

Summary
Conference on Signal Transduction of G-Protein-Coupled Receptors
Introduction
G-protein-coupled receptors (GPCRs) have a conserved structure composed of seven transmembrane helices and are generally thought to form the largest single superfamily of proteins (Bockaert & Pin, 1999; Pierce et al., 2002). GPCRs are located at the cell surface and are involved in signal transduction in almost every living organism (that is, the diploblastic metazoa, yeasts, plants, invertebrates and vertebrates). More than 1,000 GPCRs have been identified in vertebrates, and they have been classified into three main families according to their sequence homology and structural features. Family 1 is the largest, and its members have a short amino-terminal extracellular domain. This family includes a receptor for light (rhodopsin) and receptors that are activated by small ligands such as catecholamines and short peptides. Members of family 2 share a longer extracellular domain and are activated by large peptides such as glucagon and secretin. Members of family 3 include the metabotropic glutamate receptors (mGluRs) and γ-aminobutyric acid B (GABAB) receptors and have large extracellular segments that fold into ligand-binding domains.

St Odile is religiously venerated: born blind, she recovered her vision at the age of 13 when baptized in a spring at the foot of the mountain that dominates the land of Alsace and that now bears her name, Mont Sainte-Odile. Joel Bockaert and Peter Gierschik organized the XXVIIth European Symposium on Hormone and Cell Regulation entitled 'Signal transduction of G-protein-coupled receptors (GPCRs)' that took place last September in the monastery where St Odile lived and died. This was a perfect place to shed new light on many questions concerning GPCR signalling and function. (Shown is a detail of the mosaic from the Chapel of Angels, Mont Sainte-Odile, Alsace, France. Modified by M. Passama.)
The name GPCR derives from the ability of these proteins to recruit and regulate the activity of intracellular heterotrimeric G proteins. The activation of G proteins by the receptor facilitates GDP–GTP exchange on the Gα-subunit. This is followed by the dissociation of the heterotrimeric complex into the GTP-bound Gα-subunit and the Gβγ-dimer. Both the Gα-subunit and the Gβγ-dimer activate or inhibit an ever-expanding list of effectors, including adenylyl cyclases, phopholipases and ion channels. On the basis of sequence similarities in their Gα-subunits, G proteins have been grouped into four subfamilies: Gs, Gi/o, Gq and G12. Whereas stimulatory (Gs) and inhibitory (Gi/o) G proteins are implicated in the regulation of adenylyl cyclase and the gating of ion channels, members of the Gq subfamily activate phospholipase Cβ. The G12 proteins have been reported to couple GPCRs to the activation of Rho GTPases.
GPCRs have evolved to recognize a wide variety of environmental stimuli, such as ions, amino acids, nucleotides, peptides, lipids, proteins and even physical signals, including light. Because GPCRs are involved in regulating many of the body's functions, and their dysfunction can give rise to various pathologies, such as retinitis pigmentosa and nephrogenic diabetes insipidus, these receptors are the targets of more than half of the drugs used therapeutically at present. Most GPCR-related pathologies result from receptor mutations and affect either receptor expression or their coupling to the associated G protein. A further understanding of the structure and signalling mechanisms of GPCRs will be important for the successful development of new treatments. The most recent insights into the structure and signalling functions of GPCRs were presented at the Mont Sainte-Odile meeting, and here we review the most important issues and focus on the new concepts that emerged.
GPCRs and G proteins: structure and activation
Understanding how ligand binding induces a conformational change that is transmitted across the transmembrane region to the intracellular region will require knowledge of the three-dimensional structures of GPCRs. H. Jingami (Osaka, Japan) opened the meeting by reporting on the crystal structure of the ligand-binding core of mGluRs, a family of GPCRs that are essential for the development and function of the mammalian central nervous system. mGluRs are family 3 GPCRs (Bockaert & Pin, 1999) and form homodimers. Their large extracellular domain is divided into a bi-lobed ligand-binding region (LBR) composed of two subdomains, LB1 and LB2, and a cysteine-rich region that links the LBR to the seven-transmembrane region (Fig. 1). Jingami presented X-ray crystal structures of the LBR of mGluR subtype 1: two distinct non-liganded forms, and one bound to its natural agonist, glutamate. The results revealed the existence of a disulphide-linked homodimer, in which each protomer is able to adopt either an open or a closed conformation. The ligand-free dimer has either an 'open–open' or a 'closed–open' conformation (Fig. 1; Kunishima et al., 2000). The 'open–open' conformation is a resting conformation, whereas a rearrangement of the protomer interface in the 'closed–open' dimer allows it to bind to the ligand. Glutamate binding results in an increase in the proportion of active 'closed–open' dimers, and the antagonist α-methyl-4-carboxy-phenylglycine stabilizes the 'open–open' form.
Figure 1.
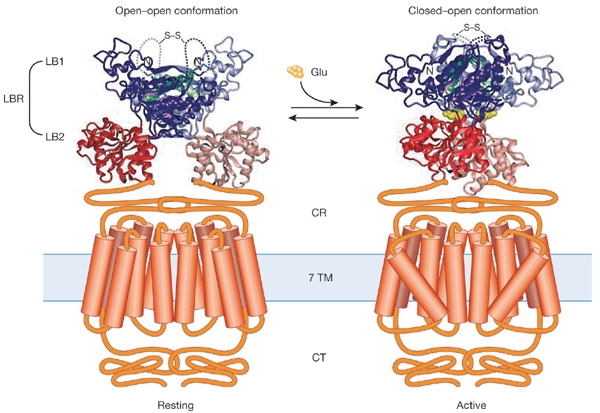
Crystal structure of the ligand-binding core of metabotropic glutamate receptor subtype 1. Metabotropic Glu receptors (mGluRs) are essential for the development and function of the mammalian central nervous system and exist as homodimers. The receptor protomers are characterized by a large extracellular domain that is divided into a ligand-binding region (LBR) and a cysteine-rich region (CR) that links LBR to the transmembrane region (7 TM). The crystal structure of the LBRs of the mGluR1 homodimer, composed of the two subdomains LB1 and LB2, is shown in the non-liganded form (left) and bound to its natural agonist, glutamate (Glu) (right). The LBRs of the homodimer can have either an open or a closed conformation. The ligand-free dimer shows either an 'open–open' resting or a 'closed–open' active conformation. Binding of the agonist stabilizes the 'closed–open' conformation, which is characterized by a change in the relative orientation of the protomers. Thus, the LB2 subdomains of the LBR protomers come closer to each other by approximately 25 Å, and this change may trigger the active state of the receptor. CT, carboxyl terminus. (Adapted from Kunishima et al. (2000) with the permission of Nature and the authors).
The activation mechanism of family 3 GPCRs was further addressed by J.-P. Pin (Montpellier, France), who developed a molecular model for the activation of GABAB receptors. These receptors are responsible for presynaptic inhibition of neurotransmitter release in the mammalian brain. They function as heterodimers that consist of the subunits GABAB1 and GABAB2, and heterodimerization has been shown to be a key requirement for functional membrane-localized GABAB receptors (Fig. 2B). Using chimaeras of GABAB1 and GABAB2 subunits, Pin showed that the LBRs of these subunits are functionally interchangeable. Conversely, a combination of the wild-type GABAB2 subunit with a GABAB1 that bears the LBR of GABAB2 led to the formation of a constitutively active dimer. These results suggest that the GABAB1 extracellular domain is necessary for binding of GABA (Fig. 2A), and that GABAB2 is responsible for G-protein interaction (Fig 2A; Galvez et al. 2001). On the basis of these findings, Pin proposed a modular organization of the GABAB receptor dimer, in which four functionally specialized domains, the LBRs and the heptahelical domains of both the GABAB1 and GABAB2 subunits cooperate. He concluded that the activation mechanism described here might be a common feature of family 3 GPCRs.
Figure 2.
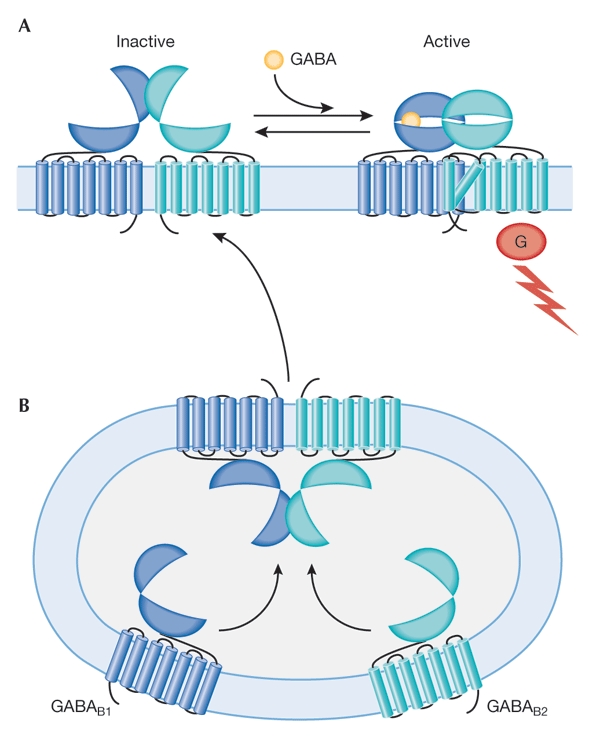
Functional expression of γ-aminobutyric acid B receptors. GABAB receptors are responsible for presynaptic inhibition of neurotransmitter release in the mammalian brain, and function as heterodimers that consist of GABAB1 and GABAB2 subunits. (A) Whereas the GABAB1 receptor is essential for responses elicited by the binding of GABA, the GABAB2 receptor mediates G-protein activation (Galvez et al., 2001). (B) GABAB1- and GABAB2-receptor subunits probably assemble in transport vesicles. Trafficking of the heterodimer to the cell surface depends on the presence of the GABAB2 receptor. G, G protein.
An exciting strategy to study the structure–function relationship of GPCRs was presented by J.L. Banères (Montpellier, France). He described the purification of a recombinant human leukotriene B4 (LTB4) receptor, BLT1, that is expressed in Escherichia coli at levels sufficient to analyse the functional properties of the receptor. This can be done by using biophysical methods such as intrinsic fluorescence spectroscopy, circular dichroism spectroscopy and neutron scattering analysis. The solubilized BLT1 protein showed functional characteristics similar to those of the native receptor with regard to dimerization and high-affinity recognition of LTB4, as well as association with and activation of G proteins. By using the cysteine scanning accessibility method, Banères analysed the exposure of residues at various positions in the receptor protein. The method is based on the replacement of amino-acid residues with cysteines, followed by modification with sulphydryl reagents. Banères reported that, on ligand stimulation, a conformational change occurred in the transmembrane regions of the BLT1 receptor, the main effect of which was a change of the topology of the cytoplasmic face of the sixth transmembrane α-helix. He also showed that only the dimerized BLT1 receptor forms a stable complex with the G protein.
G. Vassart (Bruxelles, Belgium) provided detailed insights into the mechanisms behind the ligand–receptor interaction, receptor activation and G protein coupling of a subset of GPCRs, the glycoprotein hormone receptors (GPHRs). By introducing various point mutations into the N-terminal extracellular domain and the transmembrane portions of the thyreotropin (TSH) and folliotropin receptors, Vassart's group identified key residues that are important for ligand specificity and receptor activation. Furthermore, by exploring the role of the ectodomain of the TSH receptor, they identified residues that are responsible for maintaining the receptor in an active state. Vassart proposed a model in which the non-liganded extracellular domain of the GPHRs functions as a tethered inverse agonist of the seven-transmembrane domain (Vlaeminck-Guillem et al., 2002). When bound to hormone, or when harbouring activating mutations, the extracellular domain would switch its role, acting as a tethered full agonist.
The formation of homodimers and heterodimers is thought to be important for the activation of at least some GPCRs (Angers et al., 2002), and several groups, including that of M. Bouvier (Montreal, Canada), presented results in support of this hypothesis. Using recently developed bioluminescence resonance energy transfer analysis in intact cells that express the human β1- and/or β2-adrenoceptors, Bouvier showed that dimerization of the receptors occurs in the endoplasmic reticulum soon after their biosynthesis, and he suggested that this receptor dimerization is a key requirement for receptor folding and trafficking. He pointed out that the formation of receptor heterodimers raises fascinating combinatorial possibilities that could underlie an unexpected level of pharmacological diversity.
The molecular events that follow the interaction of ligand with the receptor-binding site of GPCRs has not been extensively studied in living cells, probably because of the lack of appropriate and sufficiently powerful technology. However, interesting insights into these events were provided by M.J. Lohse (Würzburg, Germany), who presented results from studies of the activation kinetics of GPCRs. Intramolecular fluorescence resonance energy transfer analysis was performed on the α2A-adrenoceptor (a family 1 GPCR) and the parathyroid hormone (PTH) receptor (a family 2 GPCR), using cyan and yellow fluorescent proteins incorporated in the third intracellular loop and carboxy-terminal tail, respectively. This revealed that the conformational changes in family 1 and family 2 GPCRs that are induced by agonist binding involve a relative separation of the third intracellular loop from the C terminus of the receptor. The maximal activation time constants were 950 ms and 450 ms for the PTH receptor and the α2A-adrenoceptor, respectively, indicating that activation of GPCRs by hormones and neurotransmitters is much faster than previously thought. The question remains whether the conclusions that emerged from these studies can be extrapolated to other GPCRs.
Regulation of GPCR signalling
The Nunez Lecture given by M. Caron (Durham, NC, USA) focused on the basic and clinical implications of GPCR regulation. He described the mechanisms that contribute to the reduction of receptor responsiveness after persistent agonist stimulation, a process referred to as receptor desensitization. The rapid dampening of receptor signalling involves phosphorylation of the receptor, interaction of the phosphorylated receptor with arrestin and other proteins and, in many cases, removal of the receptor from the cell surface by sequestration into recycling endosomes (Fig. 3A; Claing et al., 2002). Receptor phosphorylation is either mediated by second-messenger-regulated protein kinases, such as protein kinase C and protein kinase A, or controlled by GPCR kinases (GRKs). Mice in which the genes that encode individual GRKs or arrestins have been inactivated have altered GPCR responses. This is consistent with the physiological importance of these regulatory molecules. Caron reported that inactivation of the gene that encodes GRK6 causes supersensitivity to the neurotransmitter dopamine and an exaggerated response to psychostimulants such as cocaine, even in heterozygous mice. As Caron pointed out, these findings show that unregulated desensitization can contribute to the aetiology of GPCR-based diseases, and may have a role in the plastic response to drug abuse in vivo. He concluded that GRK6 inhibitors, by prolonging and enhancing the response to dopamine, could be of value in the treatment of Parkinson's disease.
Figure 3.

Signal transduction of G-protein-coupled receptors. Functional G-protein-coupled receptors (GPCRs) exist as monomers, homodimers and/or heterodimers. (A) GPCR regulation and receptor trafficking. Rapid reduction of receptor responsiveness following persistent receptor activation by agonists (Ag) involves phosphorylation of the receptor and/or interaction of the phosphorylated receptor with arrestin and other proteins, and, in many cases, receptor internalization. Several intracellular proteins are involved in the complex process of internalization. Furthermore, internalized receptors may interact with intracellular signalling pathways. Specific proteins retain the receptor in intracellular compartments or target them to the plasma membrane. (B) Examples of receptor-activated signalling pathways. Agonist-mediated coupling of GPCRs to specific G proteins determines signalling specificity and results in the activation of various effector proteins. Members of the GTPase superfamily, such as Rho and Ras, or kinases such as tyrosine kinases and mitogen activated protein kinases (MAPKs) act as integrators at the post-G-protein level after receptor and/or G-protein activation. An increasing number of scaffolding proteins, including PDZ-domain proteins and EVH-domain proteins (such as Homer), target GPCRs to specific subcellular compartments, thereby generating signalling specificity. (C, D) Interaction of non-classical binding partners with GPCRs is important in regulating receptor function and in specifying the correct subcellular localization of GPCRs. For example, Homer3 and Homer1a proteins prevent or induce agonist-independent (constitutive) activity of mGlu1a/5 receptors in neurons (C) and the carboxy-terminal cytoplasmic tail of rhodopsin acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain t-complex testis-expressed (Tctex1) protein (D). AC, adenylyl cyclase; Ag, Agonist; ARF, ADP-ribosylation factor; ARNO, ARF nucleotide binding site opener; FAK, focal adhesion kinase; G, G protein; GRK, GPRC kinase; GIT, GRK-interacting protein; mGluR1a/5, metabotropic glutamate receptor 1a/5; PAK, p21-activated kinase; PDZ, PSD-95/Dlg1/ZO-1 domain-containing protein; PIX, PAK-interacting guanine-nucleotide-exchange factor; PKC, protein kinase C; PKA, protein kinase A; PLC, phospholipase C; PLA2, phospholipase A2; PLD, phospholipase D; PPD, phosphodiesterase; RhoGEF, Rho guanine exchange factor; RGS, regulator of G-protein signalling; Tctex1, t-complex testis-expressed.
In the second part of his presentation, Caron showed that GPCRs that are internalized as part of a stable complex with arrestin persist much longer in endocytic vesicles and are dephosphorylated and recycled to the cell surface much more slowly than GPCRs that undergo intracellular trafficking after only a transient interaction with arrestin. Caron proposed that the trafficking of arrestins together with GPCRs controls the kinetics of resensitization, and that the stable association of arrestins with GPCRs could result in constitutive downregulation of cell-surface GPCRs. This may be the basis for a pathological phenotype in certain cases, such as in nephrogenic diabetes insipidus.
Finally, Caron introduced the concept of 'constitutive desensitization', which is based on the observation that some native or mutationally activated GPCRs are constitutively phosphorylated and found as arrestin-bound complexes in endocytic vesicles, even in the absence of agonist. Removal of the phosphorylation sites that are responsible for this arrestin interaction re-establishes plasma membrane expression and agonist regulation of these receptors. Constitutive desensitization may not only be a general regulatory mechanism of GPCR function, but may also underlie certain forms of hormone resistance in man.
The data presented by L. De Vries (Castres, France) showed that GPCR signalling can be controlled not only at the level of the receptor, but also at the level of the heterotrimeric G protein. Thus, regulators of G-protein signalling (RGS) stimulate the GTPase activity of the Gα-subunits (De Vries et al., 2000) and thereby accelerate their return to the inactivated state. De Vries showed that, in the absence of agonist, RGS19 is present in clathrin-coated vesicles and is spatially segregated from the δ-opioid receptor and its respective Gi protein. On addition of agonist, activated δ-opioid receptors and Gi are co-translocated to clathrin-coated pits that contain RGS19, which subsequently turns off the Gi activity. The fact that both GRKs and arrestins are contained in clathrin-coated pits, together with the receptor, Gi and RGS19, suggests that GRKs, arrestin and RGS19 act in concert in the overall desensitization process (Fig. 3A).
The complex nature of the regulation of cell-surface GPCR function and sequestration was addressed by R.T. Premont (Durham, NC, USA). He focused on ADP-ribosylation factors (ARFs), which are known to be involved in intracellular vesicle transport, including trafficking of clathrin-coated vesicles (Fig. 3A). Premont described the isolation and characterization of GPCR-kinase-interacting proteins (GITs)—a family of GTPase-activating proteins (GAPs) for ARFs. He showed that GITs inhibit the agoniststimulated sequestration of those GPCRs that internalize in an arrestin- and clathrin-coated-vesicle-dependent manner. Furthermore, he suggested that GITs interact directly with several other signalling molecules, including GRK, focal adhesion kinase, paxillin, Rac/Cdc42, p21-activated kinase (PAK) and the PAK-interacting guanine-nucleotide-exchange factor (PIX) (Fig. 3A). Premont reported that multimers of the GIT and PIX proteins assemble and recruit other proteins into the complex. He proposed that these multiprotein complexes act as integration and branch points of the signalling pathways.
G-protein- and post-G-protein-level signal integration
Some of the presentations shed important light on the functional interaction of different G-protein signalling pathways and on the mechanisms of signal integration at the post-G-protein level. S. Offermanns (Heidelberg, Germany) reported that induction of platelet aggregation through GPCRs is an integrated response, mediated by various G proteins, including Gq, Gi and G12/G13. Using either Gαq- or Gα12/Gα13-double-deficient mouse platelets, Offermanns showed that activation and degranulation of platelets requires the activation of Gq-mediated signalling pathways, whereas changes in cell shape depend on activation of the G12 and G13 proteins. Furthermore, he reported that co-stimulation of Gi and G12/G13 results in integrin-αIIbβ3-mediated aggregation of Gαq-deficient platelets, indicating that Gq activation is not necessarily required for this process. In the second part of his presentation, Offermanns gave an example of signal integration at the level of the small GTPases of the Rho family. He discussed the role of Rho guanine exchange factors (RhoGEFs) in linking the activation of heterotrimeric G proteins of the G12 family by GPCRs to Rho and Rac signalling (Fig. 3B). The results clearly indicate that Gα12, Gα13 and two members of the RhoGEF family, PSD95/Dlg/ZO1-RhoGEF (PDZ-RhoGEF) and leukaemia-associated RhoGEF, are distributed widely and colocalize in cells of the central and peripheral nervous systems. This implies that the GPCR–Gα12/13–RhoGEF-mediated pathway is involved in regulating neural morphogenesis and function.
Another example of signal integration by small GTPases was given by W.H. Moolenaar (Amsterdam, The Netherlands). He described the effects of lysophosphatidic acid (LPA), which mediates the transient activation of RhoA and the prolonged activation of Rac, causing cell spreading, lamellipodia formation and stimulation of cell migration. Moolenaar reported that LPA-induced Rac activation is inhibited by pertussis toxin, an inhibitor of Gi/Go protein function, and requires phosphatidylinositol-3-OH kinase (PI(3)K) activity. Furthermore, he showed that LPA-induced Rac activation depends on the presence of the RhoGEF Tiam1. Thus, cells that lack Tiam1 fail to activate Rac in response to LPA. Because Tiam1-deficient cells show enhanced Rho activation, Moolenaar concluded that LPA receptors couple to a Gi–PI(3)K–Tiam1 pathway to activate Rac, with suppression of Rho activity as a consequence, and thereby stimulate cell spreading and motility.
It is well accepted that certain GPCRs can behave as potent ligand-dependent or -independent oncogenes, and it has recently been shown that the transforming capacity of GPCRs is linked to their ability to initiate mitogen-activated protein kinase (MAPK) cascades through Rho GTPase family members. S.J. Gutkind (Bethesda, MD, USA) showed that certain RhoGEFs, such as PDZ-RhoGEF, link GPCR-mediated activation of Gαq and Gα12 to the activation of Rho GTPases. Gutkind also reported on the transforming potential of a herpes virus (HHV8)-encoded, constitutively active GPCR, designated ORF74. ORF74 has been shown to initiate cellular transformation and tumorigenesis, and to induce an angiogenic phenotype that is mediated by the release of vascular endothelial growth factor. Gutkind described the endothelial-cell-specific expression of ORF74 in transgenic mice that specifically express receptors for subgroup A avian leukosis virus in the vascular endothelium. The effects of ORF74 and other oncogenes were compared, by infection of mice, with recombinant subgroup A avian sarcoma leukosis viruses. Surprisingly, expression of viral ORF74, but not of constitutively active Ras GTPases, induced vascular tumours in the endothelium.
Novel GPCR- and G-protein-associated proteins
There is increasing evidence that GPCRs and G proteins interact with different non-classical binding partners and that this interaction is important in regulating GPCR and G-protein function (Pierce et. al., 2002). One exciting example of a family of recently identified GPCR-associated proteins that regulate mGluR function was presented by L. Fagni (Montpellier, France). He showed that prolonged neural depolarization can induce expression of the Homer1a protein in cultured cerebellar granule cells. Interaction of this protein with the mGlu1a and mGlu5 receptor subtypes controls postsynaptic membrane localization of these receptors, as well as their agonist-dependent and agonist-independent (constitutive) activity (Fig. 3C; Fagni et al., 2002)
To function properly, GPCRs must be transported to their correct subcellular location, as was shown by U. Wolfrum (Mainz, Germany) for rhodopsin in retinal cells. During the microtubule-mediated transport of opsin-containing vesicles from the inner to the outer segments of photoreceptor cells, the last residues of the C-terminal cytoplasmic tail of rhodopsin act as a membrane receptor for cytoplasmic dynein, binding to the dynein light-chain t-complex testis-expressed (Tctex1) protein (Fig. 3D). Interestingly, mutations in these rhodopsin C-terminal residues are found in some patients with retinitis pigmentosa. Wolfrum further reported that newly synthesized molecules of the retinal G protein transducin associate with the Ca2+-binding protein centrin, suggesting a role for centrin in the molecular translocation of transducin to its final destination (Pulvermuller et al., 2002).
S.M. Lanier (New Orleans, LA, USA) described how the activity of heterotrimeric G proteins may be controlled by non-receptor regulatory proteins. He reported on two receptor-independent activators of G protein signalling, AGS3 and AGS4/LGN (Cismowski et al., 2001). These two proteins have similar domain structures, including a series of G protein regulatory (GPR) motifs, and are thought to regulate the signal input to G proteins. AGS3 behaves as a guanine-nucleotide-dissociation inhibitor (GDI) of Gi proteins by binding to Gαi and stabilizing its GDP-bound conformation. LGN is the mammalian counterpart of the Drosophila protein Partner of Inscuteable (Pins) and is known to be involved in the asymmetric division of neuroblasts. Lanier described the subcellular distribution of both of these proteins and proposed that they have a role in the subcellular positioning of signalling proteins. For example, he reported that LGN moves from the nucleus to the midbody structure that separates daughter cells during the later stages of mitosis, which suggests a role for G proteins in cytokinesis.
GPCR physiology and physiopathology
The physiology and function of the Frizzled receptors, a family of GPCRs that are known to be activated by members of the Wnt glycoprotein family, were central to the presentations of F. Schweisguth (Paris, France) and R.T. Moon (Seattle, WA, USA). The activation of Frizzled leads to changes in gene expression, cell behaviour, cell adhesion and cell polarity, and may thus have a role in tumorigenesis (Moon et al., 2002). Schweisguth discussed the function of Frizzled receptors in cell-fate determination during metazoan development. Their stimulation in turn activates the planar cell polarity pathway, which leads to unequal segregation of cell-fate determinants in Drosophila sensory bristles. He showed that a complex of Discs large (Dlg) and Pins responds to Frizzled signalling and is required to establish planar polarity. Moon's presentation focused on the mechanisms of Frizzled signalling and discussed the two other pathways activated by these receptors, the Wnt/β-catenin and Wnt/Ca2+ pathways (Moon et al., 2002). Several Frizzled receptors and receptor orthologues have been identified in invertebrates and vertebrates. Moon reported that rat Frizzled1 and Frizzled2 both form homodimers and heterodimers, although the functional significance is unknown.
E. Borrelli (Strasbourg, France) addressed the existence of two dopamine D2 receptor isoforms, a long (D2L) and a short (D2S) variant, and their relevance to both the control of D2-receptor-specific functions, such as the control of locomotion and pituitary gland physiology, and as conduits for drugs of abuse. Borrelli showed that isoform-specific D2L−/−- and D2S−/−-receptor-deficient mice differ in their control of locomotion. In addition, results obtained from mice that overexpress the D2 receptor isoforms revealed that the D2 receptors are involved in regulating lactotroph cell proliferation and prolactin production in the pituitary gland. Increased expression of D2S, but not D2L, dramatically reduced prolactin expression and led to MAPK induction, which resulted in pituitary hypoplasia. These findings emphasize that the ratio of D2S and D2L expression and the activation of isoform-specific pathways are important determinants of D2 receptor function in vivo.
B. Bettler (Basel, Switzerland) discussed the physiology of GABAB receptors in mice. As mentioned above, heterodimerization of the two GABAB receptor subunits, GABAB1 and GABAB2, is a prerequisite for functional membrane-localized GABAB receptors. Apparently, the GABAB2 receptor mediates GABAB1-receptor surface trafficking (Fig. 2B) and is responsible for G-protein activation (Fig. 2A), whereas the GABAB1 receptor is essential for responses elicited by the binding of GABA (Fig. 2A). Consistent with these findings GABAB1-receptor-deficient mice have lost all pre- and postsynaptic GABAB responses, show spontaneous seizures and hyperalgesia, and have a hyperactive phenotype, which is possibly due to the lack of functional GABAB-receptor-mediated inhibition of dopamine release (Schuler et al., 2001). In addition, Bettler showed that γ-hydroxybutyrate (GHB), an anaesthetic compound used as a drug of abuse (the 'rape pill') or in athletic clubs to stimulate growth hormone release, is certainly acting through GABAB receptors.
Conclusions and perspectives
The meeting in Mont Sainte-Odile provided an excellent opportunity to discuss the recent progress in structural and functional analysis of GPCR signalling. The results presented clearly indicate that the regulatory mechanisms involved extend beyond the simple 1:1:1 interaction of receptors, G proteins and effectors, and that all three of these signalling components are potential points of signal branching, convergence and integration. Dimerization of GPCRs and the binding of GPCRs and G proteins to scaffolding and regulatory proteins contribute to the spatial and/or temporal organization of signal transduction. The influence of the basal activity of both GPCRs and G proteins, and the rates of receptor-induced G protein activation and inactivation, have to be considered to understand the complex signalling events that are initiated by GPCRs. Furthermore, it should be kept in mind that downstream effectors and regulatory proteins contribute to the 'crosstalk' between different GPCRs, for example by connecting pathways, which further increases signalling complexity. A first glimpse of the combinatorial complexity of such a network was offered at this meeting.
However, many questions remain unanswered and many puzzles still need to be solved. For example, what will the crystal structures of ligand-activated GPCRs tell us? What are the structural differences between the inactive and the active conformations? How is the signalling specificity of a given GPCR determined? What is the nature of the direct interactions of scaffolding and regulatory proteins with GPCRs?
Many collaborative research approaches will be necessary to resolve these questions. Analysis in model systems, such as yeast and Drosophila, and the generation of gene-deficient and transgenic mice may help to unravel the complexities of GPCR physiology. These studies will undoubtedly lead to the identification of new therapeutic targets and, consequently, to the development of novel therapeutic agents. Let us hope that the spirit of Sainte-Odile will help us in accomplishing this task.


Acknowledgments
This meeting was made possible by a grant from the European Commission (grant number QLG3-CT-2002-30300) and was supported by several pharmaceutical companies. We apologize to the participtants whose work has not been discussed here due to space limitations.
References
- Angers S., Salahpour A. & Bouvier M. (2002) Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol., 42, 409–435. [DOI] [PubMed] [Google Scholar]
- Bockaert J. & Pin J.P. (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J., 18, 1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismowski M.J., Takesono A., Bernard M.L., Duzic E. & Lanier S.M. (2001) Receptor-independent activators of heterotrimeric G-proteins. Life Sci., 68, 2301–2308. [DOI] [PubMed] [Google Scholar]
- Claing A., Laporte S.A., Caron M.G. & Lefkowitz R.J. (2002) Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and β-arrestin proteins. Prog. Neurobiol., 66, 61–79. [DOI] [PubMed] [Google Scholar]
- De Vries L., Zheng B., Fischer T., Elenko E. & Farquhar M.G. (2000) The regulator of G protein signaling family. Annu. Rev. Pharmacol. Toxicol., 40, 235–271. [DOI] [PubMed] [Google Scholar]
- Fagni L., Worley P.F. & Ango F. (2002) Homer as both a scaffold and transduction molecule. Sci. STKE, 137, RE8. [DOI] [PubMed] [Google Scholar]
- Galvez T., Duthey B., Kniazeff J., Blahos J., Rovelli G., Bettler B., Prezeau L. & Pin J.P. (2001) Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J., 20, 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N., Shimada Y., Tsuji Y., Sato T., Yamamoto M., Kumasaka T., Nakanishi S., Jingami H. & Morikawa K. (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature, 407, 971–977. [DOI] [PubMed] [Google Scholar]
- Moon R.T., Bowerman B., Boutros M. & Perrimon N. (2002) The promise and perils of Wnt signaling through β-catenin. Science, 296, 1644–1647. [DOI] [PubMed] [Google Scholar]
- Pierce K.L., Premont R.T. & Lefkowitz R.J. (2002) Seven-transmembrane receptors. Nature Rev. Mol. Cell Biol., 3, 639–650. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A., Giessl A., Heck M., Wottrich R., Schmitt A., Ernst O.P., Choe H.W., Hofmann K.P. & Wolfrum U. (2002) Calcium-dependent assembly of centrin–G-protein complex in photoreceptor cells. Mol. Cell. Biol., 22, 2194–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler V. et al. (2001) Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABAB responses in mice lacking GABAB1. Neuron, 31, 47–58. [DOI] [PubMed] [Google Scholar]
- Vlaeminck-Guillem V., Ho S.C., Rodien P., Vassart G. & Costagliola S. (2002) Activation of the cAMP pathway by the TSH receptor involves switching of the ectodomain from a tethered inverse agonist to an agonist. Mol. Endocrinol., 16, 736–746. [DOI] [PubMed] [Google Scholar]