

Abstract
Loperamide (LOP) is a peripherally acting opioid receptor agonist used for the management of chronic diarrhea through the reduction of gut motility. The lack of central opioid effects is partly due to the efflux activity of the multidrug resistance transporter P-glycoprotein (P-gp) at the blood-brain barrier. The protease inhibitors are substrates for P-gp and have the potential to cause increased LOP levels in the brain. Because protease inhibitors, including tipranavir (TPV), are often associated with diarrhea, they are commonly used in combination with LOP. The level of respiratory depression, the level of pupil constriction, the pharmacokinetics, and the safety of LOP alone compared with those of LOP-ritonavir (RTV), LOP-TPV, and LOP-TPV-RTV were evaluated in a randomized, open-label, parallel-group study with 24 healthy human immunodeficiency virus type 1-negative adults. Respiratory depression was assessed by determination of the ventilatory response to carbon dioxide. Tipranavir-containing regimens (LOP-TPV and LOP-TPV-RTV) caused decreases in the area under the concentration-time curve from time zero to infinity for LOP (51% and 63% decreases, respectively) and its metabolite (72% and 77% decreases, respectively), whereas RTV caused increases in the levels of exposure of LOP (121% increase) and its metabolite (44% increase). In vitro and in vivo data suggest that TPV is a substrate for and an inducer of P-gp activity. The respiratory response to LOP in combination with TPV and/or RTV was not different from that to LOP alone. There was no evidence that LOP had opioid effects in the central nervous system, as measured indirectly by CO2 response curves and pupillary response in the presence of TPV and/or RTV.
Loperamide (LOP; Imodium, McNeil-PPC, Inc.) is a peripherally acting opioid receptor agonist that reduces gut motility and that is used for the management of chronic diarrhea (8, 25). The principal metabolic fate of loperamide in humans involves oxidative N-dealkylation to N-demethyl-loperamide as the principal metabolite. In human liver microsomes, cytochrome P450 3A4 (CYP3A4) appears to be the major isozyme responsible for loperamide metabolism, with minor contributions from CYP2B6 (9). At the doses used to control diarrhea, LOP has very poor penetration of the blood-brain barrier and produces no central opioid effects, such as respiratory depression, pupillary constrictions, analgesia, or changes in alertness (26). The poor central nervous system (CNS) penetration is attributed both to LOP active cellular efflux via the multidrug resistance transporter P-glycoprotein (P-gp) in the blood-brain barrier and to low systemic oral bioavailability (24). When P-gp is inhibited, LOP and its metabolites may potentially enter the brain and cause opioid-induced central neurological adverse events (AEs) (23, 24).
Current treatment for human immunodeficiency virus type 1 (HIV-1) infection consists of a combination of antiretroviral agents of different classes. Tipranavir (TPV) is a potent nonpeptidic HIV-1 and HIV-2 protease inhibitor (PI) (27, 28) that is active against laboratory strains and clinical isolates of HIV-1 that are broadly resistant to peptidic PIs (1, 12, 20, 22) and is used for therapy of treatment-experienced patients who are infected with HIV-1 (10, 17, 19). In addition to antiretroviral agents, patients are often concomitantly treated for opportunistic infections and comorbidities or to control side effects. As with other PIs, the most frequent side effect of TPV is diarrhea, which may be treated with LOP (25).
Tipranavir is a substrate for and an inducer of hepatic CYP3A (15) and may also be a substrate for P-gp. On the other hand, ritonavir (RTV) inhibits hepatic and, possibly gastrointestinal CYP3A, thereby potentially altering the systemic bioavailability of PIs like TPV metabolized by this enzyme (11). Because of this metabolic inhibition, RTV is often used to boost and maintain plasma concentrations of coadministered PIs, such as TPV (5, 16). RTV is a substrate for P-gp (13) and also a possible inhibitor of P-gp (4). If it is an inhibitor of P-gp, RTV may decrease the efflux of LOP out of the CNS, thereby increasing the CNS concentration and central opiate effects of LOP. On the other hand, if RTV inhibits only CYP3A, which would produce higher concentrations of LOP with concurrent decreases of the LOP metabolite, the concurrent use of LOP and RTV would be devoid of CNS activity (26). Because there is the potential for the use of RTV-boosted TPV (TPV-RTV) together with LOP, all three of which are substrates for P-gp, this study assessed the pharmacodynamic (PD) and pharmacokinetic (PK) interactions of LOP with TPV, RTV, and the combination TPV-RTV in HIV-1-negative, healthy adults. The respiratory response to LOP alone and after administration of TPV, RTV, and TPV-RTV were the primary end points in this study. A secondary pharmacodynamic end point was the pupillary response to LOP after administration of TPV, RTV, or TPV-RTV, as measured by the ratio between the diameter of the pupil and the diameter of the iris.
MATERIALS AND METHODS
In vitro Caco-2 cell permeability experiments.
Caco-2 cells (American Type Culture Collection, Manassas, VA) were grown as monolayers on polycarbonate filters and were cultured for 21 to 25 days, as described previously (14). Briefly, the cells were grown on Transwell (Costar, Cambridge, MA) cell culture inserts (pore size, 0.4 μm; not collagen treated) with a diameter of 12 mm. The cells were directly plated on the filters at 80,000 cells/cm2.
For apical to basolateral experiments, a [14C]tipranavir solution (∼0.15 μCi/ml or 4.9 μg/ml) was placed on the apical side of the cells, and the amount of drug that permeated the cells was determined by moving the inserts to new wells containing fresh medium. In contrast, for basolateral to apical experiments, the test solution was placed on the basolateral side and samples were taken from the apical side of the cells and replaced with fresh medium of Hank's buffered salt solution (HBSS; Gibco) with bovine serum albumin (Sigma) at discrete intervals. The monolayers were then rinsed with fresh cold HBSS and cut from the plastic well, placed into separate vials with 1.0 ml methanol, sonicated, and analyzed for drug content by liquid scintillation counting. Transport rates (J) were calculated by determining the cumulative amounts of drug that permeated as a function of time. The permeability coefficient (PCaco-2) was calculated as the ratio of the transport rate to the initial concentration of the solute in the donor chamber (C0) and the surface area of the filter (A), as described in equation 1:
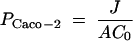 |
1 |
When an efflux transporter inhibitor (quinidine, verapamil, or LY335979) was used during the permeability experiment, it was used in all media, during the preincubation, and on both sides of the monolayer.
Study design.
A randomized, open-label, parallel-group phase I study was carried out with healthy HIV-1-negative adults over 24 days. All subjects were admitted to the study clinic for the entire duration of the study. The study sequentially evaluated the PK and PD responses after the administration of LOP alone, LOP-TPV, LOP-RTV, or LOP-TPV-RTV. The study used a TPV-RTV dose of 750 mg/200 mg, which is 50% higher than the TPV-RTV dose of 500 mg/200 mg being marketed for anti-HIV therapy. The 16-mg dose of LOP was based on the manufacturer's recommendation as the maximum dose for a 24-h period.
The subjects were randomized to each of two treatment groups: (i) group 1 received LOP→LOP-TPV→LOP-TPV-RTV and (ii) group 2 received LOP→LOP-RTV→LOP-TPV-RTV.
Subjects were randomized in blocks of two to receive either TPV 750 mg twice a day (group 1) or RTV 200 mg twice a day (group 2) for 5.5 days, followed by a 2-day period in which no study drugs were administered (days 10 and 11) and then administration of TPV 750 mg and RTV 200 mg twice a day for 10.5 days. TPV or RTV was administered for 5.5 days to ensure the achievement of steady state prior to sampling for pharmacokinetic analysis on day 9. The choice of the 10.5-day interval of drug administration for TPV and RTV was based on the need to establish steady-state TPV-RTV levels before the critical pharmacodynamic evaluations on day 22. The trial design is summarized in Fig. 1.
FIG. 1.

Study design and timeline.
The trial was conducted in compliance with the principles set forth in the Declaration of Helsinki, in accordance with the International Conference on Harmonization, the guidelines for Good Clinical Practice, and applicable regulatory requirements. All subjects gave written informed consent.
Subject selection.
Healthy (HIV-1, hepatitis B virus, and hepatitis C virus negative), nonsmoking men and women ages 18 to 60 years with body mass indices between 18 and 35 kg/m2 were eligible to participate in the study. The subjects had to have normal laboratory values (less than or equal to grade 1 on the Division of AIDS [DAIDS] Table for Grading Severity of Adult Adverse Experiences). The criteria used to exclude subjects from study were the consumption of products or medications that might potentially alter plasma levels of study medications within 14 days of study onset; a history of gastrointestinal, hepatic, or renal disorders within 60 days of study entry; a history of tobacco or alcohol abuse; and/or seated systolic blood pressure <100 mm Hg or >150 mm Hg.
Study drug administration.
There were four orally administered treatments in this study: LOP (Imodium A-D 2-mg capsules; McNeil-PPC, Inc.), TPV (Aptivus 250-mg capsules; Boehringer Ingelheim), RTV (Norvir 100-mg capsules; Abbott Laboratories), and TPV-RTV (Aptivus and Norvir).
Subjects were administered LOP as a single dose of 16 mg alone on day 1 at 9:00 a.m. The subjects were then randomly assigned to group 1 or group 2. On days 2 and 3, no study drugs were administered. On days 4 to 8 (8:00 a.m. and every 12 h thereafter) and day 9 at 8:00 a.m., group 1 subjects were administered TPV 750 mg twice a day for 5.5 days and group 2 subjects were administered RTV 200 mg twice a day for 5.5 days. On day 9, after the TPV or RTV dose at 8:00 a.m., LOP was administered at 9:00 a.m. as a single dose of 16 mg. On days 10 and 11, no study drugs were administered. From day 12 through the morning of day 22, group 1 and group 2 subjects received 10.5 days of TPV 750 mg and RTV 200 mg twice a day starting at 8:00 a.m. on day 12 and every 12 h thereafter, with the last dose taken at 8:00 a.m. on day 22. After the TPV-RTV dose at 8:00 a.m., LOP was administered to subjects in both groups 1 and 2 as a single dose of 16 mg at 9:00 a.m. Study drugs were administered with 8 fluid ounces of water, and the subjects were instructed to swallow the medication whole. When more than one medication was administered, the order of administration was TPV and/or RTV and then LOP an hour later. All administrations of study drugs were directly observed by study personnel at the study site to ensure compliance.
Sampling for pharmacokinetic analysis.
Subjects fasted for at least 12 h prior to the time that blood was drawn (days 1, 9, 11, 17, 21, and 24). On day 1, subjects were permitted to have meals 4 and 11 h after the single oral dose of LOP in the morning. On days 9, 21, and 22, the subjects were permitted to have meals 5 and 10 h after the TPV, RTV, or TPV-RTV oral dose in the morning.
Blood samples (5 ml) for plasma separation were collected for determination of the values of the pharmacokinetic parameters for LOP and the major loperamide metabolite, N-demethyl-loperamide, on day 1 (LOP alone, 16 mg [n = 24]), day 9 (LOP 16 mg plus TPV 750 mg [n = 12] and LOP 16 mg plus RTV 200 mg [n = 11]), and day 22 (LOP 16 mg plus TPV 750 mg and RTV 200 mg [n = 24]). Blood samples for LOP were collected at 10 min and 30 min and 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 7, 9, 11, 12, 24, 36, 48, and 60 h postdosing.
Blood samples (5 ml) for plasma separation were collected for the determination of the values of the pharmacokinetic parameters for TPV and RTV at steady state on day 9 (LOP 16 mg plus TPV 750 mg [n = 12] and LOP 16 mg plus RTV 200 mg [n = 11]), day 21 (TPV 750 mg and RTV 200 mg [n = 24]), and day 22 (LOP 16 mg, TPV 750 mg, and RTV 200 mg [n = 24]). Blood samples for analysis of TPV and RTV on days 9 and 22 were collected by using the same sampling regimen used for LOP, except that the samples were collected at 8, 10, and 61 h postdosing instead of at 9, 11, and 60 h postdosing and a sample collection at 5.5 h postdosing was added. On day 12, blood samples for analysis of TPV and RTV were collected at 10 min and 30 min and then at 1, 1.5, 2, 3, 4, 5, 6, 8, 10, and 12 h postdosing.
Pharmacokinetics. (i) Loperamide and N-demethyl-loperamide assay.
The analytical method used for the determination of LOP and N-demethyl-loperamide levels in human plasma was a modification of a method reported previously (7). Briefly, an aliquot of heparinized human plasma containing LOP and N-demethyl-loperamide plus loperamide-d6 (internal standard) was extracted by a liquid-liquid extraction procedure. The extracted samples were analyzed with a high-pressure liquid chromatography system with a Sciex API III mass spectrometer. Quantitation of the analytes was done by determination of the peak area ratio. Positive ions were monitored in the selected reaction-monitoring mode. A weighted quadratic regression was used to determine the concentrations of LOP and N-demethyl-loperamide. The nominal upper and lower limits of the calibration curve ranged from 25.0 pg/ml to 5,000 pg/ml.
(ii) Tipranavir and ritonavir assay.
Plasma samples were analyzed for TPV and RTV levels as described previously (15). Briefly, TPV, RTV, and the internal standards were extracted from human heparinized plasma by a two-step liquid-liquid extraction method that used an ethyl acetate-hexane mixture, followed by a hexane wash. The analytes were separated and detected with a liquid chromatography-mass spectrometry-mass spectrometry system that used a Synergi Polar RP column (2.0 by 30 mm) with a formic acid-acetic acid-acetonitrile mobile phase. Late-eluting interferences were eliminated with a low-dead-volume, step-gradient flushing system. The extraction was automated by use of a 96-well format technology. Calibration curves were obtained by using a 1/concentration2 weighted quadratic regression of the peak ratio versus the concentration. High and low calibration ranges were used to predict unknown concentrations. The high calibration curve ranged from 1,000 ng/ml to 20,000 ng/ml. The low calibration curve ranged from 25.0 ng/ml to 2,000 ng/ml.
(iii) Pharmacokinetic modeling.
The pharmacokinetic parameters for LOP, the LOP metabolite, TPV, and RTV were calculated by standard pharmacokinetic techniques (WinNonlin; Pharsight Corporation, Mountain View, CA). Drug-drug interactions were assessed on the basis of 90% confidence intervals for the geometric mean ratios of selected PK parameters, i.e., for LOP and the LOP metabolite, the area under the concentration-time curve (AUC) and the maximum concentration of drug in plasma (Cmax); for TPV-RTV, AUC, Cmax, and the concentration of drug in plasma at 12 h postdosing (Cp12 h).
Pharmacodynamics. (i) Respiration assessments.
The respiratory response to LOP alone and after administration of TPV, RTV, and TPV-RTV was the primary end point in this study. The respiratory response was measured by assessing the maximum decrease in the mean percent baseline CO2 response slope (observed at one of the examination time points during the 6-h rebreathing test) and the AUC from 0 to 6 h (AUC0-6) for the percent baseline CO2 response slope profile. The ventilatory response to CO2 was measured on days 1, 9, and 22.
Pharmacodynamic assessments were made by using the classical Read rebreathing technique to monitor the central control of ventilation (21). This technique has been shown to have the sensitivity needed to detect the respiratory depression induced by the central effects of opiates (2) and by LOP in RTV-treated healthy volunteers (26). The standard rebreathing gas mixture for the Read test of central ventilatory control (7% CO2 and 93% O2) was used in the present study.
Prior to testing on day 1, each subject's vital capacity (VC) was measured with CPX/D instrumentation (Medical Graphics Corp., St Paul, MN). At the beginning of each respiratory evaluation, a pulse oximetry device was placed on the subject's finger, the subject's nose was clamped, and the subject was asked to breathe through a mouthpiece with a three-way valve. The mouthpiece contained a pneumotach for continuous measurement of air flow and gas sampling ports for continuous side-stream measurement of partial pressure of carbon dioxide (pCO2) and oxygen (pO2). To allow the monitored ventilatory parameters to stabilize, the subject breathed room air for 2 to 5 min. The three-way valve was then switched from room air to a sealed bag of rebreathing gas with the gas volume equal to 1.5 times the subject's vital capacity. The subject rebreathed from the sealed bag until the end-tidal pCO2 (pETCO2) reached a cutoff value of 50 to 60 mm Hg. At this point, the test was terminated and the subject then breathed room air. The CPX/D machine calculated nine ventilatory parameters from the pCO2 and pO2: O2 consumption rate (VO2), CO2 production rate (VCO2), respiratory exchange rate (RER), respiration rate (RR), tidal volume (Vt), minute ventilation (VE), pETCO2, end-tidal pO2 (pETCO2), and the fractional content of O2 in inhaled gas (F1O2).
The individual PD observations used to construct AUC0-6 were derived from the rebreathing test. The rebreathing test data (VE-pETCO2 relationship) at each time point were summarized by fitting the data by the method of least-squares means to a linear regression model that relates the amount of ventilation rate (liters/min) to the pETCO2 (mm Hg). The slope of this regression was expressed relative to the baseline slope, determined from measurements taken just before administration of LOP for the same subject on the same day. The results of the rebreathing tests for the first 6 h after LOP administration were summarized by the area under the pharmacodynamic effect-time curve by using the trapezoidal rule.
(ii) Pupillary response.
The secondary pharmacodynamic end point in this study was the pupillary response to LOP after administration of TPV, RTV, or TPV-RTV, as measured by the ratio between the diameter of the pupil and the diameter of the iris. A decrease in the ratio of any magnitude was considered to be of clinical significance. Pupillary response measurements were taken on days 1, 9, and 22. The subject's left eye was photographed from a fixed distance with a 35-mm camera (Nikon DIx), attached to a chin headrest and equipped with a micro-Nikon lens and an SB-29 Macro Speedlight. The pupil and iris diameters in their largest axes were measured by using the Adobe Photoshop 7.0 software measuring tool.
Safety.
The onset, duration, and intensity (mild, moderate, or severe) of AEs were recorded. Based on the design of the study, AEs were summarized by using two treatment definitions. The type 1 definitions evaluated the AEs by nonoverlapping treatment periods, and the type 2 definitions grouped AEs by the type of study therapy. For the type 1 treatment definition grouping, the AEs were grouped by each nonoverlapping study phase as follows: LOP (days 1 to 3 for all subjects), TPV (days 4 to 8 for group 1 subjects), RTV (days 4 to 8 for group 2 subjects), TPV-LOP (days 9 to 11 for group 1 subjects), RTV-LOP (days 9 to 11 for group 2 subjects), TPV-RTV (days 12 to 21 for all subjects), TPV-RTV-LOP (days 22 to 25 for all subjects), and posttreatment (days ≥26 for all subjects). The type 2 treatment definition grouped the AEs by type of therapy: LOP alone (days 1 to 3 for all subjects), TPV alone (days 4 to 11 for group 1 subjects), RTV alone (days 4 to 11 for group 2 subjects), TPV-RTV (days 12 to 25 for all subjects), and any TPV (days 4 to 25 for group 1 and days 12 to 25 for group 2). The results of the safety data presented below primarily focus on the grouping of AEs by using the type 2 definitions, with presentation of data for the TPV-RTV and LOP alone groups.
Clinical laboratory testing for safety consisted of hematology and chemistry panels, urinalysis, and a serum pregnancy test (serum β-human chorionic gonadotropin). Standard 12-lead electrocardiograms were obtained in triplicate at screening, on day 1 prior to drug administration, and at the end of the treatment period on day 24. Pulse rate and blood pressure were determined and recorded at each visit. The DAIDS Table for Grading Severity of Adult Adverse Experiences was used to identify clinically significant (grade 3 or grade 4) laboratory test abnormalities. For laboratory tests without DAIDS grading, the Eastern Cooperative Oncology Group or the National Cancer Institute Common Toxicity Criteria grading systems were used.
Statistical analysis.
A sample size of eight subjects within each treatment group was justified by power calculations based on a paired comparison (two-sided distribution [α] = 0.05) with the ability to detect a reduction of 10% in the PD parameter AUC0-6 for respiratory response slopes while assuming a coefficient of variation of 9% with 80% power (23).
The difference in respiratory response between LOP-TPV-RTV and LOP alone was the primary comparison of the PD response, whereas the secondary comparison was the difference in respiratory response between LOP-TPV or LOP-RTV and LOP alone. The Wilcoxon signed rank test was used for the respiratory response comparisons, measured by the maximum decrease in the mean percent baseline CO2 response slope. Analysis of variance was used for AUC0-6 for the percent baseline CO2 response slope profile. A clinically relevant change in the respiratory response to CO2 was defined a priori as a 10% decrease in the area under the pharmacodynamic effect-time curve or at least a 25% decrease in at least one pharmacodynamic time point.
Descriptive statistics were used to analyze pupillary dilation and safety end points. Pharmacokinetic end points were considered secondary end points, and so descriptive statistics were deemed adequate for analysis.
RESULTS
In vitro Caco-2 cell permeability experiments.
In the presence of 0.25% (wt/vol) bovine serum albumin in HBSS, tipranavir has a very low permeability value (0.48 × 106 cm/s) comparable to that of the impermeable paracellular marker mannitol. This low permeability value may be the result of an interaction of tipranavir with an efflux transporter pump or the high level of protein binding of >99.98% for tipranavir (15). The permeability directional ratio (PDR) value of 5.9 for tipranavir is consistent with the observation that tipranavir may be a substrate for cellular efflux transporters (Table 1). A PDR value of 1 indicates passive diffusion.
TABLE 1.
Tipranavir Caco-2 permeability measurements in the presence of substrates and inhibitors
| Drug with 0.25% (wt/vol) BSAa | Permeability from A to B (cm/s [106])b | Mass balance (%) | Permeability from B to A (cm/s [106])b | Mass balance (%) | PDR |
|---|---|---|---|---|---|
| TPV | 0.48 ± 0.05 | 92.6 | 3.14 ± 0.24 | 98.4 | 5.9 |
| TPV + ritonavir 0.2 μg/ml | 0.57 ± 0.00 | 96.2 | 3.41 ± 0.18 | 95.4 | 6.0 |
| TPV + ritonavir 2.0 μg/ml | 0.55 ± 0.02 | 97.0 | 3.26 ± 0.11 | 92.1 | 5.9 |
| TPV + digoxin 30 μM | 0.48 ± 0.02 | 98.0 | 2.81 ± 0.12 | 98.5 | 5.9 |
| TPV + quinidine 100 μM | 0.78 ± 0.20 | 92.1 | 0.98 ± 0.12 | 87.5 | 1.3 |
| TPV + verapamil 100 μM | 2.01 ± 0.08 | 94.6 | 1.68 ± 0.16 | 91.0 | 0.8 |
| TPV + verapamil 100 μM | 3.08 ± 0.20 | 92.8 | 1.46 ± 0.04 | 88.3 | 0.5 |
| TPV + LY335979 1.0 μM | 0.61 ± 0.05 | 92.0 | 0.69 ± 0.08 | 89.6 | 1.1 |
BSA, bovine serum albumin.
Permeability values determined in triplicate, with means ± standard deviation reported. A to B, apical to basolateral; B to A, basolateral to apical.
The permeability of tipranavir in the presence of several P-gp inhibitors (quinidine, verapamil, and LY335979) and a P-gp substrate (digoxin) was determined (Table 1) to evaluate whether TPV is a substrate for cellular efflux pumps. Addition of digoxin to the media had no effect on the permeability of TPV, indicating that either P-gp has a higher affinity for TPV than digoxin or the two drugs bind to different sites of the protein. The PDR value for tipranavir decreased to ≤1 in the presence of P-gp inhibitors (Table 1), consistent with TPV being a substrate for a cellular efflux transporter. The noncompetitive inhibitors quinidine and LY335979 decreased the TPV PDR value by decreasing the permeability in the basolateral to apical direction, whereas the competitive inhibitor verapamil reduced the PDR by increasing the permeability in the apical to basolateral direction and not changing the permeability in the basolateral to apical direction.
Subject demographics and baseline characteristics.
Twenty-four healthy volunteers were randomized into two groups. Because four of the subjects received a different gas mixture in the rebreathing test, the pharmacodynamic evaluation was based on the data for 20 subjects. The baseline demographics for the subjects enrolled in the trial are provided in Table 2.
TABLE 2.
Baseline demographics
| Group | Age (yr)
|
Gender (no. [%])
|
Race (no. [%])
|
Ht (cm)
|
Weight (kg)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Median | Mean ± SD | Range | Male | Female | White | Black | Asian | Median | Mean ± SD | Range | Median | Mean ± SD | Range | |
| Group 1a (n = 12) | 36.0 | 36.9 ± 9.6 | 21-52 | 5 (41.7) | 7 (58.3) | 10 (83.3) | 2 (16.7) | 0 (0.0) | 168.0 | 166.9 ± 8.5 | 152-180 | 73.95 | 73.95 ± 12.29 | 51.3-95.3 |
| Group 2b (n = 12) | 28.5 | 30.1 ± 8.0 | 21-51 | 9 (75.0) | 3 (25.0) | 9 (75.0) | 2 (16.7) | 1 (8.3) | 174.0 | 173.7 ± 4.5 | 165-180 | 77.35 | 79.11 ± 13.25 | 63.0-106.6 |
| Total (n = 24) | 31.5 | 33.5 ± 9.3 | 21-52 | 14 (58.3) | 10 (41.7) | 19 (79.2) | 4 (16.7) | 1 (4.2) | 170.0 | 170.3 ± 7.5 | 152-180 | 73.95 | 76.53 ± 12.77 | 51.3-106.6 |
The sequence of treatments on days 1, 9, and 22 was LOP, LOP-TPV, and LOP-TPV-RTV.
The sequence of treatments on days 1, 9, and 22 was LOP, LOP-RTV, and LOP-TPV-RTV.
The mean baseline ventilatory characteristics (while breathing room air and prior to any drug treatment) were consistent with expected normal values. These mean baseline values were as follows: VO2 = 252 ± 13 ml/min, VCO2 = 200 ± 9 ml/min, RER = 0.83 ± 0.03, RR = 18 ± 1 breaths/min, Vt = 771 ± 73 ml, VE = 11.5 ± 1.1 ml/min, pETO2 = 118 ± 8 mm Hg, pETCO2 = 39 ± 1 mm Hg, and FIO2 = 22.6% ± 0.9%.
Respiratory response.
The difference in the respiratory response with the coadministration of LOP-TPV-RTV, LOP-TPV, and LOP-RTV, as measured by the AUC0-6 (±standard error of the mean [SEM]) for the percent baseline VE-pETCO2 response slope profile, compared to the respiratory response with the administration of LOP alone was not statistically significant. Figure 2 shows the respiratory response curves for the three comparisons over the 6-h period following administration of the study drugs. The respiratory response profiles between the LOP alone versus the LOP-TPV-RTV treatments, LOP alone versus LOP-TPV, and LOP alone versus LOP-RTV were all similar. There were no statistically significant differences in the mean percent baseline slopes over the 6 h of testing, and any differences were not considered clinically relevant. The only statistically significant change was the 27% increase from the baseline (P = 0.03) at the 2-h time point for the LOP-RTV slope.
FIG. 2.


(a) Mean percent baseline ventilatory slope (±2 SEMs) for LOP alone (n = 20) compared with that for LOP-TPV-RTV treatment (n = 20); AUC0-6 (±SEM) for the percent baseline VE-pETCO2 response slope profile was not statistically significantly different from that observed for LOP alone (633.0 ± 38.6 versus 622.4 ± 31.4 baseline slope · h; P = 0.84); (b) mean percent baseline ventilatory slope (±2 SEMs) for LOP alone (n = 10) compared with that for LOP-TPV treatment (n = 10); AUC0-6 (±SEM) for the percent baseline VE-pETCO2 response slope profile was not statistically significantly different from that observed for LOP alone (633.6 ± 59.5 versus 623.6 ± 48.5 baseline slope · h; P = 0.84); (c) mean percent baseline ventilatory slope (±2 SEMs) for LOP alone (n = 10) compared with that for LOP-RTV treatment (n = 9); AUC0-6 (±SEM) for the percent baseline VE-pETCO2 response slope profile was not statistically significantly different from that observed for LOP alone (687.8 ± 59.2 versus 621.2 ± 45.1 baseline slope · h; P = 0.21).
Pupillary response.
The mean pupil diameter-to-iris diameter ratio (±SEM) at the baseline prior to LOP administration on day 1 was not different from the mean ratio at the baseline prior to the administration of LOP-TPV-RTV on day 22 (0.59 ± 0.02 at both times). Administration of LOP alone or LOP-TPV-RTV did not affect this ratio.
There was no statistically significant difference between the pupillary response, as measured by the baseline mean pupil diameter-to-iris diameter ratio ± SEM, prior to the administration of LOP alone on day 1 versus those prior to the administration of LOP-TPV (LOP, 0.57 ± 0.03; LOP-TPV, 0.56 ± 0.03) or LOP-RTV (LOP, 0.61 ± 0.02; LOP-RTV, 0.61 ± 0.02) on day 9. With both treatments, no change in the pupil diameter-to- iris diameter ratio with time after drug treatment was observed. The mean pupillary responses for LOP-TPV and LOP-RTV were within 2 standard errors of the mean compared to that for LOP alone over the 6 h of testing.
Pharmacokinetics.
The PK parameters of LOP and its metabolite, N-demethyl-loperamide, in the presence of TPV, RTV, and TPV-RTV are shown in Table 3. Tipranavir and TPV-RTV resulted in significant decreases in the AUC from time zero to infinity (AUC0-∞) and Cmax for LOP and LOP metabolite. In contrast, these PK parameters increased in the presence of RTV coadministration (Fig. 3). Since the LOP metabolite concentrations also decreased in the presence of TPV and TPV-RTV (Table 3), the increased clearance for LOP in the presence of TPV or TPV-RTV can be attributed to a reduction in systemic bioavailability.
TABLE 3.
Effects of TPV, RTV, and TPV-RTV on LOP pharmacokineticsa
| PK parameter | LOP alone (n = 24), day 1
|
LOP-TPV (n = 12), day 9
|
LOP-RTV (n = 11), day 9
|
LOP-TPV-RTV (n = 24), day 22
|
||||
|---|---|---|---|---|---|---|---|---|
| LOP | LOP metabolite | LOP | LOP metabolite | LOP | LOP metabolite | LOP | LOP metabolite | |
| Cmax (ng/ml) | ||||||||
| Geometric mean | 3.2 | 5.5 | 1.4 | 1.9 | 5.5 | 5.3 | 1.2 | 1.1 |
| % Change | ↓58 | ↓66 | ↑83 | ↓1 | ↓61 | ↓79 | ||
| Range | 0.29-0.62 | 0.27-0.43 | 1.23-2.73 | 0.78-1.25 | 0.31-0.48 | 0.17-0.25 | ||
| AUC0-∞ (ng · h/ml) | ||||||||
| Geometric mean | 58.3 | 227.4 | 22.0 | 64.4 | 121.1 | 309.8 | 28.8 | 51.9 |
| % Change | ↓63 | ↓72 | ↑121 | ↑44 | ↓51 | ↓77 | ||
| Range | 0.27-0.51 | 0.23-0.33 | 1.53-3.19 | 1.00-2.09 | 0.40-0.61 | 0.19-0.27 | ||
| CL/F (liters/h) | ||||||||
| Geometric mean | 275 | 728 | 132 | 556 | ||||
| % Change | ↑264 | ↓52 | ↑202 | |||||
Data are expressed as geometric mean; comparisons expressed as the geometric mean difference and 90% confidence intervals about the ratio. No effect is a 0% change and a 90% confidence interval encompassing 1.00.
FIG. 3.

Geometric mean plasma loperamide concentrations alone (n = 24) and in the presence of RTV (n = 11), TPV (n = 12), and TPV-RTV (n = 24), demonstrating that RTV increases LOP concentrations during coadministration, while TPV and TPV-RTV decrease LOP concentrations during coadministration.
The effects of single-dose LOP on the steady-state PK of TPV-RTV were assessed by comparing the PK of TPV-RTV alone on day 21 to the steady-state PK of TPV-RTV plus LOP on day 22. Table 4 demonstrates that only Cp12 h for TPV was affected by LOP coadministration (decrease of 26%). For RTV, however, Cp12 h, Cmax, and AUC0-12 were decreased in the presence of LOP by 30%, 28%, and 22%, respectively (Table 4).
TABLE 4.
Changes in pharmacokinetic parameters of TPV and RTV in the presence of LOPa
| PK parametera | TPV
|
RTV
|
||||
|---|---|---|---|---|---|---|
| % Change | Geometric mean ratio | 90% CIb | % Change | Geometric mean ratio | 90% CI | |
| Cp12 h | ↓26 | 0.74 | 0.62, 0.88 | ↓30 | 0.70 | 0.55, 0.87 |
| Cmax | ↑3 | 1.03 | 0.92, 1.17 | ↓28 | 0.72 | 0.50, 1.04 |
| AUC0-12 | ↓2 | 0.98 | 0.86, 1.12 | ↓22 | 0.78 | 0.59, 1.04 |
Data are for 24 subjects.
CI, confidence interval.
Safety. (i) Adverse events.
Overall, 70.8% of the subjects (17 of 24) experienced AEs during the TPV-RTV treatment period, whereas 37.5% (9 of 24), regardless of causality, experienced AEs during the period of administration of LOP alone. The most frequently observed AEs during TPV-RTV treatment, regardless of causality, were loose stools (9 of 24 subjects), nausea (8 of 24 subjects), abdominal pain (7 of 24 subjects), headache (6 of 24 subjects), vomiting (4 of 24 subjects), dyspepsia (3 of 24 subjects), and maculopapular rash (3 of 24 subjects). With LOP alone, the most frequently observed AEs were headache (3 of 24 subjects) and constipation (2 of 24 subjects). While taking TPV-RTV, 66.7% of the subjects (16 of 24) experienced AEs considered treatment related, whereas 25% of the subjects (6 of 24) experienced AEs considered treatment related while taking LOP alone. None of the adverse events were considered serious or severe, and no subjects were withdrawn from the study because of adverse events.
(ii) Laboratory test abnormalities.
Nine subjects had grade 3 or grade 4 laboratory test abnormalities which were considered clinically significant. One subject in group 2 had a decrease in hematocrit value while receiving the LOP plus TPV-RTV treatment. Eight subjects had clinically significant increases (DAIDS grade 3 or grade 4) in alanine aminotransferase (ALT) values (four in each treatment group). Four of these eight subjects also had clinically significant elevations in aspartate aminotransferase (AST) values. No clinically significant laboratory test abnormalities were observed for total cholesterol, high-density lipoprotein, low-density lipoprotein, triglycerides, or any of the other clinical chemistry or hematology tests.
DISCUSSION
LOP and its metabolite have the potential to produce CNS opioid effects if the efflux transporter at the blood-brain barrier is inhibited (23, 26). The results of this study obtained by using a maximal dose of LOP (16 mg) and a 50% supratherapeutic dose of TPV-RTV (750 mg/200 mg) show that coadministration of LOP with TPV, RTV, or TPV-RTV did not result in clinically relevant CNS opioid effects, as determined by monitoring the subjects (HIV-1-negative, healthy individuals) for any responses known to be classic central effects of opiates, i.e., the respiratory response to CO2 and the pupillary response.
P-glycoprotein is an ATP-dependent efflux pump that transports a wide variety of agents out of cells at the blood-brain barrier, thereby restricting CNS penetration of many drugs, including LOP. LOP, TPV, and RTV are substrates for P-gp; and an interaction in efflux can cause LOP to accumulate in the brain. Sadeque et al. demonstrated in humans that quinidine-induced inhibition of P-gp resulted in LOP-induced respiratory depression (23). However, Tayrouz et al. found that 16 mg LOP plus 600 mg RTV in humans did not induce respiratory depression (26) and concluded that RTV does not substantially inhibit P-gp at the blood-brain barrier. When it is considered that the dose of RTV used in the present study was lower than that used in the study by Tayrouz et al. (26), the lack of LOP-induced CNS opioid effects with RTV administration reported here is to be expected.
TPV and TPV-RTV are substrates for P-gp efflux from cells, as demonstrated in vitro by the high PDR value (5.9) observed in the Caco-2 cell experiments. This PDR value was decreased to a value ≤1.0 when P-gp inhibitors (quinidine, verapamil, and LY335979) were added to the media. Efflux pumps, such as P-gp, are ATP-dependent transport proteins found on the apical side of Caco-2 cells. By comparing the apical to basolateral and basolateral to apical permeation of [14C]tipranavir, the affinity of tipranavir for efflux transport was estimated. The use of known inhibitors in the media confirmed the P-gp activity.
The lack of an interaction for TPV and TPV-RTV on P-gp transport at the blood-brain barrier, as measured in this study by a lack of a PD response following coadministration, was consistent with the lack of an interaction observed between TPV and digoxin in vitro, even though they are both substrates for P-gp. What was surprising in this study was the diminishment in systemic LOP exposure that was not attributable to an increased metabolic clearance. Instead, the bioavailability of LOP decreased and, because of its precursor-successor relationship, the formation of the LOP metabolite decreased whenever TPV or TPV-RTV was coadministered with LOP. This finding is consistent with the induction of P-gp by TPV, which thereby lowers the oral clearance by decreasing the fraction of LOP absorbed. This induction of P-gp by TPV occurs in the presence of RTV, a substrate for P-gp and a potential gastrointestinal P-gp inhibitor (4). Following the administration of multiple doses of TPV-RTV and the achievement of steady state, systemic RTV levels are known to be lower than the levels observed following administration of a single dose of RTV (15), consistent with P-gp induction by TPV-RTV. The clinical relevance of changes in the systemic levels of LOP is not clear, since its primary pharmacologic activity occurs in the gut.
A variety of methods for monitoring the respiratory response are used, e.g., the monitoring of transcutaneous pO2 and pCO2 (26), the steady-state technique (2), and the rebreathing technique with a variety of gas mixtures (21, 23). The choice of gas mixture must take into consideration the fact that signals from the central and peripheral chemoreceptors interact in a complex fashion to control ventilation (6). To suppress the influence of peripheral processes on VE, the rebreathing gas mixture used in this study was 7% CO2 and 93% O2. Increments in CNS pCO2 increase the sensitivity of peripheral chemoreceptors to pO2, while decrements in arterial pO2 increase the sensitivity of the central chemoreceptors to pCO2. Therefore, arterial pO2 must remain well above the normal value throughout the rebreathing test to prevent potentially confounding effects of peripheral chemoreception on VE. This is accomplished by using a rebreathing gas containing a high percentage of O2.
The Read method used in this study to assess respiratory depression from LOP included CO2 in the gas mixture and is an improvement over the method used by other researchers (2, 6). The presence of CO2 in the rebreathing gas is crucial; it serves to effectively clamp arterial pCO2 (pETCO2) at the value of venous pCO2 (tissue pCO2). Only under this condition does pETCO2 provide a reasonable measure of CNS pCO2, while the relationship between pETCO2 and VE provides a monitor of central ventilatory control (18, 21).
Overall, the safety profile in the present study was similar to that observed in previous clinical trials with TPV-RTV administered to healthy HIV-1-negative volunteers (15). No new or unexpected safety issues emerged based on comparison with the body of data available from other phase I and phase II studies of TPV-RTV in HIV-1-positive patients (29) or healthy HIV-1-negative volunteers. Diarrhea is a common side effect of all PIs, with an incidence of up to 56% (25). In a recent late-stage clinical development trial comparing treatment with TPV-RTV, amprenavir-ritonavir, saquinavir-ritonavir, or lopinavir-ritonavir, the rate of occurrence of diarrhea was similar across all groups (29) and was somewhat lower than in the present study. The 33% lower TPV-RTV dose (500 mg/200 mg) used in clinical practice is most likely the reason for this difference in incidence (29). In the present study, clinically significant laboratory test abnormalities occurred in nine subjects, including a decreased hematocrit in one subject receiving the TPV-RTV-LOP treatment. Eight subjects, four in each treatment group, had clinically significant increases in ALT values (DAIDS grade 3 and/or grade 4). Four of these eight subjects with elevated ALT values also had clinically significant elevations in AST values. Subjects with ALT and/or AST abnormalities were asymptomatic, and none of these subjects discontinued TPV-RTV.
Despite some alteration in specific PK parameters for LOP and the LOP metabolite, no clinically relevant pharmacodynamic interactions were observed in this study. Concomitant administration of LOP with TPV-RTV was as well tolerated by the study population as TPV-RTV alone. The results of the present study indicate that LOP can be safely coadministered with TPV-RTV for the management of diarrhea for the treatment of HIV-1-infected individuals with no risk of central opioid side effects.
REFERENCES
- 1.Back, N. K., A. van Wijk, D. Remmerswaal, M. van Monfort, M. Nijhuis, R. Schuurman, and C. A. Boucher. 2000. In-vitro tipranavir susceptibility of HIV-1 isolates with reduced susceptibility of other protease inhibitors. AIDS 14:101-102. [DOI] [PubMed] [Google Scholar]
- 2.Bourke, D. L., and A. Warley. 1989. The steady-state and rebreathing methods compared during morphine administration in humans. J. Physiol. 419:509-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chong, K. T., and P. J. Pagano. 1997. In vitro combination of PNU-140690, a human immunodeficiency virus type 1 protease inhibitor, with ritonavir against ritonavir-sensitive and -resistant clinical isolates. Antimicrob. Agents Chemother. 41:2367-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drewe, J., H. Gutmann, G. Fricker, M. Torok, C. Belinger, and J. Huwyler. 1999. HIV protease inhibitor ritonavir: a more potent inhibitor of P-glycoprotein than the cyclosporine analog SDZ PSC 833. Biochem. Pharmacol. 57:1147-1152. [DOI] [PubMed] [Google Scholar]
- 5.Eagling, V. A., D. J. Back, and M. G. Barry. 1997. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br J. Clin. Pharmacol. 44:190-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gold, W. M. 1994. Pulmonary function testing, p. 798-900. In J. F. Murray and J. A. Nadel (ed.), Textbook of respiratory medicine. The W. B. Saunders Co., Philadelphia, Pa.
- 7.He, H., A. Sadeque, J. C. L. Erve, A. J. J. Wood, and D. L. Hachey. 2000. Quantitation of loperamide and N-demethyl-loperamide in human plasma using electrospray ionization with selected reaction ion monitoring liquid chromatography-mass spectrometry. J. Chromatogr. B 744:323-331. [DOI] [PubMed] [Google Scholar]
- 8.Heykants, J., M. Michiels, A. Knaeps, and J. Brugmans. 1974. Loperamide (R 18 553), a novel type of antidiarrheal agent. Part 5. The pharmacokinetics of loperamide in rats and man. Arzneimittelforschung 24:1649-1653. [PubMed] [Google Scholar]
- 9.Kalgutkar, A. S., and H. T. Nguyen. 2004. Identification of an N-methyl-4-phenylpyridinium-like metabolite of the antidiarrheal agent loperamide in human liver microsomes: underlying reason(s) for lack of neurotoxicity despite the bioactivation event. Drug Metab. Dispos. 32:943-952. [PubMed] [Google Scholar]
- 10.Kohlbrenner, V., J. Gathe, G. Pierone, K. Arasteh, R. Rubio, R. LaLonde, P. Piliero, S. McCallister, S. Garfinkel, R. Chaves, G. Mukwaya, C. Dohnanyi, S. Shaw, and D. Mayers. 2003. Tipranavir/ritonavir (TPV/r) demonstrates potent efficacy in multiple protease inhibitor (PI)-experienced patients at 24 weeks (BI 1182.52), abstr. 7.2/2. 9th Eur. AIDS Conf.
- 11.Kumar, G. N., A. D. Rodrigues, A. M. Buko, and J. F. Denissen. 1996. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J. Pharmacol. Exp. Ther. 277:423-431. [PubMed] [Google Scholar]
- 12.Larder, B. A., K. Hertogs, S. Bloor, C. H. van den Eynde, W. DeCian, Y. Wang, W. W. Freimuth, and G. Tarpley. 2000. Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples. AIDS 14:1943-1948. [DOI] [PubMed] [Google Scholar]
- 13.Lee, C. G. L., M. M. Gottesman, C. O. Cardarelli, M. Ramachandra, K.-T. Jeang, S. V. Ambudkar, I. Pastan, and S. Dey. 1998. HIV-1 protease inhibitors are substrates for the MDR1 multidrug transporter. Biochemistry 37:3594-3601. [DOI] [PubMed] [Google Scholar]
- 14.Liang, E., K. Chessic, and M. Yazdanian. 2000. Evaluation of an accelerated Caco-2 cell permeability model. J. Pharm. Sci. 89:336-345. [DOI] [PubMed] [Google Scholar]
- 15.MacGregor, T. R., J. P. Sabo, S. H. Norris, P. Johnson, L. Galitz, and S. McCallister. 2004. Pharmacokinetic characterization of different dose combinations of coadministered tipranavir and ritonavir in healthy volunteers. HIV Clin. Trials 5:371-382. [DOI] [PubMed] [Google Scholar]
- 16.McCallister, S., H. Valdez, K. Curry, T. MacGregor, M. Borin, W. Freimuth, Y. Wang, and D. L. Mayers. 2004. A 14-day dose-response study of the efficacy, safety, and pharmacokinetics of the nonpeptidic protease inhibitor tipranavir in treatment-naive HIV-1-infected patients. J. Acquir. Immune Defic. Syndr. 35:376-382. [DOI] [PubMed] [Google Scholar]
- 17.Mehandru, S., and M. Markowitz. 2003. Tipranavir: a novel non-peptidic protease inhibitor for the treatment of HIV infection. Expert Opin. Investig. Drugs 12:1821-1828. [DOI] [PubMed] [Google Scholar]
- 18.Mohan, R. M., C. E. Amara, D. A. Cunningham, and J. Duffin. 1999. Measuring central-chemoreflex sensitivity in man: rebreathing and steady-state methods compared. Respir. Physiol. 115:23-33. [DOI] [PubMed] [Google Scholar]
- 19.Neubacher, D., M. Markowitz, L. Slater, R. Curry, and S. McCallister. 2003. Long-term 80-week follow-up of highly treatment-experienced (HTE) patients on tipranavir-based antiretroviral therapy (BI 1182.2), abstr. 7.2/3. 9th Eur. AIDS Conf. (EACS), 1st EACS Resist. Pharmacol. Workshop.
- 20.Poppe, S. M., D. E. Slade, K. T. Chong, R. R. Hinshaw, P. J. Pagano, M. Markowitz, D. D. Ho, H. Mo, R. R. Gorman III, T. J. Dueweke, S. Thaisrivongs, and W. G. Tarpley. 1997. Antiviral activity of the dihydropyrone PNU-140690, a new nonpeptidic human immunodeficiency virus protease inhibitor. Antimicrob. Agents Chemother. 41:1058-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Read, D. J. C. 1967. A clinical method for assessing the ventilatory response to carbon dioxide. Australas Ann. Med. 16:20-32. [DOI] [PubMed] [Google Scholar]
- 22.Rusconi, S., S. La Seta Catamancio, P. Citterio, S. Kurtagic, M. Violin, C. Balotta, M. Moroni, M. Galli, and A. d'Arminio-Monforte. 2000. Susceptibility to PNU-140690 (tipranavir) of human immunodeficiency virus type 1 isolates derived from patients with multidrug resistance to other protease inhibitors. Antimicrob. Agents Chemother. 44:1328-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadeque, A. J., C. Wandel, H. He, S. Shah, and A. J. Wood. 2000. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin. Pharmacol. Ther. 68:231-237. [DOI] [PubMed] [Google Scholar]
- 24.Schinkel, A. H., E. Wagenaar, C. Mol, and L. van Deemter. 1996. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Investig. 97:2517-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherman, D. S., and D. N. Fish. 2000. Management of protease inhibitor-associated diarrhea. Clin. Infect. Dis. 30:908-914. [DOI] [PubMed] [Google Scholar]
- 26.Tayrouz, Y., B. Ganssmann, R. Ding, R., A. Klingmann, R. Aderjan, J. Burhenne, W. E. Haefeli, and G. Mikus. 2001. Ritonavir increases loperamide plasma concentrations without evidence for P-glycoprotein involvement. Clin. Pharmacol. Ther. 70:405-414. [DOI] [PubMed] [Google Scholar]
- 27.Thaisrivongs, S., and J. W. Strohbach. 1999. Structure-based discovery of tipranavir disodium (PNU-140690E): a potent, orally bioavailable, nonpeptidic HIV protease inhibitor. Biopolymers 51:51-58. [DOI] [PubMed] [Google Scholar]
- 28.Turner, S. R., J. W. Strohbach, R. A. Tommasi, P. A. Aristoff, P. D. Johnson, H. I. Skulnick, L. A. Dolak, E. P. Seest, P. K. Tomich, M. J. Bohanon, M. M. Horng, J. C. Lynn, K. T. Chong, R. R. Hinshaw, K. D. Watenpaugh, M. N. Janakiraman, and S. Thaisrivongs. 1998. Tipranavir (PNU-140690): a potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4-hydroxy-2-pyrone sulfonamide class. J. Med. Chem. 41:3467-3476. [DOI] [PubMed] [Google Scholar]
- 29.Walmsley, S., J. Leith, C. Katlama, K. Arasteh, G. Pierone, G. Blick, A. Lazzarin, M. Johnson, C. Samuels, P. Jones, R. Chaves, A. Quinson, V. Kohlbrenner, S. McCallister, D. Mayers, and K. Curry. 2004. Pharmacokinetics and safety of tipranavir/ritonavir (TPV/r) alone or in combination with saquinavir (SQV), amprenavir (APV), or lopinavir (LPV): interim analysis of BI1182.51, abstr. WeOrB1236. XV Int. AIDS Conf.