

Abstract
The Par4 gene was first identified in prostate cells undergoing apoptosis after androgen withdrawal. PAR4 was subsequently shown to interact with, and inhibit, atypical protein kinase C isoforms, functioning as a negative regulator of the NF-κB pathway. This may explain its pro-apoptotic function in overexpression experiments. To determine the physiological role of PAR4, we have derived primary embryonic fibroblasts (EFs) from Par4−/− mice. We show here that loss of PAR4 leads to a reduction in the ability of tumour necrosis factor-α (TNF-α) to induce apoptosis by increased activation of NF-κB. Consistent with recent reports demonstrating the antagonistic actions of NF-κB and c-Jun amino-terminal kinase (JNK) signalling, we have found that Par4−/− cells show a reduced activation of the sustained phase of JNK and p38 stimulation by TNF-α and interleukin 1. Higher levels of an anti-apoptotic JNK-inhibitor protein, X-chromosome-linked inhibitor of apoptosis, in Par4−/− EFs might explain the inhibition of JNK activation in these cells.
Introduction
Programmed cell death, or apoptosis, is a physiological process that is induced in response to environmental stresses and cellular signals, and is important in development, tissue remodelling and in diseases such as cancer and neurodegeneration. Par4 was first detected in a differential screen of genes induced in prostate cells undergoing apoptosis (Sells et al., 1994). Subsequent studies showed that ectopic expression of PAR4 triggers apoptosis in NIH-3T3 cells and sensitizes prostate cancer and melanoma cells to apoptotic stimuli (Diaz-Meco et al., 1996; Berra et al., 1997; Sells et al., 1997). There is evidence that induction of PAR4 contributes to the pro-apoptotic effects of nonsteroidal anti-inflammatory drugs in human colon carcinomas (Zhang & DuBois, 2000) and to the induction of neuronal cell death by the amyloid-β protein or by trophic-factor withdrawal (Guo et al., 1998). PAR4 binds the zinc-finger domains of the atypical protein kinase C isoforms (aPKCs) through its coiled-coil carboxy terminus, thus inhibiting aPKC enzymatic activity (Diaz-Meco et al., 1996). This is important because the ability of PAR4 to induce apoptosis is abolished by the overexpression of the aPKCs (Diaz-Meco et al., 1996), which suggests that the inhibition of aPKC activity by PAR4 is a crucial event in apoptotic cell death. In fact, blocking the function of ζPKC (Diaz-Meco et al., 1996) or λ/ιPKC (Diaz-Meco et al., 1996; Murray & Fields, 1997) by using dominant-negative mutants or antisense oligonucleotides is sufficient to trigger apoptosis. In addition, embryonic fibroblasts (EFs) and mouse B cells in which the gene encoding ζPKC has been knocked out are more sensitive to apoptotic stimuli, demonstrating an anti-apoptotic role for this kinase (Leitges et al., 2001; Martin et al., 2002). Together, these results suggest a model in which PAR4 inhibits aPKCs, an event that is required for apoptosis to proceed.
The aPKCs are essential for the activation of the anti-apoptotic transcription factor NF-κB by inflammatory cytokines and the ras oncogene (Moscat & Diaz-Meco, 2000). Overexpression of PAR4 inhibits NF-κB and makes cells sensitive to the induction of apoptosis by tumour necrosis factor-α (TNF-α) (Diaz-Meco et al., 1999). These results suggest that at least one of the mechanisms by which PAR4 controls apoptosis is by blocking the aPKC–NF-κB signalling cascade. Other studies have shown that overexpression of PAR4 leads to the activation of stress-activated kinases, such as p38 (Berra et al., 1997), suggesting that inhibition of ζPKC is required for the full activation of the stress-activated MAP (mitogen-activated protein) kinases. Here, we have made Par4-deficient mice by homologous recombination and have investigated the effect of an absence of the PAR4 protein in several signalling pathways in EFs.
Results and Discussion
Generation of Par4−/− mice
To analyse the role of PAR4 in vivo, we disrupted this gene in mice by homologous recombination (Fig. 1A). Chimaeric germline-transmitting males were crossed to females and produced heterozygous F1 progeny. Intercrosses of these F1 mice were carried out to produce Par4−/− mice. Wild-type and mutant alleles were identified by Southern blot analysis, as shown in Fig. 1B. Primary cultures of EFs were obtained from heterozygous intercrosses, and it was shown that Par4−/− EFs do not express the PAR4 protein (Fig. 1C). The basal activity of aPKCs in Par4−/− EFs is increased as compared with wild-type controls, and is increased further by the addition of TNF-α to the medium (Fig. 1D). These results show that PAR4 is a negative regulator of aPKCs in vivo.
Figure 1.
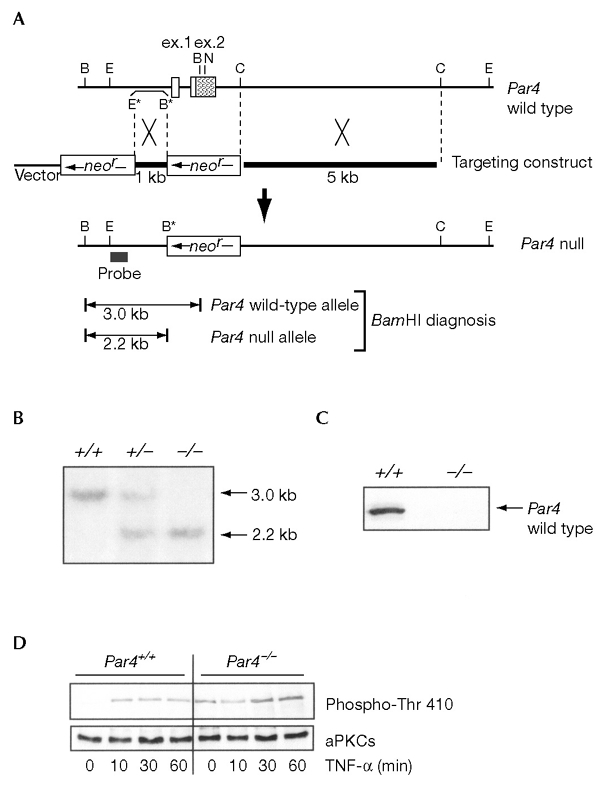
Par4−/− genotypic analysis. (A) Restriction map of the Par4 locus and generation of the Par4− mutant allele; the targeting vector was integrated into the endogenous locus by homologous recombination to produce the mutant allele (Par4 null). The two boxes in the upper part indicate exons one (ex. 1) and two (ex. 2) of Par4, and the shaded area indicates the coding region. Restriction enzyme abbreviations are as follows: B, BamHI; E, EcoRI; N, NotI; C, ClaI. Restriction sites with an asterisk were introduced to enable cloning and genotyping. (B) Southern blot analysis of litters derived from a heterozygote intercross. +/+, wild-type; +/−, heterozygous; −/−, homozygous. (C) Immunoblot analysis of extracts from wild-type (+/+) or Par4 null (−/−) embryonic fibroblasts (EFs). (D) Wild-type or Par4−/− EFs were stimulated with 10 ng ml−1 of tumour necrosis factor-α (TNF-α) for varying times, after which cell extracts were prepared and the activity of the atypical PKCs (aPKCs) was determined by immunoblotting with an anti-phospho-aPKC antibody.
Apoptosis in Par4−/− embryonic fibroblasts
Because the overexpression of PAR4 leads to an increase in the sensitivity of EFs to the pro-apoptotic actions of TNF-α (Diaz-Meco et al., 1999), we analysed whether the absence of PAR4 from Par4−/− EFs affects the ability of these cells to undergo TNF-α-induced apoptosis. Wild-type and Par4−/− EFs were incubated with TNF-α, cycloheximide or a combination of both, and the occurrence of apoptosis was detected by TUNEL (TdT-mediated dUTP nick end-labelling) analysis. Under these conditions, neither TNF-α nor cycloheximide alone was sufficient to trigger apoptosis (Fig. 2A). However, incubation with both together triggered apoptosis in wild-type and Par4−/− cells. The extent of the apoptotic response was significantly reduced in Par4−/− cells as compared with that in the control cells. This suggests that the presence of PAR4 in wild-type cells contributes to the apoptotic response. To support this finding, extracts were made from cultures of wild-type and Par4−/− EFs, and the processing of caspase-3 levels was analysed by immunoblotting. The addition of TNF-α and cycloheximide together induced the processing of caspase 3 at higher levels in wild-type EFs than in Par4−/− EFs (Fig. 2B), consistent with the TUNEL analysis.
Figure 2.
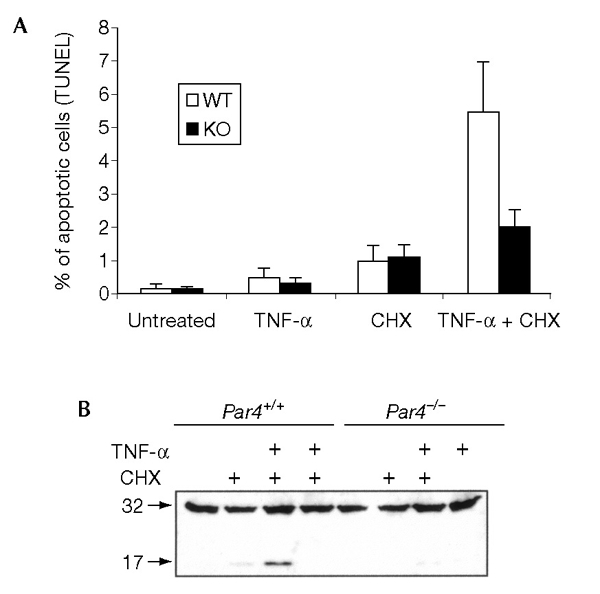
Apoptosis in Par4−/− embryonic fibroblasts. (A) Wild-type (WT) and Par4 null (KO) embryonic fibroblasts were incubated with 10 ng ml−1 of tumour necrosis factor-α (TNF-α), 10 μg ml−1 cycloheximide (CHX) or both for 72 h, after which the percentage of apoptotic cells was determined by TUNEL (TdT-mediated dUTP nick end-labelling) analysis. Results are the mean ± s.d. of three independent experiments with incubations in duplicate. (B) Cell extractions were carried out from cultures incubated for 12 h as above and the processing of caspase 3 was analysed by immunoblotting. The data shown are representative of three independent experiments.
NF-κB activation in Par4−/− embryonic fibroblasts
The aPKCs are important for NF-κB activation (Lallena et al., 1999), and overexpression of PAR4 inhibits this pathway (Diaz-Meco et al., 1999). We therefore analysed whether an absence of PAR4 has any effect on the activation of NF-κB in response to TNF-α. EFs from wild-type and Par4−/− mice were transfected with a κB-dependent luciferase reporter and then either left untreated or stimulated with suboptimal concentrations of TNF-α or interleukin-1 (IL-1). The ability of these cytokines to activate κB-dependent transcription was increased greatly in the Par4−/− cells as compared with that in the wild-type control cells (Fig. 3A). When cells were transfected with a mutant luciferase reporter lacking the κB-recognition sites, no differences were observed between wild-type and Par4−/− cells (data not shown). These results show that PAR4 is a negative regulator of NF-κB signalling. When PAR4 was re-introduced into Par4−/− EFs the enhanced activation of κB-dependent transcription in response to TNF-α (Fig. 3B) or IL-1 (not shown) was abolished. Recent studies have shown that the absence of ζPKC in EFs derived from knockout mice severely impaired κB-dependent transcription, with no effects on IκB kinase (IKK) activation (Leitges et al., 2001). As PAR4 binds and inhibits ζPKC (Diaz-Meco et al., 1996), it was of interest to determine whether the activation of IKK is affected in Par4−/− EFs. Activation of IKK, and the degradation of IκBα induced by TNF-α and IL-1, were not affected by the absence of PAR4 (see supplementary information online). In addition, the activation of NF-κB by TNF-α or IL-1, as determined by electrophoretic mobility-shift assays, was not significantly altered in Par4−/− EFs (see supplementary information online). This is consistent with observations suggesting that ζPKC regulates NF-κB activation at the IKK level in some tissues, such as lung, but not in others (Leitges et al., 2001).
Figure 3.
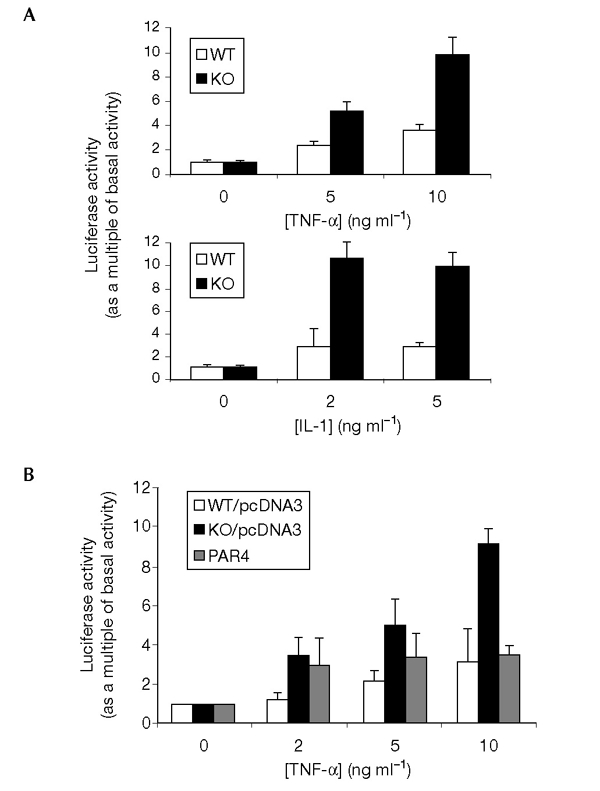
NF-κB signalling in Par4-deficient cells. (A) Wild-type and Par4−/− embryonic fibroblasts (EFs) were transfected with a κB–luciferase reporter plasmid and the control reporter plasmid pRL–CMV. After transfection (24 h) cells were treated with varying concentrations of tumour necrosis factor-α (TNF-α) or interleukin-1 (IL-1) for 6 h; cell extracts were then made and luciferase activity was analysed. (B) Wild-type and Par4−/− EFs were transfected with the same reporter plasmids as above, but Par4−/− cells were also transfected with 0.5 μg of either a PAR4 expression vector or a control plasmid (pcDNA3). Wild-type EFs were transfected, in parallel, with the control plasmid. Cells were stimulated with TNF-α and luciferase activity was analysed. Results are the mean ± s.d. of two independent experiments with incubations in duplicate.
Role of PAR4 in the activation of JNK and p38
In addition to activating NF-κB, TNF-α is a potent inducer of the c-Jun amino-terminal kinase (JNK) and p38 kinase cascades. The p38 and JNK pathways have both been implicated in the control of apoptosis and the inflammatory response. Recent evidence has shown that there is cross-talk between these signalling pathways such that inhibition of NF-κB in IKKβ−/− and RelA−/− cells is accompanied by a more sustained activation of JNK in response to TNF-α, which contributes to the induction of apoptosis by TNF-α (De Smaele et al., 2001; Tang et al., 2001). We therefore determined whether an absence of PAR4 from EFs leads to alterations in the activation of JNK and p38. Wild-type and Par4−/− EFs were stimulated with TNF-α for varying durations, after which JNK or p38 were immunoprecipitated and their activities were determined. Interestingly, stimulation of wild-type EFs with TNF-α gave a high level of JNK activation, with JNK activity peaking at 10–20 min and remaining at high levels up to 60 min (Fig. 4A). Although the early stimulation (from five to ten minutes) of JNK by TNF-α was either unaffected or only slightly affected by the absence of PAR4, JNK activity was significantly lower at 20 min in Par4−/− cells than in wild-type cells, and was completely inhibited in Par4−/− cells at 45 and 60 min (Fig. 4A). Stimulation of p38 was also sustained in TNF-α-treated cells (Fig. 4A). We also detected a reduction in TNF-α-induced p38 activation in Par4−/− cells, although there was less of a reduction than that seen for JNK activity in these cells (Fig. 4A). Furthermore, when stimulated with IL-1, a less sustained activation of JNK and p38 was seen in Par4−/− EFs as compared with that in the wild-type control EFs (Fig. 4B). Immunoblot analyses with anti-JNK and anti-p38 antibodies showed that there was no significant difference in JNK and p38 protein levels between wild-type and Par4−/− cells (see supplementary information online). When the expression of the JNK upstream-activator, MAPK kinase 4 (MKK4), was analysed, its stimulation in Par4−/− cells was less sustained than in wild-type EFs (see supplementary information online). Interestingly, from the point of view of the specificity of PAR4 function, the activation of both the p38 and the JNK kinase cascades by osmotic shock was not affected by the absence of PAR4 (see supplementary information online). These results suggest that an absence of PAR4 selectively inhibits the sustained activation of JNK and p38 in response to TNF-α and IL-1. When PAR4 was ectopically expressed in Par4−/− cells the sustained activation of JNK was restored (Fig. 5).
Figure 4.
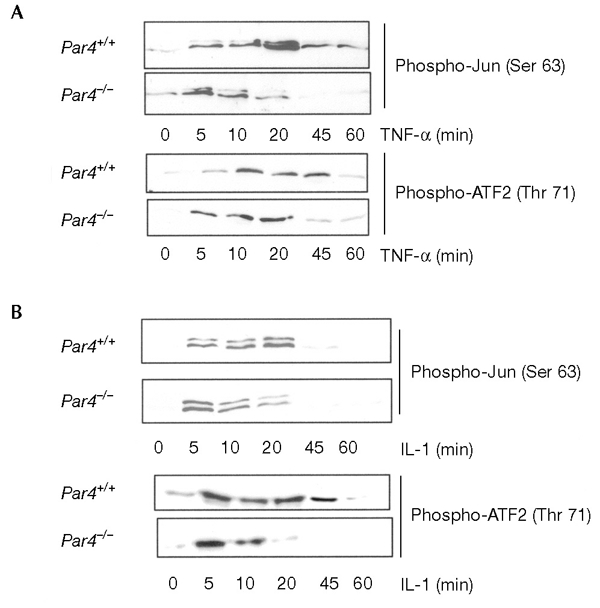
Mitogen-activated protein kinase signalling in Par4−/− embryonic fibroblasts. Wild-type and Par4−/− embryonic fibroblasts were stimulated for varying lengths of time with 10 ng ml−1 of tumour necrosis factor-α (TNF-α) (A) or interleukin-1 (IL-1) (B), after which cell extracts were prepared and the activity of c-Jun amino-terminal kinase (JNK; determined by level of Ser 63 phosphorylation of Jun) or p38 (determined by level of Thr 71 phosphorylation of activating transcription factor 2 (ATF 2) were analysed in immunoprecipitates as described in Methods.
Figure 5.

Expression of Par4 restores normal c-Jun amino-terminal kinase signalling in Par4-deficient cells. Wild-type cells were transduced with the pBabe retroviral vector and Par4-deficient embryonic fibroblasts (EFs) with either pBabe or pBabe–PAR4 retroviral vectors. Cells were then stimulated with tumour necrosis factor-α (TNF-α) at 10 ng ml−1 for 30 min and c-Jun amino-terminal kinase (JNK) activity was determined with an antibody specific for phospho-JNK. Levels of JNK and PAR4 expression were analysed using antibodies against these proteins. Representative data from at least three independent experiments are shown.
Enhanced expression of XIAP in Par4−/− EFs
One possible explanation for the ability of NF-κB to affect the activation of JNK by TNF-α is that the expression of the anti-apoptotic protein X-chromosome-linked inhibitor of apoptosis (XIAP) is reduced in the absence of NF-κB activation in RelA−/− and IKKβ−/− cells (Tang et al., 2001). Ectopic expression of XIAP in RelA−/− cells, but not of other NF-κB-dependent genes, restores the transient mode of JNK activation that is observed in wild-type cells (Tang et al., 2001). Because Par4−/− cells show enhanced NF-κB-dependent transcription, it is possible that these cells express increased levels of XIAP, which may account for the lower level of sustained JNK activation seen in these cells. To determine whether this hypothesis is correct, wild-type and Par4−/− EFs were stimulated for varying durations with TNF-α, and the levels of XIAP were determined by immunoblot analysis. The data shown in Fig. 6A show that stimulation of wild-type EFs with TNF-α induces XIAP expression after 45 min, as previously reported. XIAP levels were constitutively increased in Par4−/− cells (Fig. 6A). Therefore, the downregulation of JNK in Par4−/− cells is consistent with alterations in the levels of XIAP. Interestingly, the increased XIAP levels in Par4−/− cells were detected even when these cells were treated with cycloheximide (Fig. 6B). Thus, the presence of cycloheximide decreases both actin and XIAP levels in wild-type and Par4−/− cells (Fig. 6B, lanes 2 and 5), but the levels of XIAP, although reduced when compared with those in the untreated cultures (Fig. 6B, lanes 5 and 6, compared with lane 4), remain elevated in Par4−/− cells (Fig. 6B, lanes 5 and 6) as compared with those in wild-type cells (Fig. 6B, lanes 2 and 3). This may explain why Par4−/− EFs are more resistant to apoptosis than wild-type cells in response to TNF-α even in the presence of cycloheximide (Fig. 2A and B). It should be noted that the concentrations of cycloheximide used in these experiments only partially inhibit protein synthesis, maintaining the differences between the two cell types in regard to the levels of anti-apoptotic proteins such as XIAP.
Figure 6.
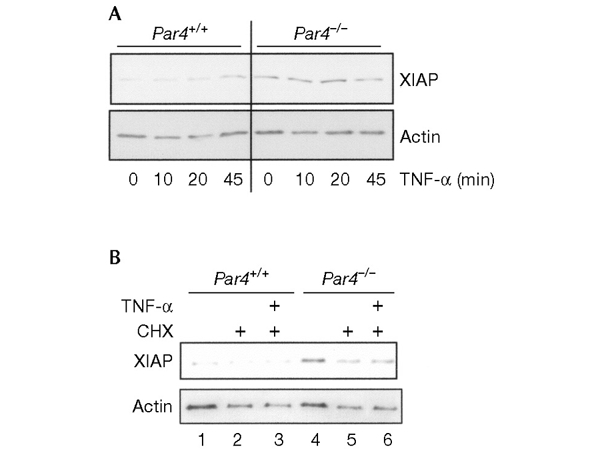
X-chromosome-linked inhibitor of apoptosis induction in Par4−/− embryonic fibroblasts. (A) Wild-type and Par4-deficient embryonic fibroblasts (EFs) were incubated with tumour necrosis factor-α (TNF-α) at 10 ng ml−1 for varying lengths of time, cell extracts were made, and the levels of X-chromosome-linked inhibitor of apoptosis (XIAP) determined by immunoblotting. (B) Wild-type or Par4-deficient EFs were incubated with cycloheximide (CHX) at 10 μg ml−1 with or without TNF-α at 10 ng ml−1 for 12 h. Levels of XIAP were determined by immunoblotting. Levels of actin expression are also shown, as a control. Representative data from three independent experiments are shown.
PAR4 is not required for activation of ERK
Previous studies have suggested that the aPKCs are essential activators of extracellular-signal-related protein kinase (ERK) (Berra et al., 1995; Schonwasser et al., 1998) and that the overexpression of PAR4 leads to strong inhibition of the ERK pathway (Berra et al., 1997). However, although in B cells from ζPKC−/− mice there is a clear reduction in the activation of ERK (Martin et al., 2002), the absence of ζPKC does not affect the activation of ERK in EFs in response to several stimuli (Leitges et al., 2001). Therefore, we analysed whether the absence of PAR4 in Par4−/− EFs affected ERK activation. In contrast to the stimulation of JNK in these cells, the activation of ERK by TNF-α, platelet-derived growth-factor (PDGF) or serum was not affected by the absence of PAR4 (see supplementary information online).
Absence of ζPKC leads to enhanced activation of JNK
As PAR4 is an inhibitor of ζPKC and, according to the data presented in this study, is a regulator of the sustained activation of JNK, it is possible that the absence of ζPKC might lead to a hyperactivation of JNK. To address this possibility, we treated either wild-type or ζPKC-deficient primary cultures of EFs with TNF-α for varying durations, after which JNK activity was determined. The absence of ζPKC correlates with enhanced activation of JNK by TNF-α (Fig. 7A). When the activity of p38 was analysed, it was clear that the loss of ζPKC leads to a more sustained increase in TNF-αstimulated p38 activity (Fig. 7B). Similar results were obtained in cells activated by IL-1 (data not shown). These findings are consistent with PAR4 being a negative regulator of ζPKC and with the fact that the loss of PAR4 leads to the impairment of the sustained activation of JNK and p38 by TNF-α and IL-1. When ζPKC was ectopically expressed in ζPKC−/− cells, the increase in the sustained activation of JNK (Fig. 7C) and p38 (data not shown) in response to TNF-α was diminished in comparison with the increase detected in wild-type cells. It should be noted that the anti-ζPKC antibody used in this experiment to monitor the expression of transduced ζPKC cross-reacts with λ/ιPKC.
Figure 7.
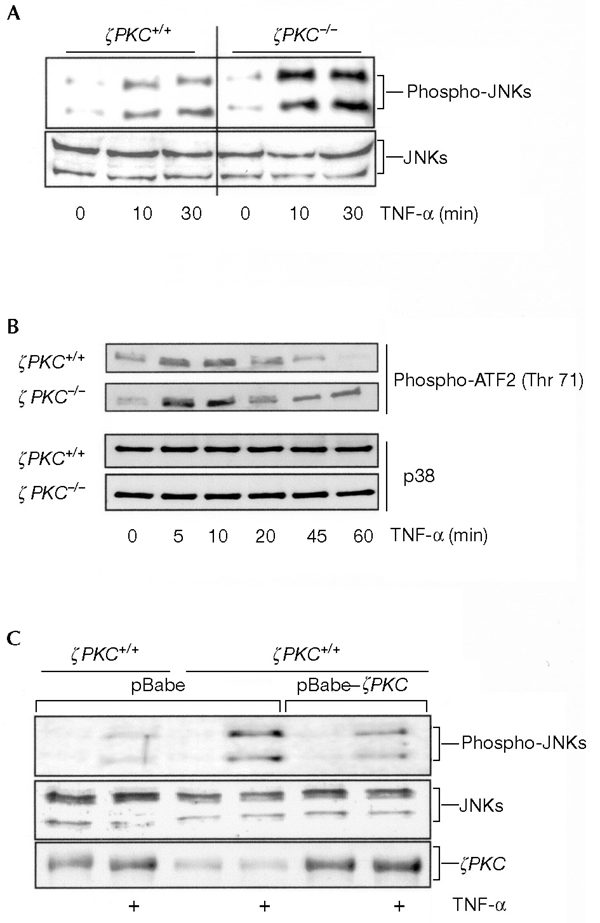
Mitogen-activated protein kinase signalling in embryonic fibrobalsts null for the ζ isoform of protein kinase C. Wild-type or ζ protein kinase C (ζPKC)-deficient embryonic fibroblasts (EFs) were incubated with tumour necrosis factor-α (TNF-α) at 10 ng ml−1 for varying durations, cell extracts were prepared and c-Jun amino-terminal kinase (JNK) (A) and p38 (B) activities were determined as described in Methods. Levels of JNK and p38 are shown as a control. Representative data from at least three independent experiments are shown. (C) Wild-type cells were transduced with the retroviral pBabe vector, whereas ζPKC-deficient EFs were transduced with either pBabe or pBabe–ζPKC retroviral vectors. Cells were stimulated with TNF-α at 10 ng ml−1 for 30 min and JNK activity was determined as above. Levels of JNK and ζPKC were determined using appropriate antibodies. Representative data from three independent experiments are shown.
Speculation
These results show that PAR4 is an important regulator of apoptosis due to its ability to modulate the aPKCs and NF-κB, which in turn regulate the levels of several anti-apoptotic genes, including XIAP. This is particularly important as XIAP has been reported to control JNK activation, which contributes to the activation of apoptosis (De Smaele et al., 2001; Tang et al., 2001). The inactivation of NF-κB by PAR4 increases JNK activation in response to TNF-α and IL-1 (Reuther-Madrid et al., 2002). Previous studies have suggested that the activation of p38 is not affected (De Smaele et al., 2001; Tang et al., 2001; Reuther-Madrid et al., 2002). However, we show here that the loss of PAR4 affects not only the activation of JNK, but also that of p38. These results suggest that PAR4 and the aPKCs modulate the extent of JNK activation through NF-κB, whereas the regulation of p38 takes place through NF-κB-independent signals. Further studies are needed to determine whether other anti-apoptotic genes that may target p38 are constitutively activated in Par4−/− cells.
Future experiments will provide information about the effect that the absence of PAR4 has on the physiology of whole animals and tissues. PAR4 was first identified as having an important function in prostate cancer cells (Sells et al., 1994). Therefore, we can speculate that loss of the Par4 gene may induce neoplastic alterations in the prostate in both mice and humans. A link between PAR4 and cancer has already been reported (Barradas et al., 1999; Nalca et al., 1999): tumour transformation leads to a dramatic downregulation of PAR4 protein levels (Barradas et al., 1999), which, consistent with the data reported here, correlates with an increased stimulation of the NF-κB pathway in transformed cells (Diaz-Meco et al., 1999). However, it remains to be shown that downregulation of PAR4 is commonly observed in human tumours or autoimmune diseases.
Methods
Reagents and antibodies.
Reagents were purchased as follows: TNF-α (Promega), cycloheximide (Sigma), and PDGF (Upstate Biotechnology). Polyclonal antibodies anti-PAR4, anti-IKKγ, anti-ERK, anti-p38, anti-IκBα, anti-IKKβ, anti-MKK4 and anti-actin, and monoclonal antibodies anti-phospho-ERK and anti-phospho-JNK (antibody H1) were purchased from Santa Cruz. Monoclonal anti-caspase-3 and anti-XIAP antibodies, and the polyclonal anti-JNK antibody were purchased from Becton Dickinson. Polyclonal anti-phospho-c-Jun (Ser 63), anti-phospho-ATF-2 (Thr 71), anti-phospho-MKK4 (Thr 261) and anti-phospho-PKC (Thr 410) antibodies were from Cell Signaling. All antibodies were used in accordance with the manufacturers' instructions.
Generation of Par4−/− mice.
Mice genetically deficient for Par4 were produced using standard procedures. A genomic DNA segment containing murine Par4 was identified by screening a commercial library (Down-to-the-Well, Genome Systems; 129Sv genetic background) by PCR using primers complementary to exon two of Par4. The construct that eliminates exons one and two of Par4 (Fig. 1A) was linearized and electroporated into R1 embryonic stem (ES) cells (129Sv). One ES cell clone was used to produce chimaeric male mice, which were crossed with CD1 females, and the progeny from this cross were genotyped by Southern blotting. EFs were isolated from embryos at 13.5 days post-coitum from crosses between Par4+/− mice of mixed genetic background CD1/129Sv (a ratio of 3:1).
Western blotting and in vitro kinase and electrophoretic mobility-shift assays.
Cells were lysed with cell extraction buffer (Cell Signaling). Soluble fractions were immunoprecipitated with antibodies, as indicated in Results and Discussion. The immunoprecipitates were washed extensively, separated by SDS–polyacrylamide gel electrophoresis and transferred to Nitrocellulose-ECL membranes (Amersham Biosciences). The proteins present in the immunoprecipitates were detected by chemiluminescence using an Amersham Biosciences ECL Kit. In vitro kinase assays for JNK and p38 were carried out in accordance with the manufacturer's instructions (Cell Signaling). For IKK, activity was determined in anti-IKKγ immunoprecipitates using glutathione S-transferase–IκBα1-54 as described (Leitges et al., 2001). Electrophoretic mobilityshift assays were carried out as described (Martin et al., 2002). Cell cultures were transduced with pBabe, pBabe–PAR4 or pBabe–ζPKC retro-viral constructs as described (Barradas et al., 1999).
Apoptosis assay.
Subconfluent EFs were incubated with TNF-α (10 ng ml−1), cycloheximide (10 μg ml−1) or both for 72 h, after which the percentage of apoptotic cells was determined by TUNEL analysis using the Mebstain Apoptosis Kit II (MBL International).
Luciferase assays.
Subconfluent cultures of EFs, either wild-type or Par4−/−, were transfected using Lipofectamine Plus (Invitrogen) with 100 ng of a multimerized κB–luciferase reporter-gene plasmid (described previously in Leitges et al., 2001) and 2 ng of the control reporter pRL–CMV (Promega). In some experiments, cells were co-transfected with pCDNA3–HA–PAR4 (Lallena et al., 1999). After 24 h, cells were stimulated for 6 h with varying concentrations of TNF-α, extracts were prepared and the levels of luciferase activity were determined using the Dual-Luciferase Reporter Assay System (Promega) in accordance with the manufacturer's instructions.
Supplementary data are available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor769-s1.mov).
Supplementary Material
supplementary information
Acknowledgments
J.M. is the recipient of the Ayuda Investigación Juan, March 2001. I.G.-C. recipient of a fellowship from the Ministerio de Educación y Ciencia. Work in J.M.'s lab was supported by grants SAF1999-0053, 2FD97-1429, and SAF2000-0175 from the Ministerio de Ciencia y Tecnologia (MYCT), and 08.1/0060/2000 from the Comunidad Autonoma de Madrid, and by an institutional grant from Fundación Ramón Areces. Work in M.S.'s lab was funded by grant SAF2002-03402 from the MYCT and a consortium between the Consejo Superior de Investigaciones Científicas and Pharmacia Corporation.
References
- Barradas M., Monjas A., Diaz-Meco M.T., Serrano M. & Moscat J. (1999) The downregulation of the pro-apoptotic protein Par4 is critical for Ras-induced survival and tumour progression. EMBO J., 18, 6362–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra E., Diaz-Meco M.T., Lozano J., Frutos S., Municio M.M., Sanchez P., Sanz L. & Moscat J. (1995) Evidence for a role of MEK and MAPK during signal transduction by protein kinase Cζ. EMBO J., 14, 6157–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra E., Municio M., Sanz L., Frutos S., Diaz-Meco M. & Moscat J. (1997) Positioning atypical protein kinase C isoforms in the UV-induced apoptotic signaling cascade. Mol. Cell. Biol., 17, 4346–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smaele E., Zazzeroni F., Papa S., Nguyen D.U., Jin R., Jones J., Cong R. & Franzoso G. (2001) Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature, 414, 308–313. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M.T., Municio M.M., Frutos S., Sanchez P., Lozano J., Sanz L. & Moscat J. (1996) The product of par4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell, 86, 777–786. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M.T., Lallena M.J., Monjas A., Frutos S. & Moscat J. (1999) Inactivation of the inhibitory κB protein kinase/nuclear factor-κB pathway by Par4 expression potentiates tumour necrosis factor-α-induced apoptosis. J. Biol. Chem., 274, 19606–19612. [DOI] [PubMed] [Google Scholar]
- Guo Q., Fu W., Xie J., Luo H., Sells S.F., Geddes J.W., Bondada V., Rangnekar V.M. & Mattson M.P. (1998) Par4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer disease. Nature Med., 4, 957–962. [DOI] [PubMed] [Google Scholar]
- Lallena M.J., Diaz-Meco M.T., Bren G., Pay C.V. & Moscat J. (1999) Activation of IκB kinase β by protein kinase C isoforms. Mol. Cell. Biol., 19, 2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M., Sanz L., Martin P., Duran A., Braun U., Garcia J.F., Camacho F., Diaz-Meco M.T., Rennert P.D. & Moscat J. (2001) Targeted disruption of the ζPKC gene results in the impairment of the NF-κB pathway. Mol. Cell, 8, 771–780. [DOI] [PubMed] [Google Scholar]
- Martin P., Duran A., Minguet S., Gaspar M.L., Diaz-Meco M.T., Rennert P., Leitges M. & Moscat J. (2002) Role of ζPKC in B-cell signaling and function. EMBO J., 21, 4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J. & Diaz-Meco M.T. (2000) The atypical protein kinase Cs. Functional specificity mediated by specific protein adapters. EMBO Rep., 1, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray N.R. & Fields A.P. (1997) Atypical protein kinase C ι protects human leukemia cells against drug-induced apoptosis. J. Biol. Chem., 272, 27521–27524. [DOI] [PubMed] [Google Scholar]
- Nalca A., Qiu S.G., El-Guendy N., Krishnan S. & Rangnekar V.M. (1999) Oncogenic Ras sensitizes cells to apoptosis by Par4. J. Biol. Chem., 274, 29976–29983. [DOI] [PubMed] [Google Scholar]
- Reuther-Madrid J.Y., Kashatus D., Chen S., Li X., Westwick J., Davis R.J., Earp H.S., Wang C-Y. & Baldwin A.S. Jr (2002) The p65/RelA subunit of NF-κB suppresses the sustained anti-apoptotic activity of Jun kinase induced by tumour necrosis factor. Mol. Cell. Biol., 22, 8175–8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonwasser D.C., Marais R.M., Marshall C.J. & Parker P.J. (1998) Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol., 18, 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells S.F., Wood D.P. Jr, Joshi-Barve S.S., Muthukumar S., Jacob R.J., Crist S.A., Humphreys S. & Rangnekar V.M. (1994) Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ., 5, 457–466. [PubMed] [Google Scholar]
- Sells S.F. et al. (1997) Expression and function of the leucine zipper protein Par4 in apoptosis. Mol. Cell. Biol., 17, 3823–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Minemoto Y., Dibling B., Purcell N.H., Li Z., Karin M. & Lin A. (2001) Inhibition of JNK activation through NF-κB target genes. Nature, 414, 313–317. [DOI] [PubMed] [Google Scholar]
- Zhang Z. & DuBois R.N. (2000) Par4, a proapoptotic gene, is regulated by NSAIDs in human colon carcinoma cells. Gastroenterology, 118, 1012–1017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information