

The dramatic rise in the prevalence of antibiotic resistance among bacteria currently poses a serious threat to public health worldwide. Of particular concern are infections caused by methicillin-resistant Staphylococcus aureus (MRSA), penicillin-resistant Streptococcus pneumoniae, vancomycin-resistant Enterococcus (62), and Mycobacterium tuberculosis (60), since many of these organisms are resistant to several classes of established antibiotics (60, 62). This situation is driving the search for novel antibacterial agents that inhibit targets that are essential to bacteria and that are not affected by mechanisms of resistance to current chemotherapeutic agents (18, 19). In this regard, the aminoacyl-tRNA synthetase (AaRS) enzymes have been a focus of recent research for antibacterial drug discovery. These enzymes play crucial roles in protein biosynthesis by catalyzing the synthesis of aminoacyl-tRNAs (aa-rRNA). Once these enzymes are inhibited, protein biosynthesis is halted, which in turn results in the attenuation of bacterial growth under both in vitro and infectious conditions (80). Consequently, these enzymes are interesting antibacterial drug targets.
An important example of the clinical application of an AaRS inhibitor is provided by the antibiotic mupirocin (marketed as Bactroban), which selectively inactivates bacterial isoleucyl-tRNA synthetase (IleRS). This product is currently the world's most widely used topical antibiotic for the control of MRSA (12). In recent years, reports of compounds (both natural and synthetic) which inhibit AaRS have increased. While many of these new inhibitors also affect the counterpart human enzymes, there has been some success in identifying molecules that are specific for bacterial AaRS enzymes and that also exhibit antibacterial activities against several species in experimental infections (37, 54, 71, 75). However, mupirocin is currently the only clinically available AaRS inhibitor and therefore acts as the paradigm for the prospective clinical development and deployment of future AaRS inhibitors.
Several suggestions for the prioritization of targets in the current process of antibiotic discovery and development have been made (63). The design of antibiotics which minimize the potential for the subsequent emergence of resistance is paramount (23, 32, 33, 63, 78). An opportunity to minimize drug-resistant organisms occurs when resistance emergence is accompanied by substantial reductions in the biological fitness of bacteria (3, 27, 59). Consequently, it is perceived that unfit drug-resistant mutants would be unable to survive upon withdrawal of an antibacterial drug and that policies involving antibiotic cycling would provide a means of eliminating drug-resistant mutants from natural populations (3, 27, 51, 58, 59). However, drug targets for which this approach may be appropriate have not yet been clearly identified, since in most cases compensatory evolution restores the fitness of drug-resistant mutants without the concomitant loss of resistance (2, 3). Nevertheless, recent novel findings in our laboratory suggest that AaRS could represent therapeutic targets where the loss of fitness associated with resistance could be compatible with antimicrobial restriction policies to eliminate resistance (46, 48).
Another opportunity to search for new drugs with reduced potential to select drug-resistant variants concerns the identification of novel agents with the capacity for multiple-target inhibition (23, 63, 76). Simultaneous inhibition of more than one molecular target renders the emergence of resistance less likely because mutations are required in all targets to confer resistance to the drug (23). The β-lactam antibiotics provide an excellent example to support this contention (76), since they are multisite inhibitors which target bacterial penicillin binding proteins (PBPs). Resistance to β-lactam antibiotics rarely results from target site modification and is more commonly due to expression of β-lactamases, efflux mechanisms, or β-lactam-resistant PBPs acquired by horizontal gene transfer (76). Approaches that have been considered or that are currently under investigation include dual or multitargeting of DNA topoisomerase IV and DNA gyrase (78); cell wall biosynthetic ATP-dependent amino acid ligases (33); MurA and AroA (32); DNA gyrase and dihydropteroate synthetase (1); and DNA gyrase, DNA topoisomerase IV, and rRNA (43). Due to the existence of homologous sequences in phylogenetically related synthetases (Table 1), there is an important opportunity to develop single molecules which simultaneously inhibit multiple AaRS enzymes. These molecules could prove essential in limiting the emergence of both chromosomally and horizontally acquired forms of resistance to AaRS inhibitors.
TABLE 1.
Structural classification of aminoacyl-tRNA synthetase (49)
| Characteristic | Class I | Class II |
|---|---|---|
| Conserved characteristics | ||
| Active site structure | Rossman fold | Antiparallel β-sheet |
| Motifs | HIGH, KMSKS | Motif 1, Motif 2, Motif 3 |
| ATP conformation | Extended | Bent |
| Enzyme subclass | Subclass Ia [subunit structure(s)] | Subclass IIa [subunit structure(s)] |
| IleRS (α) | Ala-RS (α, α4) | |
| LeuRS (α) | ProRS (α2) | |
| ValRS (α) | HisRS (α2) | |
| CysRS (α) | SerRS (α2) | |
| MetRS (α, α2) | ThrRS (α2) | |
| ArgRS (α) | GlyRS (α2, α2β2) | |
| Subclass Ib (subunit structure) | Subclass IIb (subunit structure) | |
| GlnRS (α) | AsnRS (α2) | |
| GluRS (α) | AspRS (α2) | |
| LysRS-I (α) | LysRS-II (α2) | |
| Subclass Ic (subunit structure) | Subclass IIc (subunit structure) | |
| TyrRS (α2) | Phe-RS (α2β2) | |
| TrpRS (α2) |
In this minireview, we examine the prospects for the development of several recently reported AaRS inhibitors as chemo-therapeutic agents. The chemistry of these inhibitors will not be described in detail, as comprehensive reviews of these compounds from chemical perspectives have already been published (54, 71). Consequently, we examine the modes of action and potential mechanisms of clinical resistance to these compounds. We also discuss ways in which novel strategies aimed at reducing the emergence of resistance to these compounds can be exploited.
AMINOACYL-tRNA SYNTHETASES AS NOVEL ANTIBACTERIAL TARGETS
Properties of AaRS for drug discovery and development.
The AaRS enzymes represent ideal targets for antibacterial drug discovery, since they meet various criteria defined by the pharmaceutical industry. (i) Despite their universal distribution in cellular organisms, considerable evolutionary divergence has occurred between prokaryotic and eukaryotic enzymes, and these differences can be exploited for the development of antibiotics that selectively inhibit bacterial rather than human enzymes (49, 72). (ii) The respective synthetases are highly conserved across different bacterial pathogens, which makes possible the discovery of broad-spectrum antibiotics (49, 72). (iii) The full complement of 20 synthetases is found in most bacterial pathogens and may represent 20 independent antibacterial targets (49, 72). (iv) These enzymes are soluble, stable, and easy to purify in large quantities from recombinant expression systems and can be assayed by one or more conventional methods which are amenable to high-throughput screening (37, 61). (v) X-ray crystal structures for most of the synthetases have been solved and provide a platform for rational drug design (49, 71).
Enzyme mechanisms and consequences of inhibition.
Aminoacylation proceeds via a two-step reaction, as follows:
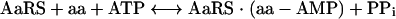 |
(1) |
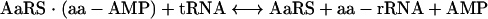 |
(2) |
where aa is an amino acid, and aa-AMP is aminoacyl-AMP.
In the first step (stage 1), reaction between amino acid and ATP yields an activated, hydrolyzable, aminoacyl-adenylate intermediate (49). Subsequently (stage 2), the amino acid moiety is transferred to its cognate tRNA and the resulting amino-acyl-tRNA acts as a substrate for polypeptide synthesis, which occurs on the ribosome (49). Inhibition of either one of these enzymatic stages leads to the accumulation of uncharged tRNA molecules, which bind to ribosomes, causing the interruption of polypeptide chain elongation (20, 21, 66, 90). Induction of the stringent response by relA then occurs (20). This results in the biosynthesis of guanosine tetra- and pentapeptides, which inhibit the activity of RNA polymerase and downregulate several energetically demanding processes, including the biosynthesis of stable RNA, DNA, protein, and peptidoglycan (Fig. 1) (20, 21, 66, 90). By perturbing several metabolically important processes, the chemical inhibition of AaRS leads to the cessation of bacterial growth and the attenuation of virulence in vivo (80).
FIG. 1.

Pleiotropic effects associated with activation of the stringent response following inhibition of aminoacyl-tRNA synthetases (20). PL, phospholipid; PG, peptidoglycan.
Enzyme structure and classification of enzymes.
The 20 AaRSs are divided into two major classes based on the architecture of their catalytic domains (Table 1) (31, 49). All class I enzymes possess an active site characterized by a Rossman dinucleotide binding fold, which consists of alternating α helices and β-pleated sheets that are essential for ATP binding (31, 49). This ATP binding cleft is further demarcated by the HIGH and KMSKS consensus motifs characteristic of class I enzymes (49). In contrast, class II enzymes posses an unusual catalytic domain that is composed of alternating antiparallel α folds and contain three characteristic signature sequences, designated motifs 1, 2, and 3 (Table 1) (49). Enzymes within each division are additionally classified into three subclasses based on additional structural similarities and the ability to recognize other chemically related amino acids (49). In view of the apparent structural homologies between related enzymes, there is an important opportunity to develop multitarget inhibitors directed at conserved motifs found in the relevant synthetases.
Editing domain of AaRS as a potential antibacterial drug target.
The inability to differentiate accurately between structurally related amino acids often leads to errors of amino acid misactivation (noncognate aa-AMP) or misacylation (noncognate aa-tRNA) and requires the activity of an editing domain which is separate from the catalytic domain of AaRS enzymes (49). Since the editing domain is also essential for ensuring the fidelity of the genetic code (49) and maintenance of maximum growth rates (5), this site may act as a further point against which synthetase-directed inhibitors can be developed. Additionally, evidence that editing domains of bacterial and mammalian systems recognize noncanonical amino acids by different mechanisms (83) further supports the suggestion that editing domains may represent an additional selective target site for inhibitors of bacterial AaRS.
MUPIROCIN
Mechanism of mupirocin inhibition.
Mupirocin (Fig. 2, structure 1) is a natural product of Pseudomonas fluorescens and is the only commercially available antibiotic that inhibits bacterial AaRS. It shares no structural homology with other antibiotics in clinical practice and consists of a short fatty acid (nonanoic acid) ester linked to monic acid (22), with the tail portion closely resembling the isoleucyl moiety of the isoleucyl-adenylate reaction intermediate (Ile-AMP) (Fig. 2, structure 2). Early work by Yanagisawa et al. (93) demonstrated that mupirocin acts as a bifunctional inhibitor of IleRS with respect to both isoleucine and ATP binding. This observation has now been confirmed by using the X-ray crystal structures of IleRS bound with either mupirocin or Ile-AMP (Fig. 3) (48, 65). In bacteria inhibition of protein synthesis by mupirocin is reversed by increasing concentrations of l-isoleucine, a finding which confirms that IleRS is the intracellular target of mupirocin (45).
FIG. 2.

Chemical structures of mupirocin (structure 1), isoleucyl-AMP (Ile-AMP; structure 2), and mupirocin derivatives (structures 3 and 4).
FIG. 3.

(A) Comparative views of bound structures of mupirocin (top) and reactive intermediate (red; bottom) and superposition of the structures (middle) in the IleRS of S. aureus. Details of the binding pockets for mupirocin (B) and the acylated intermediate (C) and the positions of mupirocin resistance mutations are also shown. (Reprinted from reference 48.)
Both mupirocin and the reactive intermediate bind in very similar regions within the enzyme (Fig. 3), with the epoxide ring of mupirocin occupying the same domain as the phosphate group in the intermediate (48). The moiety that morphologically resembles the hydrophobic side chain of l-isoleucine is recognized by the same hydrophobic pocket that binds l-isoleucine (48, 65). Likewise, the dihydrotetrahydropyran ring of mupirocin also binds in a similar domain to the ribose moiety of Ile-AMP (48, 65). Since the binding models reveal that the domain for mupirocin binding overlaps with that for the Ile-AMP intermediate, it is predicted that mupirocin binds to the same conformer that is required for Ile and ATP binding (17, 48). This is well supported by steady-state kinetics which shows that mupirocin is a competitive inhibitor of both Ile and ATP binding (17). However, an important difference in inhibitor binding occurs in the region of the extended nonanoic acid moiety of mupirocin, which is essentially unoccupied in the X-ray models containing the reactive intermediate (Fig. 3) (48). This nonanoic acid moiety fits neatly into a strongly hydrophobic pocket that is lined by valine 588. Hydrogen bond interaction between the carboxylic acid group of mupirocin and the amide of valine 588 is important for stabilizing mupirocin within the IleRS. In shorter-chain alkyl variants of mupirocin (17) and, indeed, monic acid, the inability to form this H-bond interaction results in diminished inhibitory activity (4, 48).
Antimicrobial activity and clinical use of mupirocin.
Mupirocin is primarily active against gram-positive pathogens, including S. aureus (MICs = 0.06 to 0.12 μg/ml) and Streptococcus pyogenes (MICs = 0.12 to 0.5 μg/ml), and is relatively inactive against anaerobic streptococci, enterococci, and most gram-negative bacteria, with the exception of Haemophilus influenzae, Neisseria spp., and Bordetella pertussis (79).
Mupirocin is rapidly hydrolyzed in blood plasma to form monic acid, which is inactive against bacteria, and therefore, its clinical use is restricted to topical applications (44, 79). Formulated as 2% mupirocin ointment in a water-miscible polyethylene glycol base (for application to skin) or 2% cream in a soft paraffin base (for nasal application), mupirocin is indicated for eradication of S. aureus, including MRSA, from the anterior nares; treatment of skin and surgical site infections (SSIs); and the control of MRSA outbreaks (25). By eradicating nasal carriage, mupirocin prevents SSIs in high-risk groups, including patients undergoing continuous ambulatory peritoneal dialysis, hemodialysis, or cardiac surgery (28). Furthermore, the use of mupirocin has been shown to reduce the health care costs associated with the treatment of MRSA infections (28, 69).
Molecular mechanisms of resistance to mupirocin.
The widespread clinical use of mupirocin has resulted in the emergence of resistance among methicillin-susceptible S. aureus, MRSA, and glycopeptide-intermediate S. aureus (30, 36, 47). Two principal mupirocin resistance phenotypes have been identified (39). High-level resistance (MICs = >512 μg/ml) results from acquisition of the mupA gene, which is often plasmid borne and encodes an alternate synthetase (IleRS-2) that is refractory to mupirocin (25, 39). Low-level mupirocin resistance (MICs = 8 to 256 μg/ml) arises from point mutations within the chromosomally encoded IleRS gene (ileS) (4, 47, 48).
There is no clear evidence for the origin of the mupA determinant in S. aureus. However, different authors have speculated that it was acquired either from a eukaryotic source (15) or from Bacillus anthracis (14). Although current clinical resistance to mupirocin is not mediated by the mupirocin-resistant IleRS-2 from P. fluorescens (which produces mupirocin), this gene represents another possible source of resistance to mupirocin in S. aureus (92). There is no evidence to suggest that antibiotic efflux or drug inactivation systems are associated with resistance to mupirocin in S. aureus (57, 81, 91).
The emergence of high-level, mupA-mediated resistance to mupirocin is associated with therapeutic failure (64). However, until recently, low-level mupirocin resistance was considered clinically unimportant, since topical applications of mupirocin result in concentrations (20 mg mupirocin/ml) which would appear sufficient to inhibit these mutants (25, 41). However, in clinical isolates this mechanism of resistance appears to be more prevalent than high-level resistance (30, 36, 77), and the emergence of these mutants appears to increase failure rates for mupirocin therapy (29, 64, 85). Therefore, despite the apparently high concentrations of mupirocin that can be achieved following topical application, there would appear to be niches where antibiotic concentrations are lower, thereby permitting survival of strains exhibiting low-level resistance to mupirocin (85).
Impact of chromosomal mupirocin resistance alleles.
In clinical isolates of S. aureus expressing low-level resistance to mupirocin, single amino acid changes have been detected at only two sites (i.e., V588F or V631F) (4, 35, 47). In contrast, the stepwise selection of high-level mupirocin-resistant mutants, which presumably carried multiple genetic changes in ileS, was demonstrated in vitro a number of years ago (82). This suggests that, in addition to V588F and V631F, other loci could mediate resistance to mupirocin in S. aureus. Recently, we identified and genetically characterized a set of novel mutations which conferred mupirocin resistance (Fig. 3; Table 2) (48). In addition to the clinically occurring mutations, a single substitution of G593V resulted in low-level resistance to mupirocin (MICs = 1 to 2 μg/ml). However, elevated resistance to mupirocin (MICs = 16 to 128 μg/ml) required double mutations within the IleRS and involved combinations of first-step mutations (i.e., V588F-V631F and G593V-V631F) or additional combinations involving second-site changes of R816C, H67Q, and F563L (Table 2). These mutations occurred at sites that are highly conserved across the eubacterial kingdom and are important for substrate binding but also interactions with mupirocin (Fig. 3) (48). For example, H67Q affected the tetrapeptide consensus (HMGH) involved in ATP binding, while the F563L change caused considerable disruption to the isoleucyl binding pocket (48). Since mutants with these mutations have not been described clinically, it seems plausible that substantial reductions in bacterial fitness may prevent their survival (48).
TABLE 2.
First- and second-step and compensatory mutations associated with mupirocin resistance and their associated effects on resistance and competitive fitness in S. aureusa
| Mutant and amino acid substitution in IleRSb | Mupirocin MIC (μg/ml) | Competitive fitness |
|---|---|---|
| Wild type | 0.016 | 1.00 |
| First-step mupirocin-resistant mutants | ||
| G593V | 1 | 0.92 ± 0.02 |
| G593V | 2 | 0.94 ± 0.05 |
| V631F | 2 | 0.99 ± 0.08 |
| V631F | 2 | 1.00 ± 0.03 |
| V588F | 8 | 0.94 ± 0.04 |
| V588F | 8 | 0.95 ± 0.01 |
| V588F | 8 | 0.93 ± 0.01 |
| V588F | 8 | 0.96 ± 0.05 |
| V588F | 16 | 0.99 ± 0.03 |
| V588F | 16 | 0.94 ± 0.04 |
| Second-step mupirocin-resistant mutants | ||
| G593V, V631F | 16 | 0.63 ± 0.03 |
| V631F, V588F | 32 | 0.57 ± 0.03 |
| V588F, R816C | 64 | 0.32 ± 0.06 |
| V588F, F563L | 128 | 0.24 ± 0.08 |
| V588F, H67Q | 128 | 0.33 ± 0.01 |
| Fitness-restored mutants | ||
| G593V, V631F, A196V | 2 | 0.99 ± 0.02 |
| G593V, V631F, A196V | 2 | 0.94 ± 0.07 |
| G593V, V631F, E190K | 2 | 1.00 ± 0.00 |
| G593V, V631F, E190K | 2 | 1.00 ± 0.01 |
| V631F, V588F, none | 32 | 1.01 ± 0.05 |
| V588F, R816C, E195K | 4 | 0.95 ± 0.01 |
| V588F, R816C, A196V | 8 | 0.93 ± 0.08 |
| V588F, F563L, none | 8 | 0.98 ± 0.01 |
| V588F, H67Q, Q67H | 8 | 0.96 ± 0.05 |
| V588F, H67Q, H396Q | 8 | 0.79 ± 0.07 |
| V588F, H67Q, H396Q | 16 | 0.79 ± 0.07 |
The table is based on data presented elsewhere (48).
The mutations in IleRS occurring at each stepwise selection are shown in boldface.
While the competitive fitness of single-step mupirocin-resistant mutants was not substantially affected by their mupirocin resistance alleles (Table 2), it was shown that all higher-level resistant mutants carrying double mutations in IleRS were unfit both in laboratory media and in a murine skin abscess model of S. aureus infection (48). Further microbiological characterization revealed that mupirocin resistance substantially affected the exponential growth rates of high-level mupirocin-resistant mutants, causing their counterselection. This information is consistent with the clinical appearance of mupirocin-resistant S. aureus strains carrying only a single mutation (i.e., V588F or V631F) in IleRS and the absence in the clinic of higher-level resistant mutants which contain multiple genetic changes (4, 35, 47).
Laboratory investigations of other unfit antibiotic-resistant mutants have shown that in the absence of continued antibiotic selection, bacterial fitness can be restored by compensatory mutations or backmutation of the resistance allele (2, 3). In the former situation, this usually occurs without affecting the level of antibiotic resistance (2, 3). However, a quite unusual observation was made with unfit mupirocin-resistant mutants, whereby third-site compensatory mutations generally suppressed the effect of the mupirocin resistance alleles (Table 2). Essentially, fitness-restored mupirocin-resistant mutants were more susceptible to mupirocin than their unfit progenitor strains. It therefore appears that compensatory reconfiguration of IleRS not only restored the substrate binding pocket for Ile-AMP in unfit mutants but also simultaneously reproduced the domain to which mupirocin is bound.
In view of the overlapping nature of the substrate and mupirocin binding domains (Fig. 3), restoration of fitness and suppression of resistance upon compensatory evolution is not unexpected. Since AaRS inhibitors generally compete for the substrate binding pockets or adjacent domains, it is likely that in other synthetases target alterations which mediate resistance would be associated with substantial reductions in fitness. In the following sections we describe prospective AaRS inhibitor candidates for clinical development and examine where appropriate whether fitness costs are associated with resistance to these molecules.
OTHER NATURAL PRODUCTS WHICH INHIBIT tRNA SYNTHETASES
In addition to mupirocin, several biosynthetic products which inhibit AaRS have been discovered (24, 54). These include indolmycin (which targets TrpRS; Fig. 4, structure 6), chuangxinmycin (which targets TrpRS; Fig. 4, structure 7), borrelidin (which targets ThrRS; Fig. 4, structure 8), granaticin (which targets LeuRS; Fig. 4, structure 9), furanomycin (which targets IleRS; Fig. 4, structure 10), ochratoxin A (which targets PheRS; Fig. 4, structure 11), and cispentacin (which targets ProRS; Fig. 4, structure 12). Unfortunately, many of these older antibiotics have not been developed as chemotherapeutic agents since they either exhibit poor antibacterial activity or lack specificity for the bacterial target (24). Nevertheless, the diversity of molecular structures presented by natural product inhibitors (Fig. 4), coupled with in silico modeling of X-ray crystal structures for target enzymes, may provide a scaffold for the structure-based design of novel inhibitors with antibacterial action. For example, recent crystallographic analysis of borrelidin bound to Escherichia coli ThrRS revealed a hydrophobic noncatalytic domain which perturbs the catalytic conformation of ThrRS upon inhibitor binding (74). This domain may yet prove to be an interesting target for inhibitor development, in view of the fact that structural alterations which confer resistance to borrelidin also impose fitness burdens in E. coli, which result from reduced binding of ATP and threonine (68). Borrelidin is currently undergoing investigation as a candidate for the treatment of malaria (67) and/or antiangiogenesis (53). Another agent undergoing clinical development is the semisynthetic derivative of cispentacin (Fig. 4, structure 12) known as PLD-118 (70). This antibiotic has been shown to inhibit IleRS of Candida albicans (94) and is being developed as part of an oral formulation for use against candidal infections (70). We are unaware of studies on the activity of PLD-118 against bacteria. Indolmycin and chuangxinmycin are especially interesting therapeutic candidates which could prove applicable in controlling current drug-resistant bacterial pathogens. These antibiotics are considered in more detail below.
FIG. 4.

Chemical structures of l-tryptophan (structure 5), indolmycin (structure 6), chuangxinmycin (structure 7), borrelidin (structure 8), granaticin (structure 9), furanomycin (structure 10), ochratoxin A (structure 11), and cispentacin (structure 12).
Indolmycin.
The antibiotic indolmycin (Fig. 4, structure 6), which is a biosynthetic derivative of l-tryptophan (Fig. 4, structure 5), was first isolated from Streptomyces griseus (ATCC 12648) and patented by Pfizer in 1965 (8, 34). Indolmycin is a potent and selective inhibitor of bacterial tryptophanyl-tRNA synthetase (50% inhibitory concentration [IC50] = 9.25 nM for E. coli TrpRS) and displays minimal activity against the eukaryotic counterpart (IC50 = 4.04 mM for bovine liver TrpRS) (52, 89). However, it is relatively inactive against several common bacterial pathogens, including streptococci, enterococci, and members of the family Enterobacteriaceae (52). Although indolmycin is transported into susceptible bacteria via the aromatic amino acid system, narrow-spectrum antibacterial activity results from the lipophilic character of the molecule, which restricts its ability to penetrate certain bacterial cell envelopes (86). Efforts to synthesize analogs of indolmycin with enhanced hydrophilic properties were, however, unsuccessful (87). Moreover, indolmycin perturbs the metabolism of tryptophan in rat liver by inhibiting enzymes involved in the catabolism of this amino acid (88). Consequently, initial interests in developing indolmycin for human use were abandoned.
Indolmycin has also been described by Takeda Pharmaceuticals (Japan) as TAK-083, a natural product of Streptomyces sp. strain HC-1 (52) and was shown to have potent activity against clinical isolates of Helicobacter pylori (MICs = 0.008 to 0.031 μg/ml) (52). Accordingly, TAK-083 was patented by Takeda as part of a novel formulation for the prevention and treatment of H. pylori infections and associated diseases, such as gastritis and gastric ulcers (8). Indeed, supporting biological data demonstrated that oral administration of indolmycin was more efficacious than clarithromycin and amoxicillin in clearing an H. pylori infection in a Mongolian gerbil model (52). Since H. pylori shows preferential uptake of hydrophobic compounds, the potent activity of indolmycin against this organism can be explained (11).
Early microbiological studies demonstrated that indolmycin was active against staphylococci following its rapid accumulation through the tryptophan uptake system (86). The antistaphylococcal properties of indolmycin have recently been reinvestigated in our laboratory, where we found that indolmycin (MICs = 0.125 to 2 μg/ml) had activities comparable to those of fusidic acid and mupirocin against a panel of clinical isolates of MRSA (46). Indolmycin might therefore represent a candidate for development as a topical anti-infective for the treatment of staphylococcal infections. Topical applications of indolmycin to the skin are unlikely to compromise the agent's potential as an anti-H. pylori drug.
In contrast to the bacteriostatic activity exhibited against S. aureus, indolmycin is bactericidal against H. pylori, suggesting that in H. pylori it has other antibacterial effects, in addition to inhibiting TrpRS. This is consistent with the observation that indolmycin-resistant mutants could not be selected in H. pylori (52), whereas mutants of S. aureus could be readily obtained following selective plating (46). A single mutation (i.e., H43N) in staphylococcal TrpRS which conferred higher-level resistance to indolmycin (MIC = 128 μg/ml) imposed substantial fitness burdens (a 40 to 44% reduction in fitness) on S. aureus (46). Indolmycin-resistant mutants which presumably contained mutations that affected the uptake system for this antibiotic also emerged, but they were susceptible to lower drug concentrations (MICs = 8 to 32 μg/ml) and exhibited only a minor loss of fitness (a 7% reduction) (46).
The histidine residue at position 43 in TrpRS is directly involved in binding to tryptophan as well as stabilization of the Trp-adenylate intermediate (73). It is therefore likely that the loss of this key histidine residue accounts for the significant reduction in bacterial fitness in S. aureus. If this mutation also arose in vivo, then organisms expressing higher-level resistance to indolmycin might be counterselected in the absence of indolmycin selection pressure; i.e., their carriage might cease upon removal of the topical medication. However, the emergence of permease mutants may be a cause for concern. In addition, the acquisition of a transferable indolmycin-resistant TrpRS cannot be ruled out in view of its presence in the producing strain (56).
Chuangxinmycin.
The antimicrobial natural product chuangxinmycin (Fig. 4, structure 7) bears some structural resemblance to l-tryptophan and indolmycin and also specifically inhibits bacterial TrpRS (IC50 = 30 nM for E. coli TrpRS) (16). This antibiotic was initially reported to possess antibacterial activity against a number of gram-positive and gram-negative organisms and to show in vivo efficacy in murine models of infection involving Shigella dysenteriae and E. coli (16). Preliminary clinical data indicated that chuangxinmycin was effective for the treatment of septicemia and urinary and biliary infections caused by E. coli. In spite of chuangxinmycin's early promise as a chemotherapeutic agent, this antibiotic has not yet been developed for medical use (16). We are unaware of resistance studies performed with chuangxinmycin.
SYNTHETIC AND SEMISYNTHETIC tRNA SYNTHETASE INHIBITORS
Chemical derivatives of mupirocin.
Because mupirocin demonstrates activity against S. aureus and some other bacterial pathogens (see above) which cause systemic infections, there has been much interest in developing mupirocin analogs which display systemic properties (8). GlaxoSmithKline reported on the synthesis of a series of β-diketone acrylate bioisoteres of mupirocin that retained activity against S. aureus, S. pyogenes, S. pneumoniae, H. influenzae, and Moraxella catarrhalis (9). Subcutaneous injection of the compound shown in Fig. 2 (structure 3) was reported to be effective in eradicating a S. aureus systemic infection (9).
A series of oxazole isosteres bearing nitroheterocycles were also developed as stable mimics of mupirocin (e.g., Fig. 2, structure 4) (13). Activity against mupA-positive mupirocin-resistant S. aureus was observed, despite the lack of significant inhibition of IleRS-2 from these strains (13). Modes of action other than inhibition of IleRS-2 were therefore suggested as the likely explanation for the activities of these compounds against highly mupirocin-resistant cells. In a murine S. aureus infection model, the molecule represented by structure 4 (Fig. 2) demonstrated in vivo efficacy following administration by the oral and subcutaneous routes, with doses of 1.2 mg/kg of body weight and 3.2 mg/kg, respectively, being required for the protection of 50% of mice from death (13). Despite success in synthesizing analogs of mupirocin with systemic antibacterial activity, there are no reports on the clinical investigation of these compounds.
Analogs of reaction intermediates as antibiotics.
Several research groups have sought to use aminoacyl-adenylate reaction intermediates as chemical platforms for constructing inhibitors directed at different synthetases. Central to this approach is the rationale that analogs of reaction intermediates are likely to bind to their corresponding synthetase with a high affinity and in a manner that is analogous to the substrate. This approach may therefore yield antibacterial inhibitors which display potent activities against the target. Conventional strategies for synthesizing these molecules have sought to replace the hydrolysable acyl-phosphate moiety of the aminoacyl-ade-nylate intermediate with chemically stable, nonhydrolyzable groups (54, 71).
Analogs of the prolyl adenylate (Fig. 5, structure 13) containing sulfonamide, a commonly used linker group, were reported to be potent against E. coli ProRS (IC50 = 4.3 nM) (40). Unfortunately, these molecules also inhibited the human ProRS (IC50 = 0.6 nM), which illustrates a common problem in developing reaction mimics as antibiotics. In addition, several of these analogs lack whole-cell antibacterial activity, possibly due to poor penetration of the bacterial cell envelope (54, 75).
FIG. 5.

Chemical structures of l-prolyl-sulfamoyl adenosine (structure 13), CB432 (structure 14), quinolinone derivative (structure 15), REP8839 (structure 16), phenylthiourea sulfonamide derivative (structure 17), and SB-203207 (structure 18).
Analogs with potent antibacterial activity and selectivity for bacterial IleRS were described by Cubist Pharmaceuticals (8, 75). One of these compounds (designated CB-432; Fig. 5, structure 14) preferentially inhibited bacterial IleRS (IC50s = 1 nM and 570 nM for IleRS from E. coli and human sources, respectively) and was most potent against S. pyogenes (MIC = 0.5 μg/ml) (75). In the presence of increasing concentrations of l-isoleucine, the antibacterial activity of CB-432 was reduced, consistent with IleRS as the intracellular target of this antibiotic (75). However, in mice systemically infected with S. pyogenes, doses beyond those which could be applied to humans were required for efficacy (75). Subsequently, it was found that CB-432 was highly serum bound, resulting in low bioavailability (75). These data are, however, reassuring because they indicate that reaction intermediate mimics with potent antibacterial properties might be developed for clinical use.
Inhibitor molecules obtained by high-throughput screening.
Dramatic progress in automated high-throughput screening technology is currently acting as an important approach for discovering low-molecular-weight molecules which perturb the function of AaRS enzymes (37). Chemical optimization of these hits has led to the generation of multiple series of pharmacophores which inhibit bacterial AaRS enzymes and which represent clinical candidates. Some of the most promising antibiotics thus far include molecules which inhibit MetRS and PheRS (54, 71).
GlaxoSmithKline described a series of quinolinone derivatives that inhibited the activity of S. aureus MetRS by competition with a methionine substrate (38). One of these lead compounds (Fig. 5, structure 15) selectively inhibited bacterial MetRS (IC50 = 12 nM for S. aureus MetRS) and was potent against S. aureus (MIC = 0.12 μg/ml) and Enterococcus spp. (MIC = 0.06 μg/ml) (38, 50). This compound was also efficacious in a groin abscess S. aureus infection model in rats (50). Similar to the classical AaRS inhibitor, mupirocin, compound 15 (Fig. 5) also stimulated the stringent response pathway in S. aureus (50). A mutation in MetRS involving G49S conferred a fourfold decrease in susceptibility to compound 15 (Fig. 5). However, the impacts of these changes on microbiological fitness are unknown (50).
In 2003, Replidyne Inc. disclosed the in-licensing of the GlaxoSmithKline tRNA synthetase program, which included the quinolinone series of inhibitors. Accordingly, Replidyne reported the synthesis of a fluorovinylthiophene analog (designated REP8839; Fig. 5, structure 16), which was potent against S. aureus MetRS (IC50 = 3.5 nM) (J. Guiles, T. Tarasow, J. Qui, I. Critchley, K. Stone, C. Young, U. Ochsner, N. Janjic, and R. Jarvest, Abstr. 44th Intersci. Conf. Antimicrob. Agents Chemother., abstr. F-727, 2004). This correlated with good antimicrobial activity against this organism (MIC90 = 0.06 μg/ml), vancomycin-resistant Enterococcus faecalis (MIC90 = 0.015 μg/ml), and S. pyogenes (MIC90 = 0.06 μg/ml) (I. Critchley, C. Young, K. Stone, U. Ochsner, C. Dang, and N. Janjic, Abstr.44th Intersci. Conf. Antimicrob. Agents Chemother., abstr. F-729, 2004). REP8839 is currently in advanced preclinical studies as a topical agent for controlling the eradication of S. aureus and associated superficial infections.
Although quinolinone derivatives are not structurally related to methionine, three-dimensional quantitative structure-activity relationships have shown that these molecules compete with methionine for important binding interactions in the amino acid hydrophobic binding pocket of MetRS (55). This has important implications for the development of antibiotic resistance. Indeed, it was recently reported that mutations in a region that overlaps the catalytic domain of MetRS (55) substantially reduced the biological fitness of REP8839-resistant mutants (U. Ochsner, C. Young, K. Stone, D. Gentry, N. Janjic, and I. Critchley, Abstr. 44th Intersci. Conf. Antimicrob. Agents Chemother., abstr. F-730, 2004). Such unfit resistant mutants carried a G54S mutation in MetRS and were resistant to elevated concentrations of the drug (MIC = 32 μg/ml). It is therefore plausible that REP8839-resistant mutants might be controlled clinically by the scheduled withdrawal of antibiotic treatment. However, since a second MetRS enzyme (MetRS-2), which is resistant to REP8839 and other quinolinone derivatives, has been detected among current clinical isolates of S. pneumoniae, B. anthracis, and Clostridium perfringens (14, 38), the possibility of horizontal transfer of resistance via MetRS-2 to staphylococci cannot be ruled out.
Broad-spectrum activity against gram-negative and gram-positive pathogens was described for the PheRS inhibitor phenylthiazolylurea-sulfonamides (Fig. 5, structure 17) (10). Inhibition of PheRS derived from E. coli and H. influenzae (IC50 = 15 nM) as well as S. pneumoniae (IC50 = 50 nM) was associated with significant activity against S. aureus, S. pneumoniae, H. influenzae, and M. catarrhalis, with MICs ranging from 0.4 to 0.8 μg/ml (10). However, inclusion of phenylalanine in the culture medium resulted in the partial reduction of antimicrobial activity, which was most pronounced against S. aureus (10-fold) and modest against all other target pathogens (2-fold) (10). Nevertheless, intravenous applications of this substance demonstrated efficacy in an S. aureus sepsis model in mice fed phenylalanine-free diets (10). Efficacy was also shown in a sepsis model for S. pneumoniae in rats with normal levels of phenylalanine in blood plasma (10).
Activation of the stringent response by the phenylthiazolylurea-sulfonamide inhibitor was shown to be an important feature of the drug's antimicrobial action (10). Indeed, this compound showed reduced activity against an E. coli relA mutant (MIC = 0.2 μg/ml), which is defective in the stringent response pathway (Fig. 1), compared to that against its relA-positive parent (MIC = 0.003 μg/ml) (10). By using E. coli strain carrying a mutation (A294G) in the phenylalanine binding pocket, decreased susceptibility to phenylthiazolylurea-sulfonamide based compounds was shown (10). The impact of this resistance allele on microbiological fitness was not reported. Nevertheless, if mutations within the phenylalanine binding pocket are required to mediate resistance to compound 17 (Fig. 5), then an associated fitness cost can be envisaged.
APPROACHES TO MINIMIZE RESISTANCE TO AMINOACYL-tRNA SYNTHETASE INHIBITORS
Antimicrobial usage strategies involving AaRS inhibitors.
Antimicrobial restriction has been proposed as an approach to control the emergence and prevalence of drug resistance in the clinic (42). However, there are limited data available to support its benefit (42). There is also much ongoing debate over which antimicrobials may be suitable for this process and how it should be conducted in the clinic (42). At the molecular genetic level, the observations that compensatory evolution may fix drug resistance alleles in bacterial populations in the absence of selection pressure (2) and that horizontally acquired resistance is often associated with moderate or no fitness costs threaten the prospects for antimicrobial restriction policies. In terms of fitness-compensated mutants with unaffected resistance levels, these organisms are likely to predominate upon reintroduction of the previously withdrawn drug. As part of the ongoing research to decide which antibiotics might minimize resistance emergence through temporary deployment schemes, we propose that AaRS inhibitors are good candidates for such investigations. Indeed, we have described that the development of resistance to these agents is accompanied by reductions in bacterial fitness which can lead to the counterselection of unfit mutants. Thus, it is plausible that unfit mutants that contain severely distorted synthetases and that are resistant to high levels of drug will not become established in a clinical setting. This situation may already be restricting the development and maintenance of high-level endogenous resistance to mupirocin in the clinic (see above). Indeed, Walker et al. (84) recently described that the restricted use of mupirocin resulted in decreased prevalence of both low- and high-level (mupA-mediated) resistance to mupirocin among MRSA isolates in the clinic. However, the role that fitness costs may have played in the reduced prevalence of resistant strains was not investigated.
Multisynthetase inhibitors.
The existence of structurally conserved catalytic residues across related synthetases (Table 1) provides a realistic opportunity for the discovery of a single molecule that simultaneously inhibits multiple enzymes. Such molecules could be of major clinical importance, since bacteria would require simultaneous point mutations within each drug target to become resistant, which is an unlikely event. However, if such mutants were to emerge as a result of poor compliance or the use of suboptimal concentrations of antibiotic, it is likely that their survival in the clinic would be affected by cumulative reductions in bacterial fitness. Multisynthetase inhibitors may also reduce the likelihood of resistance emergence through the horizontal acquisition of alternate enzymes, as the simultaneous acquisition of these would be required to confer complete resistance. However, multisite inhibitors may be susceptible to other classical strategies of antimicrobial resistance, including target gene upregulation, reduced permeability, efflux, and drug modification systems, each of which may promote resistance (7).
An early indication that a single pharmacophore might be exploited to develop a multienzyme inhibitor was provided by ochratoxin A (Fig. 4, structure 11), an inhibitor of PheRS. When the phenyl moiety of this molecule is replaced with valine, inhibition of ValRS occurs (26). The development of ochratoxin A is, however, affected by its lack of selectivity for bacterial synthetases (26). Similarly, the natural product SB-203207 (Fig. 5, structure 18) inhibits both bacterial IleRS (IC50 = 14 nM) and eukaryotic IleRS (IC50 = 4.2 nM) (6). However, replacement of isoleucyl with a leucyl moiety afforded an inhibitor of bacterial LeuRS (IC50 = 16 nM) which also interacted with bacterial ValRS (IC50 = 290 nM), but it was less active against the bacterial IleRS (IC50 = 910 nM) (6). These examples depict a central challenge in discovering a single molecule which binds to the target synthetases with similar potencies. Nevertheless, the increasing database of X-ray crystal structures of both bacterial and mammalian enzymes could enable the structure-based design of a single molecule which selectively perturbs several bacterial AaRSs.
CONCLUSIONS
Bacterial AaRSs present a number of opportunities for the identification of novel inhibitors that may be candidates for development as antibiotics. The prospect of discovering and developing single molecules which simultaneously inhibit multiple AaRS offers one method for minimizing the subsequent emergence of resistance. However, even if this cannot be achieved, increasing amounts of data, primarily obtained from studies with mupirocin, indicate that the fitness costs associated with the development of resistance to AaRS inhibitors may be disadvantageous in the clinical setting, such that the long-term survival of these mutants in the absence of antibiotic selection pressure may be limited. Thus, antibiotic rotation programs may be successful as a means of controlling the possibility of emerging clinical resistance to AaRS inhibitors.
REFERENCES
- 1.Alovero, F., M. Nieto, M. R. Mazzieri, R. Then, and R. H. Manzo. 1998. Mode of action of sulfanilyl fluoroquinolones. Antimicrob. Agents Chemother. 42:1495-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson, D. I. 2003. Persistence of antibiotic resistant bacteria. Curr. Opin. Microbiol. 6:452-456. [DOI] [PubMed] [Google Scholar]
- 3.Andersson, D. I., and B. R. Levin. 1999. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2:489-493. [DOI] [PubMed] [Google Scholar]
- 4.Antonio, M., N. McFerran, and M. J. Pallen. 2002. Mutations affecting the Rossman fold of isoleucyl-tRNA synthetase are correlated with low-level mupirocin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 46:438-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bacher, J. M., V. de Crecy-Lagard, and P. R. Schimmel. 2005. Inhibited cell growth and protein functional changes from an editing-defective tRNA synthetase. Proc. Natl. Acad. Sci. USA 102:1697-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banwell, M. G., C. F. Crasto, C. J. Easton, A. K. Forrest, T. Karoli, D. R. March, L. Mensah, M. R. Nairn, P. J. O'Hanlon, M. D. Oldham, and W. Yue. 2000. Analogues of SB-203207 as inhibitors of tRNA synthetases. Bioorg. Med. Chem. Lett. 10:2263-2266. [DOI] [PubMed] [Google Scholar]
- 7.Barker, K. F. 1999. Antibiotic resistance: a current perspective. Br. J. Clin. Pharmacol. 48:109-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaulieu, D., and K. A. Ohemeng. 1999. Patents on bacterial tRNA synthetase inhibitors: January to March 1999. Expert Opin. Ther. Targets 9:1021-1028. [Google Scholar]
- 9.Bennett, I., N. J. Broom, R. Cassels, J. S. Elder, N. D. Masson, and P. J. O'Hanlon. 1999. Synthesis and antibacterial properties of beta-diketone acrylate bioisosteres of pseudomonic acid A. Bioorg. Med. Chem. Lett. 9:1847-1852. [DOI] [PubMed] [Google Scholar]
- 10.Beyer, D., H. P. Kroll, R. Endermann, G. Schiffer, S. Siegel, M. Bauser, J. Pohlmann, M. Brands, K. Ziegelbauer, D. Haebich, C. Eymann, and H. Brotz-Oesterhelt. 2004. New class of bacterial phenylalanyl-tRNA synthetase inhibitors with high potency and broad-spectrum activity. Antimicrob. Agents Chemother. 48:525-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bina, J. E., R. A. Alm, M. Uria-Nickelsen, S. R. Thomas, T. J. Trust, and R. E. Hancock. 2000. Helicobacter pylori uptake and efflux: basis for intrinsic susceptibility to antibiotics in vitro. Antimicrob. Agents Chemother. 44:248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyce, J. M. 2001. MRSA patients: proven methods to treat colonization and infection. J. Hosp. Infect. 48(Suppl. A):S9-S14. [DOI] [PubMed] [Google Scholar]
- 13.Broom, N. J., R. Cassels, H. Y. Cheng, J. S. Elder, P. C. Hannan, N. Masson, P. J. O'Hanlon, A. Pope, and J. M. Wilson. 1996. The chemistry of pseudomonic acid. 17. Dual-action C-1 oxazole derivatives of pseudomonic acid having an extended spectrum of antibacterial activity. J. Med. Chem. 39:3596-3600. [DOI] [PubMed] [Google Scholar]
- 14.Brown, J. R., D. Gentry, J. A. Becker, K. Ingraham, D. J. Holmes, and M. J. Stanhope. 2003. Horizontal transfer of drug-resistant aminoacyl-transfer-RNA synthetases of anthrax and gram-positive pathogens. EMBO Rep. 4:692-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown, J. R., J. Zhang, and J. E. Hodgson. 1998. A bacterial antibiotic resistance gene with eukaryotic origins. Curr. Biol. 8:R365-R367. [DOI] [PubMed] [Google Scholar]
- 16.Brown, M. J., P. S. Carter, A. S. Fenwick, A. P. Fosberry, D. W. Hamprecht, M. J. Hibbs, R. L. Jarvest, L. Mensah, P. H. Milner, P. J. O'Hanlon, A. J. Pope, C. M. Richardson, A. West, and D. R. Witty. 2002. The antimicrobial natural product chuangxinmycin and some synthetic analogues are potent and selective inhibitors of bacterial tryptophanyl tRNA synthetase. Bioorg. Med. Chem. Lett. 12:3171-3174. [DOI] [PubMed] [Google Scholar]
- 17.Brown, M. J. B., L. M. Mensah, M. L. Doyle, N. J. P. Broom, N. Osbourne, A. K. Forrest, C. M. Richardson, P. J. O'Hanlon, and A. J. Pope. 2000. Rational design of femtomolar inhibitors of isoleucyl tRNA synthetase from a binding model for pseudomonic acid-A. Biochemistry 39:6003-6011. [DOI] [PubMed] [Google Scholar]
- 18.Bush, K. 2004. Antibacterial drug discovery in the 21st century. Clin. Microbiol. Infect. 10(Suppl. 4):10-17. [DOI] [PubMed] [Google Scholar]
- 19.Bush, K., M. Macielag, and M. Weidner-Wells. 2004. Taking inventory: antibacterial agents currently at or beyond phase 1. Curr. Opin. Microbiol. 7:466-476. [DOI] [PubMed] [Google Scholar]
- 20.Cashel, M., D. R. Gentry, V. J. Hernandez, and D. Vinella. 1996. The stringent response, p. 1458-1496. In F. C. Neidhardt (ed.), Escherichia coli and Salmonella: cellular and molecular microbiology, vol. 1, 2nd ed. ASM Press, Washington, D.C. [Google Scholar]
- 21.Cassels, R., B. Oliva, and D. Knowles. 1995. Occurrence of the regulatory nucleotides ppGpp and pppGpp following induction of the stringent response in staphylococci. J. Bacteriol. 177:5161-5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chain, E. B., and G. Mellows. 1977. Pseudomonic acid. Part 1. The structure of pseudomonic acid A, a novel antibiotic produced by Pseudomonas fluorescens. J. Chem. Soc. Perkins Trans. I 1:294-309. [PubMed] [Google Scholar]
- 23.Chopra, I. 1998. Research and development of antibacterial agents. Curr. Opin. Microbiol. 1:495-501. [DOI] [PubMed] [Google Scholar]
- 24.Chopra, I., L. Hesse, and A. J. O'Neill. 2002. Exploiting current understanding of antibiotic action for discovery of new drugs. Appl. Bacteriol. Symp. Ser. 92:4S-15S. [PubMed] [Google Scholar]
- 25.Cookson, B. D. 1998. The emergence of mupirocin resistance: a challenge to infection control and antibiotic prescribing practice. J. Antimicrob. Chemother. 41:11-18. [DOI] [PubMed] [Google Scholar]
- 26.Creppy, E. E., M. Mayer, D. Kern, M. Schlegel, P. S. Steyn, R. Vleggaar, and G. Dirheimer. 1981. In vitro inhibition of yeast valyl-tRNA synthetase by the valine homologue of ochratoxin A. Biochim. Biophys. Acta 656:265-268. [DOI] [PubMed] [Google Scholar]
- 27.Curtis, N. A., R. L. Eisenstadt, S. J. East, R. J. Cornford, L. A. Walker, and A. J. White. 1988. Iron-regulated outer membrane proteins of Escherichia coli K-12 and mechanism of action of catechol-substituted cephalosporins. Antimicrob. Agents Chemother. 32:1879-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davey, P. 1998. Eradication of nasal carriage of Staphylococcus aureus—is it cost-effective? J. Hosp. Infect. 40(Suppl. B):S31-S37. [DOI] [PubMed] [Google Scholar]
- 29.Decousser, J. W., P. Pina, J. C. Ghnassia, J. P. Bedos, and P. Y. Allouch. 2003. First report of clinical and microbiological failure in the eradication of glycopeptide-intermediate methicillin-resistant Staphylococcus aureus carriage by mupirocin. Eur. J. Clin. Microbiol. Infect. Dis. 22:318-319. [DOI] [PubMed] [Google Scholar]
- 30.Deshpande, L. M., A. M. Fix, M. A. Pfaller, and R. N. Jones. 2002. Emerging elevated mupirocin resistance rates among staphylococcal isolates in the SENTRY Antimicrobial Surveillance Program (2000): correlations of results from disk diffusion, Etest and reference dilution methods. Diagn. Microbiol. Infect. Dis. 42:283-290. [DOI] [PubMed] [Google Scholar]
- 31.Eriani, G., M. Delarue, O. Poch, J. Gangloff, and D. Moras. 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347:203-206. [DOI] [PubMed] [Google Scholar]
- 32.Eschenburg, S., W. Kabsch, M. L. Healy, and E. Schonbrunn. 2003. A new view of the mechanisms of UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) and 5-enolpyruvylshikimate-3-phosphate synthase (AroA) derived from X-ray structures of their tetrahedral reaction intermediate states. J. Biol. Chem. 278:49215-49222. [DOI] [PubMed] [Google Scholar]
- 33.Eveland, S. S., D. L. Pompliano, and M. S. Anderson. 1997. Conditionally lethal Escherichia coli murein mutants contain point defects that map to regions conserved among murein and folyl poly-gamma-glutamate ligases: identification of a ligase superfamily. Biochemistry 36:6223-6229. [DOI] [PubMed] [Google Scholar]
- 34.Floss, H. G. 1981. Biosynthesis of some aromatic antibiotics, p. 236-261. In J. W. Corcoran (ed.), Antibiotic biosynthesis, vol. 4. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 35.Fujimura, S., Y. Tokue, and A. Watanabe. 2003. Isoleucyl-tRNA synthetase mutations in Staphylococcus aureus clinical isolates and in vitro selection of low-level mupirocin-resistant strains. Antimicrob. Agents Chemother. 47:3373-3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujimura, S., and A. Watanabe. 2003. Survey of high- and low-level mupirocin-resistant strains of methicillin-resistant Staphylococcus aureus in 15 Japanese hospitals. Chemotherapy 49:36-38. [DOI] [PubMed] [Google Scholar]
- 37.Gallant, P., J. Finn, D. Keith, and P. Wendler. 2000. The identification of quality antibacterial drug discovery targets: a case study with aminoacyl-tRNA synthetases. Expert Opin. Ther. Targets 4:1-9. [Google Scholar]
- 38.Gentry, D. R., K. A. Ingraham, M. J. Stanhope, S. Rittenhouse, R. L. Jarvest, P. J. O'Hanlon, J. R. Brown, and D. J. Holmes. 2003. Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae with two distinct methionyl-tRNA synthetase genes. Antimicrob. Agents Chemother. 47:1784-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbart, J., C. R. Perry, and B. Slocombe. 1993. High-level mupirocin resistance in Staphylococcus aureus—evidence for 2 distinct isoleucyl-transfer RNA-synthetases. Antimicrob. Agents Chemother. 37:32-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heacock, D., C. J. Forsyth, K. Shiba, and K. Musiter-Forsyth. 1996. Synthesis and aminoacyl-tRNA synthetase inhibitory of prolyl adenylate analogs. Bioorg. Chem. 24:273-289. [Google Scholar]
- 41.Henkel, T., and J. Finlay. 1999. Emergence of resistance during mupirocin treatment: is it a problem in clinical practice? J. Chemother. 11:331-337. [DOI] [PubMed] [Google Scholar]
- 42.Hodges, B. M., and R. L. White. 2001. Antimicrobial cycling: the future or a fad? Ann. Pharmacother. 35:1224-1232. [DOI] [PubMed] [Google Scholar]
- 43.Hubschwerlen, C., J. L. Specklin, D. K. Baeschlin, Y. Borer, S. Haefeli, C. Sigwalt, S. Schroeder, and H. H. Locher. 2003. Structure-activity relationship in the oxazolidinone-quinolone hybrid series: influence of the central spacer on the antibacterial activity and the mode of action. Bioorg. Med. Chem. Lett. 13:4229-4233. [DOI] [PubMed] [Google Scholar]
- 44.Hudson, I. R. B. 1994. The efficacy of intranasal mupirocin in the prevention of staphylococcal infections—a review of recent experience. J. Hosp. Infect. 27:81-98. [DOI] [PubMed] [Google Scholar]
- 45.Hughes, J., and G. Mellows. 1978. Inhibition of isoleucyl-transfer ribonucleic acid synthetase in Escherichia coli by pseudomonic acid. Biochem. J. 176:305-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hurdle, J. G., A. J. O'Neill, and I. Chopra. 2004. Anti-staphylococcal activity of indolmycin, a potential topical agent for control of staphylococcal infections. J. Antimicrob. Chemother. 54:549-552. [DOI] [PubMed] [Google Scholar]
- 47.Hurdle, J. G., A. J. O'Neill, and I. Chopra. 2004. The isoleucyl-tRNA synthetase mutation V588F conferring mupirocin resistance in glycopeptide-intermediate Staphylococcus aureus is not associated with a significant fitness burden. J. Antimicrob. Chemother. 53:102-104. [DOI] [PubMed] [Google Scholar]
- 48.Hurdle, J. G., A. J. O'Neill, E. Ingham, C. Fishwick, and I. Chopra. 2004. Analysis of mupirocin resistance and fitness in Staphylococcus aureus by molecular genetic and structural modeling techniques. Antimicrob. Agents Chemother. 48:4366-4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ibba, M., and D. Soll. 2000. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69:617-650. [DOI] [PubMed] [Google Scholar]
- 50.Jarvest, R. L., J. M. Berge, V. Berry, H. F. Boyd, M. J. Brown, J. S. Elder, A. K. Forrest, A. P. Fosberry, D. R. Gentry, M. J. Hibbs, D. D. Jaworski, P. J. O'Hanlon, A. J. Pope, S. Rittenhouse, R. J. Sheppard, C. Slater-Radosti, and A. Worby. 2002. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against gram-positive pathogens. J. Med. Chem. 45:1959-1962. [DOI] [PubMed] [Google Scholar]
- 51.John, J. F., Jr., and L. B. Rice. 2000. The microbial genetics of antibiotic cycling. Infect. Control Hosp. Epidemiol. 21:S22-S31. [DOI] [PubMed] [Google Scholar]
- 52.Kanamaru, T., Y. Nakano, Y. Toyoda, K. I. Miyagawa, M. Tada, T. Kaisho, and M. Nakao. 2001. In vitro and in vivo antibacterial activities of TAK-083, an agent for treatment of Helicobacter pylori infection. Antimicrob. Agents Chemother. 45:2455-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawamura, T., D. Liu, M. J. Towle, R. Kageyama, N. Tsukahara, T. Wakabayashi, and B. A. Littlefield. 2003. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J. Antibiot. 56:709-715. [DOI] [PubMed] [Google Scholar]
- 54.Kim, S., S. W. Lee, E. C. Choi, and S. Y. Choi. 2003. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl. Microbiol. Biotechnol. 61:278-288. [DOI] [PubMed] [Google Scholar]
- 55.Kim, S. Y., and J. Lee. 2003. 3-D-QSAR study and molecular docking of methionyl-tRNA synthetase inhibitors. Bioorg. Med. Chem. 11:5325-5331. [DOI] [PubMed] [Google Scholar]
- 56.Kitabatake, M., K. Ali, A. Demain, K. Sakamoto, S. Yokoyama, and D. Soll. 2002. Indolmycin resistance of Streptomyces coelicolor A3(2) by induced expression of one of its two tryptophanyl-tRNA synthetases. J. Biol. Chem. 277:23882-23887. [DOI] [PubMed] [Google Scholar]
- 57.Kumar, A., and H. P. Schweizer. 2005. Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv. Drug Deliv. Rev. 57:1486-1513. [DOI] [PubMed] [Google Scholar]
- 58.Lavin, B. S. 2000. Antibiotic cycling and marketing into the 21st century: a perspective from the pharmaceutical industry. Infect. Control Hosp. Epidemiol. 21:S32-S35. [DOI] [PubMed] [Google Scholar]
- 59.Lenski, R. E. 1997. The cost of antibiotic resistance—from the perspective of a bacterium, p. 131-140. In Antibiotic resistance: origins, evolution, selection and spread, vol. 207. Wiley, Chichester, United Kingdom.
- 60.Loddenkemper, R., D. Sagebiel, and A. Brendel. 2002. Strategies against multidrug-resistant tuberculosis. Eur. Respir. J. Suppl. 36:66S-77S. [DOI] [PubMed] [Google Scholar]
- 61.Macarron, R., L. Mensah, C. Cid, C. Carranza, N. Benson, A. J. Pope, and E. Diez. 2000. A homogeneous method to measure aminoacyl-tRNA synthetase aminoacylation activity using scintillation proximity assay technology. Anal. Biochem. 284:183-190. [DOI] [PubMed] [Google Scholar]
- 62.Menichetti, F. 2005. Current and emerging serious gram-positive infections. Clin. Microbiol. Infect. 11(Suppl. 3):22-28. [DOI] [PubMed] [Google Scholar]
- 63.Miesel, L., J. Greene, and T. A. Black. 2003. Genetic strategies for antibacterial drug discovery. Nat. Rev. Genet. 4:442-456. [DOI] [PubMed] [Google Scholar]
- 64.Mody, L., C. A. Kauffman, S. A. McNeil, A. T. Galecki, and S. F. Bradley. 2003. Mupirocin-based decolonization of Staphylococcus aureus carriers in residents of 2 long-term care facilities: a randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 37:1467-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakama, T., O. Nureki, and S. Yokoyama. 2001. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J. Biol. Chem. 276:47387-47393. [DOI] [PubMed] [Google Scholar]
- 66.Ogilvie, A., K. Wiebauer, and W. Kersten. 1975. Stringent control of ribonucleic acid synthesis in Bacillus subtilis treated with granaticin. Biochem. J. 152:517-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Otoguro, K., H. Ui, A. Ishiyama, M. Kobayashi, H. Togashi, Y. Takahashi, R. Masuma, H. Tanaka, H. Tomoda, H. Yamada, and S. Omura. 2003. In vitro and in vivo antimalarial activities of a non-glycosidic 18-membered macrolide antibiotic, borrelidin, against drug-resistant strains of Plasmodia. J. Antibiot. 56:727-729. [DOI] [PubMed] [Google Scholar]
- 68.Paetz, W., and G. Nass. 1973. Biochemical and immunological characterization of threonyl-tRNA synthetase of two borrelidin-resistant mutants of Escherichia coli K12. Eur. J. Biochem. 35:331-337. [DOI] [PubMed] [Google Scholar]
- 69.Perl, T., and J. Golub. 1998. New approaches to reduce Staphylococcus aureus nosocomial infection rates: treating S. aureus nasal carriage. Ann. Pharmacother. 32:7-16. [DOI] [PubMed] [Google Scholar]
- 70.Petraitis, V., R. Petraitiene, A. M. Kelaher, A. A. Sarafandi, T. Sein, D. Mickiene, J. Bacher, A. H. Groll, and T. J. Walsh. 2004. Efficacy of PLD-118, a novel inhibitor of Candida isoleucyl-tRNA synthetase, against experimental oropharyngeal and esophageal candidiasis caused by fluconazole-resistant C. albicans. Antimicrob. Agents Chemother. 48:3959-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pohlmann, J., and H. Brotz-Oesterhelt. 2004. New aminoacyl-tRNA synthetase inhibitors as antibacterial agents. Curr. Drug Targets Infect. Disord. 4:261-272. [DOI] [PubMed] [Google Scholar]
- 72.Raczniak, G., M. Ibba, and D. Soll. 2001. Genomics-based identification of targets in pathogenic bacteria for potential therapeutic and diagnostic use. Toxicology 160:181-189. [DOI] [PubMed] [Google Scholar]
- 73.Retailleau, P., X. Huang, Y. Yin, M. Hu, V. Weinreb, P. Vachette, C. Vonrhein, G. Bricogne, P. Roversi, V. Ilyin, and C. W. Carter, Jr. 2003. Interconversion of ATP binding and conformational free energies by tryptophanyl-tRNA synthetase: structures of ATP bound to open and closed, pre-transition-state conformations. J. Mol. Biol. 325:39-63. [DOI] [PubMed] [Google Scholar]
- 74.Ruan, B., M. L. Bovee, M. Sacher, C. Stathopoulos, K. Poralla, C. S. Francklyn, and D. Soll. 2005. A unique hydrophobic cluster near the active site contributes to differences in borrelidin inhibition among threonyl-tRNA synthetases. J. Biol. Chem. 280:571-577. [DOI] [PubMed] [Google Scholar]
- 75.Schimmel, P., J. Tao, and J. Hill. 1998. Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 12:1599-609. [PubMed] [Google Scholar]
- 76.Schmid, M. B. 2001. New targets and strategies for identification of novel classes of antibiotics, p. 197-208. In D. Hughes and D. I. Andersson (ed.), Antibiotic development and resistance. Taylor and Francis, New York, N.Y.
- 77.Schmitz, F. J., E. Lindenlauf, B. Hofmann, A. C. Fluit, J. Verhoef, H. P. Heinz, and M. E. Jones. 1998. The prevalence of low- and high-level mupirocin resistance in staphylococci from 19 European hospitals. J. Antimicrob. Chemother. 42:489-495. [DOI] [PubMed] [Google Scholar]
- 78.Strahilevitz, J., and D. C. Hooper. 2005. Dual targeting of topoisomerase IV and gyrase to reduce mutant selection: direct testing of the paradigm by using WCK-1734, a new fluoroquinolone, and ciprofloxacin. Antimicrob. Agents Chemother. 49:1949-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sutherland, R., R. J. Boon, K. E. Griffin, P. J. Masters, B. Slocombe, and A. R. White. 1985. Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob. Agents Chemother. 27:495-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tao, J., P. Wendler, G. Connelly, A. Lim, J. Zhang, M. King, T. Li, J. A. Silverman, P. R. Schimmel, and F. P. Tally. 2000. Drug target validation: lethal infection blocked by inducible peptide. Proc. Natl. Acad. Sci. USA 97:783-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thomas, D. G., J. M. Wilson, M. J. Day, and A. D. Russell. 1999. Structural changes induced by mupirocin in Staphylococcus aureus cells. Int. J. Antimicrob. Agents 13:9-14. [DOI] [PubMed] [Google Scholar]
- 82.Thomas, D. G., J. M. Wilson, M. J. Day, and A. D. Russell. 1999. Mupirocin resistance in staphylococci: development and transfer of isoleucyl tRNA synthetase-mediated resistance in vitro. J. Appl. Microbiol. 86:715-722. [DOI] [PubMed] [Google Scholar]
- 83.von der Haar, F., H. Gabius, and F. Cramer. 1981. Target directed drug synthesis: the aminoacyl-tRNA synthetases as possible targets. Angew. Chem. 20:217-302. [Google Scholar]
- 84.Walker, E. S., F. Levy, M. Shorman, G. David, J. Abdalla, and F. A. Sarubbi. 2004. A decline in mupirocin resistance in methicillin-resistant Staphylococcus aureus accompanied administrative control of prescriptions. J. Clin. Microbiol. 42:2792-2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watanabe, H., H. Masaki, N. Asoh, K. Watanabe, K. Oishi, S. Kobayashi, A. Sato, R. Sugita, and T. Nagatake. 2001. Low concentrations of mupirocin in the pharynx following intranasal application may contribute to mupirocin resistance in methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 39:3775-3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Werner, R. G. 1980. Uptake of indolmycin in gram-positive bacteria. Antimicrob. Agents Chemother. 18:858-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Werner, R. G., and A. L. Demain. 1981. Directed biosynthesis of new indolmycins. J. Antibiot. 34:551-554. [DOI] [PubMed] [Google Scholar]
- 88.Werner, R. G., and W. Reuter. 1979. Interaction of indolmycin in the metabolism of tryptophan in rat liver. Arzneimittelforschung 29:59-63. [PubMed] [Google Scholar]
- 89.Werner, R. G., L. F. Thorpe, W. Reuter, and K. H. Nierhaus. 1976. Indolmycin inhibits prokaryotic tryptophanyl-tRNA ligase. Eur. J. Biochem. 68:1-3. [DOI] [PubMed] [Google Scholar]
- 90.Wilson, J. M., B. Oliva, R. Cassels, P. J. O'Hanlon, and I. Chopra. 1995. SB 205952, a novel semisynthetic monic acid analog with at least two modes of action. Antimicrob. Agents Chemother. 39:1925-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wright, G. D. 2005. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 57:1451-1470. [DOI] [PubMed] [Google Scholar]
- 92.Yanagisawa, T., and M. Kawakami. 2003. How does Pseudomonas fluorescens avoid suicide from its antibiotic pseudomonic acid? Evidence for two evolutionarily distinct isoleucyl-tRNA synthetases conferring self-defense. J. Biol. Chem. 278:25887-25894. [DOI] [PubMed] [Google Scholar]
- 93.Yanagisawa, T., J. T. Lee, H. C. Wu, and M. Kawakami. 1994. Relationship of protein structure of isoleucyl-tRNA synthetase with pseudomonic acid resistance of Escherichia coli. A proposed mode of action of pseudomonic acid as an inhibitor of isoleucyl-tRNA synthetase. J. Biol. Chem. 269:24304-24309. [PubMed] [Google Scholar]
- 94.Ziegelbauer, K., P. Babczinski, and W. Schonfeld. 1998. Molecular mode of action of the antifungal beta-amino acid BAY 10-8888. Antimicrob. Agents Chemother. 42:2197-2205. [DOI] [PMC free article] [PubMed] [Google Scholar]