inhibition of IKKα phosphorylation is consistent with the observed defect in p100 processing detected in the PKC-β-deficient B cells (Saijo et al., 2002). However, the small decrease in IKKβ phosphorylation in these cells is difficult to reconcile with the lack of IKK activity (Su et al., 2002). It might be that in B cells this phosphorylation event is necessary but not sufficient for IKK activation. Alternatively, it is possible that, as in mammary epithelial cells (Cao et al., 2001), IKKα in B cells contributes to the BCR-mediated IKK activity (Fig. 2).
The stimulation of BCR leads to the recruitment of signalling molecules into membrane lipid rafts, an event that resembles the activation of T cells and seems to be important for the efficient transmission of signalling events. Notably, the lack of PKC-β in B cells severely impairs the accumulation of IKKα and IKKβ in lipid rafts in response to stimulation of BCR, and might be connected to the observed defects in signalling (Su et al., 2002). A concern with these results, however, is that the recruitment of other signalling molecules such as BCR itself, Syk or PLC-γ2 to the lipid rafts might also be impaired in the pkcβ−/− B cells. It might therefore be that the absence of PKC-β results in a more general defect in complex formation, and that the decreased activation of IKK measured in these mutant B cells is a secondary consequence of a decrease in assembled transducing complexes. Interestingly, there is a strong parallel between B and T cells with regard to the involvement of PKC in NF-κB activation and in raft complex formation. In mature T cells the Ca2+-insensitive, DAG-activatable PKC isoform, PKC-θ, has been shown to be essential for NF-κB activation through the T-cell receptor (TCR) (Sun et al., 2000). Interestingly, PKC-θ is recruited to the rafts upon TCR activation, along with Bcl-10, which interacts with the adapter protein CARMA1 which is constitutively located in T-cell rafts (Gaide et al., 2002; Wang et al., 2002). Bcl-10 KO mice display an impaired activation of NF-κB in T cells and also in B cells (Ruland et al., 2001). Because no defects in B cells have been reported in the PKC-θ KO mice, it is possible that PKC-β has an equivalent role in B cells to that of PKC-θ in T cells. However, it is still not completely clear how PKC-θ regulates Bcl-10 in T cells and whether PKC-β acts upstream of that adapter in B cells.
Whereas PKC-β is a member of the classical subfamily of the PKC family of isotypes, PKC-ζ and PKC-λ/ι, which are insensitive to Ca2+ and DAG, are the two members of the atypical subfamily (Moscat & Diaz-Meco, 2000). The characterization of PKC-ζ KO mice indicates that the role of this PKC isoform within the immune system is also specific to B-cell function (Martin et al., 2002) and that it most probably acts through NF-κB (Leitges et al., 2001). B cells from pkcζ−/− mice show increased spontaneous apoptosis and impaired survival in response to IgM crosslinking, whereas both peripheral T cells and thymocytes seem to develop and proliferate normally. Interestingly, the defective survival of B cells in these mice correlates with a defective induction, after BCR activation, of several NF-κB-dependent genes, including that encoding Bcl-xL. Consistent with this finding is the observation that pkcζ−/− mice are unable to mount an optimal T-cell-dependent immune response, in spite of the fact that as adults they exhibit no major defects in the subpopulations of B cells (Martin et al., 2002). This indicates that the deficiency in the immune response in this KO is intrinsic to B-cell signalling rather than being a more conspicuous failure in B-cell maturation. The impairment in B-cell signalling observed in pkcζ−/− mice might also explain the detection of significant alterations in the development of secondary lymphoid organs, especially in Peyer's patches, in young mice (Leitges et al., 2001). This morphological phenotype, although less penetrant, generally weaker and restricted to young animals, is reminiscent of the alterations observed in several other NF-κB-pathway mutant mice (Gerondakis et al., 1999; Gugasyan et al., 2000). However, the fact that the microarchitecture of the pkc-ζ−/− spleens was preserved (Leitges et al., 2001) in this case reinforces the notion that PKC-ζ is important for B-cell function but possibly not for the normal physiology of the spleen stroma.
The lack of PKC-ζ in B cells and EFs does not affect the ability of TNF-α or IgM to activate the IKK complex or the DNA-binding activity of NF-κB, respectively (Leitges et al., 2001; Martin et al., 2002). However, the ability of TNF-α or IL-1 to stimulate NF-κB-dependent transcription in pkc-ζ−/− EFs is significantly impaired (Leitges et al., 2001). This suggests that, like IKKα and NIK in fibroblasts, PKC-ζ might control the transcriptional activity of NF-κB (Fig. 2). In fact, PKC-ζ can directly and efficiently phosphorylate RelA and, more importantly, TNF-α-induced phosphorylation of RelA in vivo was shown to be seriously inhibited in pkcζ−/− EFs (Leitges et al., 2001). It is likely that PKC-ζ is linked to more upstream components in the pathways that are stimulated by TNF-α, RANK and IL-1, through the interaction of its adapter, p62, with receptor-interacting protein (RIP) and TNF-R-associated factor-6 (TRAF6), linkers known to act in NF-κB receptor signalling (Moscat & Diaz-Meco, 2000). However, in B cells the role of p62 as a potential link between PKC-ζ and components of the BCR complex has not yet been addressed. Interestingly, after challenge with IgM, PKC-ζ, but not the other atypical PKC-λ/ι, is phosphorylated in its T-loop, which is required for PKC-ζ activity (Martin et al., 2002). It therefore seems that BCR activation selectively targets PKC-ζ.
Finally, another group of PKC KO mice has added valuable information on the role of these kinases in the control of the immune response. Recently, two independent laboratories have characterized mice in which the gene encoding PKC-δ has been disrupted (Mecklenbrauker et al., 2002; Miyamoto et al., 2002). PKC-δ is a Ca2+-independent, DAGsensitive isoform that was previously implicated as a positive regulator of apoptosis in several cell systems. Interestingly, the loss of PKC-δ leads to increased B-cell proliferation and auto-immunity, with significantly augmented production of IgG antibodies. From the point of view of cell signalling, the data in both studies are not entirely consistent: whereas one suggests that pkcδ−/− mice do not show a generalized enhancement of signalling events (Mecklenbrauker et al., 2002), the other detected increased proliferation of the pkcδ−/− B cells in response to several stimuli (Miyamoto et al., 2002). In any case, this enhanced response cannot be accounted for by increased NF-κB activation (at least as determined by electrophoretic mobilityshift assays), although other aspects of the NF-κB pathways, such as the potential roles of non-canonical cascades during PKC-δ signalling, should be investigated.
Pending questions
The past few years have brought many discoveries about the mechanisms by which NF-κB is regulated in B cells and other systems, as well as about the involvement of the different PKC isoforms in this pathway. However, several important questions remain unsolved. For example, it is not clear how the cell discriminates between signals going through the different subunits of the IKK complex, or how IKKα in some systems acts as an IκB kinase whereas its substrates, in others, could be p100 or even RelA. It is likely that different adapters, yet to be discovered, might help to establish the necessary specificity. Other outstanding questions are related to the mechanism by which PKC-ζ regulates the transcriptional activity of RelA. Mapping the sites targeted by PKC-ζ is required if we are to understand how this kinase functions in relation to other potential RelA kinases such as PKA (Zhong et al., 1998). It will also be important to determine how PKC-δ is activated in B cells, as well as the mechanisms by which it represses B-cell function. Is it turned on in response to membrane receptor signals, or is it just a constitutively active switch that keeps B-cell activation below a potentially damaging threshold? In addition, we still do not know how PKC-β activates the IKK complex. Is it a direct action or mediated by as yet unknown raft-recruited adapters?
In summary, the use of KO mice to study different elements in these pathways has revealed their functional relevance. It remains to be established how they work and how they are interconnected. Understanding these mechanisms will help in the discovery of better therapies for human diseases such as rheumatoid arthritis, lupus and other autoimmune disorders.



Acknowledgments
J.M. is the recipient of the Ayuda Investigación Juan March 2001.
References
- Caamano J.H. et al. (1998) Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med., 187, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y. et al. (2001) IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell, 107, 763–775. [DOI] [PubMed] [Google Scholar]
- Claudio E., Brown K., Park S., Wang H., & Siebenlist U. (2002) BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nature Immunol., 3, 958–965. [DOI] [PubMed] [Google Scholar]
- Coope H.J. et al. (2002) CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J., 21, 5375–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejardin E. et al. (2002) The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity, 17, 525–535. [DOI] [PubMed] [Google Scholar]
- Fagarasan S. et al. (2000) Alymphoplasia (aly)-type nuclear factor κB-inducing kinase (NIK) causes defects in secondary lymphoid tissue chemokine receptor signaling and homing of peritoneal cells to the gut-associated lymphatic tissue system. J. Exp. Med., 191, 1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G. et al. (1998) Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med., 187, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futterer A., Mink K., Luz A., Kosco-Vilbois M.H. & Pfeffer K. (1998) The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity, 9, 59–70. [DOI] [PubMed] [Google Scholar]
- Gaide O. et al. (2002) CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-κB activation. Nature Immunol., 3, 836–843. [DOI] [PubMed] [Google Scholar]
- Gauld S.B., Dal Porto J.M. & Cambier J.C. (2002) B cell antigen receptor signaling: roles in cell development and disease. Science, 296, 1641–1642. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Grossmann M., Nakamura Y., Pohl T. & Grumont R. (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene, 18, 6888–6895. [DOI] [PubMed] [Google Scholar]
- Ghosh S. & Karin M. (2002) Missing pieces in the NF-κB puzzle. Cell, 109 (Suppl.), S81–S96. [DOI] [PubMed] [Google Scholar]
- Gugasyan R. et al. (2000) Rel/NF-κB transcription factors: key mediators of B-cell activation. Immunol. Rev., 176, 134–140. [DOI] [PubMed] [Google Scholar]
- Horwitz B.H., Scott M.L., Cherry S.R., Bronson R.T. & Baltimore D. (1997) Failure of lymphopoiesis after adoptive transfer of NF-κB-deficient fetal liver cells. Immunity, 6, 765–772. [DOI] [PubMed] [Google Scholar]
- Hu Y. et al. (2001) IKKα controls formation of the epidermis independently of NF-κB. Nature, 410, 710–714. [DOI] [PubMed] [Google Scholar]
- Israel A. (2000) The IKK complex: an integrator of all signals that activate NF-κB? Trends Cell Biol., 10, 129–133. [DOI] [PubMed] [Google Scholar]
- Kaisho T. et al. (2001) IκB kinase α is essential for mature B cell development and function. J. Exp. Med., 193, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. (1998) The NF-κB activation pathway: its regulation and role in inflammation and cell survival. Cancer J. Sci. Am., 4, S92–S99. [PubMed] [Google Scholar]
- Karin M. & Ben-Neriah Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol., 18, 621–663. [DOI] [PubMed] [Google Scholar]
- Kayagaki N. et al. (2002) BAFF/BlyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity, 10, 515–524. [DOI] [PubMed] [Google Scholar]
- Leitges M. et al. (1996) Immunodeficiency in protein kinase Cβ-deficient mice. Science, 273, 788–791. [DOI] [PubMed] [Google Scholar]
- Leitges M. et al. (2001) Targeted disruption of the ζPKC gene results in the impairment of the NF-κB pathway. Mol. Cell, 8, 771–780. [DOI] [PubMed] [Google Scholar]
- Martin P. et al. (2002) Role of ζ PKC in B-cell signaling and function. EMBO J., 21, 4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecklenbrauker I., Saijo K., Zheng N.Y., Leitges M. & Tarakhovsky A. (2002) Protein kinase Cδ controls self-antigen-induced B-cell tolerance. Nature, 416, 860–865. [DOI] [PubMed] [Google Scholar]
- Miyamoto A. et al. (2002) Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cδ. Nature, 416, 865–869. [DOI] [PubMed] [Google Scholar]
- Moscat J. & Diaz-Meco M.T. (2000) The atypical protein kinase Cs. Functional specificity mediated by specific protein adapters. EMBO Rep., 1, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petro J.B. & Khan W.N. (2001) Phospholipase C-γ2 couples Bruton's tyrosine kinase to the NF-κB signaling pathway in B lymphocytes. J. Biol. Chem., 276, 1715–1719. [DOI] [PubMed] [Google Scholar]
- Petro J.B., Rahman S.M., Ballard D.W. & Khan W.N. (2000) Bruton's tyrosine kinase is required for activation of IκB kinase and nuclear factor κB in response to B cell receptor engagement. J. Exp. Med., 191, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings D.J. (1999) Bruton's tyrosine kinase controls a sustained calcium signal essential for B lineage development and function. Clin. Immunol., 91, 243–253. [DOI] [PubMed] [Google Scholar]
- Ruland J. et al. (2001) Bcl10 is a positive regulator of antigen receptor-induced activation of NF-κB and neural tube closure. Cell, 104 33–42. [DOI] [PubMed] [Google Scholar]
- Saijo K. et al. (2002) Protein kinase Cβ controls nuclear factor κB activation in B cells through selective regulation of the IκB kinase α. J. Exp. Med., 195, 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann B. et al. (2001) An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science, 293, 2111–2114. [DOI] [PubMed] [Google Scholar]
- Senftleben U. et al. (2001a) Activation by IKKα of a second, evolutionarily conserved, NF-κB signaling pathway. Science, 293, 1495–1499. [DOI] [PubMed] [Google Scholar]
- Senftleben U., Li Z.W., Baud V. & Karin M. (2001b) IKKβ is essential for protecting T cells from TNFα-induced apoptosis. Immunity, 14, 217–230. [DOI] [PubMed] [Google Scholar]
- Sizemore N., Lerner N., Dombrowski N., Sakuria H. & Stark G.R. (2001) Distinct roles of the IκB kinase α and β subunits in liberating nuclear factor κB (NF-κB) from IκB and in phosphorylating the p65 subunit of NF-κB. J. Biol. Chem., 277, 3863–3869. [DOI] [PubMed] [Google Scholar]
- Solan N.J., Miyoshi H., Carmona E.M., Bren G.D. & Paya C.V. (2002) RelB cellular regulation and transcriptional activity are regulated by p100. J. Biol. Chem., 277, 1405–1418. [DOI] [PubMed] [Google Scholar]
- Su T.T. et al. (2002) PKC-β controls IκB kinase lipid raft recruitment and activation in response to BCR signaling. Nature Immunol., 3, 780–786. [DOI] [PubMed] [Google Scholar]
- Sun Z. et al. (2000) PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature, 404, 402–407. [DOI] [PubMed] [Google Scholar]
- Takeda K. et al. (1999) Limb and skin abnormalities in mice lacking IKKα. Science, 284, 313–316. [DOI] [PubMed] [Google Scholar]
- Tan J.E., Wong S.C., Gan S.K., Xu S. & Lam K.P. (2001) The adaptor protein BLNK is required for B cell antigen receptor-induced activation of nuclear factor-κB and cell cycle entry and survival of B lymphocytes. J. Biol. Chem., 276, 20055–20063. [DOI] [PubMed] [Google Scholar]
- Thompson J.S. et al. (2001) BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science, 293, 2108–2111. [DOI] [PubMed] [Google Scholar]
- Wang D. et al. (2002) A requirement for CARMA1 in TCR-induced NF-κB activation. Nature Immunol., 3, 830–835. [DOI] [PubMed] [Google Scholar]
- Xiao G., Harhaj E.W. & Sun S.C. (2001) NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell, 7, 401–409. [DOI] [PubMed] [Google Scholar]
- Yin L. et al. (2001) Defective lymphotoxin-beta receptor-induced NF-κB transcriptional activity in NIK-deficient mice. Science, 291, 2162–2165. [DOI] [PubMed] [Google Scholar]
- Zhong H., Voll R.E. & Ghosh S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell, 1, 661–671. [DOI] [PubMed] [Google Scholar]