

Abstract
Hepatitis C virus (HCV) is a major cause of chronic liver disease, cirrhosis, and hepatocellular carcinoma worldwide. HCV has a positive-strand RNA genome of about 9.4 kb in size, which serves as a template for replication and for translation of a polyprotein of about 3,000 amino acids. The polyprotein is cleaved co- and posttranslationally by cellular and viral proteases into at least 10 different mature proteins. One of these proteins, nonstructural protein 3 (NS3), has serine protease and NTPase/RNA helicase activity. Arginine 467 in the helicase domain of NS3 (arginine 1493 in the polyprotein) can be methylated by protein arginine methyltransferase 1 (PRMT1). Here we report that the methylation of NS3 inhibits the enzymatic activity of the helicase. Furthermore, we found that PRMT1 activity itself is regulated by protein phosphatase 2A (PP2A). PP2A inhibits PRMT1 enzymatic activity and therefore increases the helicase activity of NS3. This is important, because we found an increased expression of PP2A in cell lines with inducible HCV protein expression, in transgenic mice expressing HCV proteins in hepatocytes, and in liver biopsy samples from patients with chronic hepatitis C. Interestingly, up-regulation of PP2A not only modulates the enzymatic activity of an important viral protein, NS3 helicase, but also interferes with the cellular defense against viruses by inhibiting interferon-induced signaling through signal transducer and activator of transcription 1 (STAT1). We conclude that up-regulation of PP2A might be crucial for the efficient replication of HCV and propose PP2A as a potential target for anti-HCV treatment strategies.
Infections with hepatitis C virus (HCV) become chronic in most patients, and chronic hepatitis C (CHC) can progress to cirrhosis and hepatocellular carcinoma (9, 17, 23, 29). To persist in the host, the virus has to evade the immune system. We have shown previously that HCV interferes with alpha interferon (IFN-α)-induced signaling through the Jak-STAT pathway (5, 10, 14). The interferon system is an important component of the host response against viruses, and mice with deficiencies of IFN receptors or of signal transducer and activator of transcription 1 (STAT1) are highly susceptible to viral infections (2, 11, 26). IFN-α/β binding to its receptor activates members of the Jak family of tyrosine kinases, which then phosphorylate STAT1, STAT2, and STAT3 on a single tyrosine residue. Phosphorylated STATs form dimers, translocate into the nucleus, bind to promoter elements of interferon-stimulated genes, and activate the transcription of interferon-stimulated genes (7). This activation cycle is terminated by tyrosine dephosphorylation in the nucleus, followed by the decay of dimers and the nuclear export of STATs (8, 35). The pathway is tightly controlled by a number of inhibitory proteins (20, 33), among them protein inhibitor of activated STAT1 (PIAS1) (25). PIAS1 inhibits the last step in the Jak-STAT pathway, i.e., DNA binding. The complex formation between STAT1 and PIAS1 is regulated by an important posttranslational modification of STAT1, arginine methylation (30). Methylation of STAT1 is catalyzed by protein arginine methyltransferase 1 (PRMT1) and protects STAT1 from binding and inactivation by PIAS1 (30).
We have previously reported that HCV inhibits IFN-α-induced signaling at the level of STAT DNA binding (5, 14). Further analysis of the mechanism responsible for HCV interference with STAT signaling led to two a priori independent observations made both with extracts from liver cells of HCV transgenic mice and with liver biopsy samples from patients with CHC: (i) STAT1 was hypomethylated and bound by PIAS1, and (i) protein phosphatase 2Ac (PP2Ac) expression was increased (10). PP2A is a heterotrimeric protein phosphatase consisting of a 36-kilodalton catalytic C subunit (PP2Ac), a 65-kilodalton structural A subunit, and a variable regulatory B subunit. PP2A is expressed in all cell types, is primarily a serine/threonine phosphatase, and is involved in a wide range of cellular processes, including cell cycle regulation, cell morphology, development, signal transduction, translation, apoptosis, and stress response (15, 27). An involvement of PP2A in the regulation of PRMT1 has not been reported before. However, we found that expression of a constitutively active form of PP2Ac in human hepatoma cells (Huh7) resulted in hypomethylation of STAT1 and inhibition of IFN-α-induced signaling (10), demonstrating that PP2Ac is upstream in a signaling pathway that regulates STAT1 methylation. We therefore further investigated the role of PP2A in the regulation of STAT1 methylation. In the present paper, we report that PP2Ac interacts directly with PRMT1 and that it inhibits the enzymatic activity of PRMT1.
Interestingly, PRMT1 is also responsible for arginine methylation of NS3, a nonstructural HCV protein with protease and helicase activity (31). The NS3 helicase-NTPase domain consists of the 442 C-terminal amino acids of NS3 and belongs to the DEAD (Asp-Glu-Ala-Asp) box RNA helicase family (19, 21, 24). The domain has probably multiple functions, including RNA-stimulated NTPase activity, RNA binding, and unwinding of RNA regions with extensive secondary structure. Mutational analysis of NS3 helicase revealed that arginines 1490 and 1493 (position in the polyprotein) are essential for enzymatic activity (16). Interestingly, arginine 1493 (arginine 467 of the helicase domain) has been shown to be posttranslationally modified by methylation through PRMT1 (31). However, the functional consequence of this modification is not known. Here we show that arginine methylation inhibits the enzymatic activity of NS3. Furthermore, we show that PRMT1 itself is negatively regulated by PP2A. Therefore, by increasing the expression level of PP2Ac, HCV can indirectly regulate the helicase activity of NS3.
Taken together, our results support an important role of PP2A in the regulation of the viral life cycle of HCV. By inducing PP2Ac overexpression, HCV achieves both an inhibition of IFN-α signaling and an increase in NS3 helicase activity.
MATERIALS AND METHODS
Reagents, antibodies, and plasmids.
Human IFN-α was obtained from Hoffmann LaRoche (Basel, Switzerland). Purified PP2Ac and okadaic acid (OA) were purchased from Upstate (LucernaChem, Luzern, Switzerland) and Sigma (Fluka Chemie GmbH, Buchs, Switzerland), respectively. His-Bax was purchased from Santa Cruz (LabForce AG, Nunningen, Switzerland). Glutathione S-transferase (GST) columns (MicroSpin GST purification module), IPTG (isopropyl-β-D-thiogalactopyranoside), and 3H-AdoMet (specific activity, 15 Ci/mmol) were obtained from Amersham Biosciences (Amersham Pharmacia Biotech Europe GmbH, Dübendorf, Switzerland). pGEX-HRMT1L2 (human PRMT1) was a generous gift from Pamela A. Silver. The NS3 helicase domain (NS3h) construct His5n-Hel-His5c was a gift from David Frick. It contains amino acids 166 to 631 of NS3 (amino acids 1197 to 1663 of the polyprotein) and is derived from the genotype 1a isolate H77 (accession number, AAB66324). A detailed description of the construct has been published (13).
For protein expression, transfected bacteria cells were grown overnight in LB medium supplemented with ampicillin or kanamycin until they reached an optical density of 0.6 to 0.7. GST-PRMT1 expression was induced with 1 mM IPTG for 3 h at 30°C. NS3h expression was induced with 1 mM IPTG for 3 h at 37°C. After lysis of the bacteria, the fusion proteins were purified with a GST MicroSpin column (Amersham) or a Ni-nitrilotriacetic acid spin kit (QIAGEN AG, Basel, Switzerland) according to the manufacturers' instructions. Purified NS3h was dialyzed using a Float-A-Lyzer system (Socochim, Lausanne, Switzerland). Purified proteins were then concentrated and buffers exchanged in a storage buffer (50 mM NaCl, 50 mM Tris, 1 mM EDTA) with iCON concentrator (Pierce).
Cell lines.
The generation of HA-PP2Ac cells was described previously (10). The cells were cultured in 10% calf serum/minimum essential medium supplemented with neomycin (Invitrogen, Basel, Switzerland). U-2 OS human osteosarcoma-derived, tetracycline (tet)-regulated cell lines UHCV-57.3 and UHCV-32, which inducibly express the entire HCV polyprotein derived from an HCV H consensus cDNA (18, 28), and UGFP-20 cells, which inducibly express green fluorescent protein, have been described previously (32).
Preparation of extracts from cells.
Cell extracts for Western blots and electrophoretic mobility shift assays were prepared as described previously (10).
RNA isolation, reverse transcription, and SYBR-PCR.
Total RNA was isolated from the cells using a Perfect RNA Eukaryotic Mini kit (Eppendorf, Vaudaux-Eppendorf, Basel, Switzerland) according to the manufacturer's instructions. RNA was reverse transcribed by Moloney murine leukemia virus reverse transcriptase (Promega, Promega Biosciences Inc., Wallisellen, Switzerland) in the presence of random hexamers (Promega) and deoxynucleoside triphosphate. The reaction mixture was incubated for 5 min at 70°C and then for 1 h at 37°C. The reaction was stopped by heating to 95°C for 5 min. SYBR-PCR was performed based on SYBR green fluorescence (SYBR green PCR master mix, Applied Biosystems, Foster City, CA). To prevent genomic DNA amplification, the primers for RPL19 and PP2Ac were designed across exon-intron junctions. The primers for RPL19 were 5′ GATGCCGGAAAAACACCTTG 3′ and 5′ TGGCTGTACCCTTCCGCTT 3′. The primers for PP2Ac were 5′ CCACAGCAAGTCACACATTGG 3′ and 5′ CAGAGCACTTGATCGCCTACAA 3′. The change in cycle threshold (CT) value (ΔCT) was derived by subtracting the CT value for ribosomal protein L19 (RPL19), which served as an internal control, from the CT values for PP2Ac. All reactions were run in duplicate by use of an ABI 7000 sequence detection system (Applied Biosystems). The difference in mRNA expression levels between HCV and non-HCV cells was expressed as an n-fold increase according to the formula 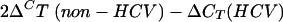 .
.
Immunoprecipitation and immunoblotting.
Cell lysates were incubated with anti-PP2Ac (Upstate), anti-PRMT1 (Abcam), anti-BIP (BD Biosciences, Basel, Switzerland), anti-NS4B (made by Darius Moradpour), or anti-GST (Amersham) antibodies overnight at 4°C. Protein A-Sepharose (Sigma) was added, and the samples were incubated for 3 h at 4°C on a rotating wheel. After sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transfer onto a nitrocellulose membrane (Schleicher & Schuell, Bottmingen, Switzerland), proteins were detected with anti-PP2Ac (Upstate) or anti-His antibodies (Santa Cruz). For loading amount control, the nitrocellulose membrane was stained using Blot-FastStain (GenoTech, Cell Concepts GmbH, Umkirch, Germany) according to the manufacturer's instructions.
GST-PRMT1 binding to PP2Ac.
One ml of bacterial cell lysates expressing GST-PRMT1 was loaded onto a GST column (Amersham) and incubated for 10 min at room temperature. After centrifugation at 2,500 rpm for 1 min, 40 μg of total proteins from Huh7 cells was loaded onto the column to a final volume of 120 μl, and then the binding reaction was performed overnight at 4°C on a rotating wheel. The column was then washed four times with cold phosphate-buffered saline. For elution, 120 μl of reduced glutathione was used for 1 h at 4°C on a rotating wheel. To verify the specificity of the binding of PP2Ac to GST-PRMT1, the membrane was stripped and reblotted for BIP.
Methylation assay.
An in vitro methylation assay was performed according to a protocol previously described by Mowen et al. in 2001 (30) with some modifications. For Fig. 1 C, 50 μg of Huh7 lysate was incubated in the presence of 6 μg of GST-PRMT1 and 4 μl of 14C-AdoMet in a final reaction volume of 80 μl for 2 h at 37°C. The reaction was stopped by adding 20 μl of sample loading buffer and boiling for 5 min. The proteins were separated on an 8% SDS-polyacrylamide gel. The lower part of the gel was cut out and stained with Coomassie blue to check for equal loading.
FIG. 1.

PP2Ac binds directly to PRMT1 and inhibits its enzymatic activity. (A) Whole-cell lysate from Huh7 cells was incubated overnight at 4°C in a GST column that was left empty (left lane) or in one that was preloaded with GST-PRMT1 (right lane). The unbound proteins were recovered as flowthrough, and the bound proteins were eluted with reduced glutathione. PP2Ac was detected by Western blotting (upper panels). As a control, the membranes were then stripped and reprobed with antibodies against the immunoglobulin binding protein BIP (lower panels). The arrow with the asterisk shows the size of BIP. (B) Purified PP2Ac (0.2 units) or His-Bax (100 ng) was incubated with 10 μg of purified GST-PRMT1. Antibodies to GST were used to immunoprecipitate bound proteins. PP2Ac or His-Bax was detected by Western blotting (with anti-PP2Ac or anti-His, respectively). (C) Whole-cell lysate from Huh7 cells was incubated with 14C-AdoMet alone (lane 1), with 14C-AdoMet plus GST-PRMT1 (lane 2), or with 14C-AdoMet plus GST-PRMT1 plus 0.1 units (lane 3), 0.2 units (lane 4), 0.3 units (lane 5), or 0.4 units (lane 6) of purified PP2Ac. The proteins were separated on an SDS-polyacrylamide gel, and methylation of proteins was detected by autoradiography. To control for equal loading, a 1-cm strip at the bottom of the gel was cut and stained with Coomassie blue (middle panel). The strongest band in the autoradiography (indicated with an asterisk) was quantified for each lane by use of NIH imaging software (lower panel).
For methyltransferase activity measurements in UHCV-57.3 cells (Fig. 2C), the reaction was performed by using 10 μg of whole-cell lysate in the presence of 3 μl of 3H-AdoMet for 2 h at 37°C. The reaction was then stopped by adding 20 μl of sample loading buffer, and the reaction volume was boiled for 5 min and separated on an 8% SDS-polyacrylamide gel. The upper part of the gel was fixed for 30 min in a solution of isopropanol:water:acetic acid (25:65:10) and then amplified in Amplify (Amersham) with gentle agitation for 30 min. The gel was dried and then exposed to X-ray Hyperfilm (Amersham) for 5 days. The lower part of the gel was cut out and stained with Coomassie blue to check for equal loading.
FIG. 2.

Expression of HCV proteins induces PP2Ac. UHCV-57.3 and UGFP-20 cells (controls that express green fluorescent protein) were cultured for 24 h in the presence or absence of tetracycline. (A) PP2Ac mRNA was quantified with real-time RT-PCR. The ratio of the amounts of PP2Ac mRNA in derepressed (minus tet) and repressed cells (plus tet) is shown. Expression of viral proteins increases PP2Ac mRNA levels about twofold. The graph shows the mean (with the standard error of the mean) of two independent experiments. No error bar is shown for UHCV-57.3 cells, because the two values (2.22 and 2.21) were too close. (B) PP2Ac protein (arrow) expression in UHCV-32 cells was examined by Western blotting (left panel). Cells were cultured in presence (left lane) or absence (right lane) of tetracycline. Membranes were probed with a mixture of antibodies to PP2Ac (arrow) and NS4B (asterisk; faint upper band in right lane). The PP2Ac bands were quantified with NIH imaging software (lower panel). As a loading control, the membrane was stained with Blot-FastStain (right panel). (C) UHCV-57.3 cells were cultured in presence (left lane) or absence (right lane) of tetracycline for 24 h. Whole-cell lysates were then incubated for 2 h with radioactively labeled AdoMet. Protein methylation was detected by autoradiography. Equal loading was controlled by cutting the lower part of the gel and staining with Coomassie blue (lower panel). (D) Whole-cell extracts were prepared from two culture plates each of Huh7 cells and of Huh7 cells harboring an HCV replicon (Huh7.5 cells). The samples were analyzed by Western blotting using an antibody against PP2Ac (arrow). Loading was controlled by staining the membrane with Blot-FastStain (lower panel). Huh7.5 cells (lanes 3 and 4) had increased PP2Ac protein expression compared with Huh7 cells (lanes 1 and 2).
For in vitro methylation of purified NS3 helicase (Fig. 3), 30 μg of His-Hel-His (13) was incubated with 2 μg of GST-PRMT in the presence of 6 μl of 14C-AdoMet for 2 h at 37°C. The reaction was then stopped by adding 20 μl of sample loading buffer, and the reaction volume was boiled for 5 min and separated on an 8% SDS-polyacrylamide gel.
FIG. 3.

PRMT1 methylates NS3 on its helicase domain, and PP2Ac inhibits the enzymatic activity of PRMT1. Ten μg of purified NS3 helicase domain was incubated with 14C-AdoMet alone (lane 1), 14C-AdoMet with 6 μg of GST-PRMT1 (lane 2), or 14C-AdoMet with 6 μg of GST-PRMT1 plus 0.2 units of PP2Ac (lane 3).
DNA-DNA substrate.
To prepare the double-stranded DNA (dsDNA) substrate for the unwinding assay, a short DNA oligonucleotide, 5′-TGG TAC TCC TCA CAC CTG GGC GGC GGT TAA-3′, was 32P radiolabeled using the T4 polynucleotide kinase (Promega). Unincorporated ATP was removed through a Mini Quick Spin column. The labeled oligonucleotide was mixed with an equal amount the unlabeled complementary strand, 5′-GAC TAC GTA CTG TTA ACC GCC GCC CAG GTG TGA GGA GTA CCA GGC CAG ATC TGC-3′. The mixture was heated to 95°C for 3 min and left to cool slowly at room temperature.
DNA helicase assay.
Purified methylated (Met-NS3h) or unmethylated (NS3h) His-Hel-His (100 nM) (13) was incubated with the double-stranded DNA substrate (10 nM) in reaction buffer (25 mM MOPS [morpholinepropanesulfonic acid; pH 6.2], 0.1% Tween 20, 3 mM MgCl2) for 15 min at 23°C. The reactions were started by the addition of 5 mM ATP and then stopped at the indicated time points by the addition of a glycerol loading buffer containing 50 nM of a capture oligonucleotide (5′-TTA ACC GCC GCC CAG GTG TGA GGA GTA CCA-3′), 20 mM EDTA, and 0.5% SDS. The samples were analyzed by 8% nondenaturating polyacrylamide gel electrophoresis and quantitated by use of a PhosphorImager (Molecular Dynamics).
Purified methylated NS3h was obtained by incubating 30 μg of His-Hel-His (13) with 1 μg, 2 μg, or 3 μg of purified GST-PRMT in the presence of 9 mM AdoMet for 2 h at 37°C. To prepare the unmethylated NS3h, 30 μg of His-Hel-His was incubated with 2 μg of GST-PRMT1 for 2 h at 37°C (but without AdoMet).
ATPase assay.
Reactions were done at 37°C in reaction buffer (25 mM MOPS [pH 6.2], 0.1% Tween 20, 3 mM MgCl2, 5 mM ATP) with 100 nM of methylated or unmethylated NS3h. ATP quantification was done after 10 min with an ATP determination kit (Biaffin GmbH & Co KG, Kassel, Germany) according to the manufacturer's instructions.
Treatment of HCV replicon cell lines with the PP2A inhibitor okadaic acid.
HuH-7.5 cells harboring a subgenomic HCV replicon (3, 4) (kindly provided by Charles M. Rice, The Rockefeller University, New York, NY) were treated for 18 h with 5 μl/ml dimethyl sulfoxide, 25 nM OA, 2 μM 2′-C-methyladenosine (6) (a specific inhibitor of the HCV RNA-dependent RNA polymerase; kindly provided by Steven S. Carroll, Merck & Co., Inc., West Point, PA), or 1,000-U/ml human IFN-α2a (Roferon-A; Roche). Preliminary dose titrations indicated that 25 nM OA strongly inhibited PP2A activity with only minimal toxicity after 18 h. Subsequently, total cellular RNA was extracted with Trizol (Invitrogen) following the manufacturer's instructions. Denaturing agarose gel electrophoresis and Northern blot analyses were performed according to standard protocols. 32P-labeled cDNA fragments representing the entire HCV nonstructural region and a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe (12) as an internal control were employed simultaneously for Northern blot hybridization.
PP2Ac siRNA treatment of HuH-7.5 cells harboring a subgenomic HCV replicon.
HuH-7.5 cells (106) were transfected using Lipofectamine 2000 from Invitrogen according to the manufacturer's instructions with 10 μl of a 20-pmol/μl solution of PP2Ac SMARTpool small interfering RNA (siRNA) duplexes from Dharmacon or with a corresponding amount of a nonsilencing siRNA duplex from QIAGEN. Four hours after the transfection, cells were recovered for 48 h in Dulbecco's minimal essential medium supplemented with 10% fetal calf serum. Total RNA was isolated and reverse transcribed. RNA quantification was done by SYBR-PCR using specific primers for PP2Ac, HCV, and GAPDH. The primers for HCV were 5′ CGCTGCTTCTGCTTTCG 3′ and 5′ CACCCCTGCTCATAACC 3′. The primers for GAPDH were 5′ CAAGCTGTGGGCAAGGT 3′ and 5′ GGAAGGCCATGCCAGTGA 3′. The primers for PP2Ac and further details of the method are described above (see “RNA isolation, reverse transcription, and SYBR-PCR.”)
PP2A phosphatase activity assay.
Assessment of PP2A activity was performed according to the manufacturer's instructions with whole-cell extracts by use of a serine/threonine phosphatase assay system (Promega) with PPTase-2A buffer containing 250 mM imidazole (pH 7.2), 1 mM EGTA, 0.1% β-mercaptoethanol, and 0.5 mg/ml bovine serum albumin.
RESULTS
PP2Ac binds directly to PRMT1 and inhibits its enzymatic activity.
We have shown previously that expression of a constitutive active form of PP2Ac in Huh7 cells results in a strongly reduced methylation of STAT1 (10). Since STAT1 methylation is catalyzed by PRMT1 (30), we first wanted to test if PP2Ac can bind to PRMT1. To that end, whole-cell extracts from Huh7 cells were loaded onto a GST-PRMT1 column, and the flowthrough as well as the bound proteins were analyzed by Western blotting. As shown in Fig. 1A, PP2Ac was specifically bound by PRMT1 (whereas the ubiquitously expressed protein BIP was not). This interaction was direct and did not require additional proteins present in the whole-cell extract, because purified PP2Ac was also bound by GST-PRMT1 (Fig. 1B). As a control, purified His-Bax fusion protein was incubated with GST-PRMT1 but could not be coimmunoprecipitated (Fig. 1B). Next, we tested if PP2Ac binding to PRMT1 inhibits its methyltransferase catalytic activity. Whole-cell lysates from Huh7 cells were incubated with the radioactively labeled methyl group donor 14C-AdoMet. In the in vitro methylation assay used, the transfer of 14C-labeled methyl groups to cellular proteins by endogenous PRMT1 can be detected by autoradiography after separation of the proteins on an SDS-polyacrylamide gel (Fig. 1C, lane 1). The addition of purified GST-PRMT1 increases the methylation of proteins (Fig. 1C, lane 2). Importantly, the methylation of proteins in the lysate can be inhibited in a dose-dependent way by the addition of purified PP2Ac (Fig. 1C, lanes 3 to 6). We conclude that PP2Ac directly binds and inhibits PRMT1.
Expression of HCV proteins in cells up-regulates PP2Ac expression and inhibits cellular methyltransferases.
We have shown previously an increased expression of the catalytic subunit of protein phosphatase 2A (PP2Ac) in liver extracts from HCV transgenic mice and in liver biopsy samples from patients with CHC (10). The significant increase of PP2Ac expression in these samples was surprising, since it has been reported that PP2Ac expression is very tightly regulated by an autoregulatory loop (1). To further study the effect of HCV proteins on PP2Ac expression, we used a well-characterized cell line that inducibly expresses the entire open reading frame of HCV (UHCV-57.3) (32). HCV protein expression in these cells can be repressed by adding tetracycline to the culture medium. In the absence of tetracycline, UHCV-57.3 cells express all HCV proteins (32). As shown in Fig. 2A, which depicts results of real-time quantitative reverse transcription-PCR (RT-PCR) analysis, HCV protein expression induced a twofold increase in the PP2Ac mRNA concentration. By contrast, no upregulation of PP2Ac was found in UGFP-20 cells, which inducibly express the green fluorescent protein as a nonrelevant control. This enhanced mRNA expression resulted in an increased PP2Ac protein expression level (Fig. 2B). Because PP2Ac expression levels are usually tightly controlled in cells, we speculated that the increase in PP2Ac expression induced by HCV protein expression is biologically relevant. We therefore measured the consequences of increased PP2Ac expression on methylation of cellular substrates using the methylation assay. Indeed, when cell extracts of repressed and derepressed UHCV-57.3 cells were incubated with 3H-AdoMet, an overall reduction of endogenous methylase activity was found in HCV protein-expressing cells (Fig. 2C). Considering the in vitro assay results shown in Fig. 1C, we believe that the most likely explanation for this result is that HCV protein-induced overexpression of PP2Ac inhibits PRMT1. HCV-induced overexpression of PP2Ac was also found in HCV replicon cells (Fig. 2D).
PP2Ac inhibits NS3 methylation by PRMT1.
Next, we tested whether GST-PRMT1 can methylate NS3. To this end, we expressed and purified a NS3 helicase protein (NS3h). Incubation of NS3h with purified GST-PRMT1 in the presence of 14C-AdoMet led to methylation of NS3h (Fig. 3, lane 2). This result is in agreement with a previous report that NS3 is methylated on arginine 467 of the NS3 helicase domain (corresponding to position 1493 of the polyprotein) (31). Importantly, the addition of purified PP2Ac to the in vitro methylation assays inhibited the arginine methylation of the NS3 helicase domain (Fig. 3, lane 3).
Methylation of NS3 inhibits its helicase activity.
To assess the effect of NS3 methylation on its helicase activity, we used methylated and unmethylated purified NS3 helicase (amino acids 166 to 631 of NS3) in a helicase assay (22). The substrate for the reaction was a radioactively labeled double-stranded DNA. Heating the dsDNA at 95°C for 5 min or unwinding the dsDNA by enzymatic activity of NS3 helicase produced a single-stranded DNA (Fig. 4). Methylated NS3 helicase was obtained by incubating purified NS3 helicase with 1 μg, 2 μg, or 3 μg of purified PRMT1 in the presence of the methyl donor AdoMet (9 mM) for 2 h at 37°C. To exclude an unspecific inhibition of NS3 helicase by PRMT1, the sample with the unmethylated NS3 helicase was also incubated with purified PRMT1 (2 μg) without the methyl donor AdoMet. When tested in the helicase assay, the unmethylated NS3 helicase was more active than the methylated samples (Fig. 4A, lane 2 versus lanes 3 to 5). The difference in the levels of unwinding activity was evident at all time points during a time course experiment (Fig. 4B). We then measured the levels of ATP consumption by the methylated and unmethylated NS3 helicases during a 10-min helicase assay experiment. The more active unmethylated NS3 helicase consumed 65% of the ATP, whereas the less active methylated NS3 helicase samples consumed only 55% of the ATP. The difference was statistically significant (Fig. 4C).
FIG. 4.
Methylation of NS3 helicase inhibits its enzymatic activity. (A) Samples of 30 μg of purified NS3 helicase were incubated with 1 μg, 2 μg, or 3 μg of GST-PRMT1 and 9 nM AdoMet for 2 h at 37°C (lanes 3 to 5). The unmethylated NS3 helicase sample was also incubated with 2 μg of GST-PRMT1 for 2 h, but without AdoMet (lane 2). The samples were then used in a helicase assay with double-stranded DNA oligonucleotides as a substrate (lane 1). After 10 min, most of the dsDNA oligonucleotides were unwound to single-stranded DNA (ssDNA) by the unmethylated NS3 helicase (lane 2). In the cases of the methylated (Met-) NS3 helicase samples, about half the dsDNA substrates were still unwound after 10 min (lanes 3 to 5). (B) Time course experiment with unmethylated and methylated NS3 helicase. The enzymes were incubated with dsDNA substrate for the times indicated in the figure. In the samples with methylated NS3 helicase, a large proportion of the dsDNA substrate remains unwound (lanes 5 to 9). In the first and the last lanes, the substrate dsDNA and single-stranded DNA obtained by heating dsDNA, respectively, are shown. (C) ATP hydrolysis assay. Methylated or unmethylated NS3 helicase (100 nM) was incubated with 5 mM ATP. The amount of the unconsumed ATP was measured after 10 min. The unmethylated enzyme (lane 1) consumed 65%, and the methylated enzyme (lane 2) consumed about 55% of the input ATP.
We conclude that methylation of NS3 by PRMT1 inhibits its helicase activity. Theoretically, this conclusion could be tested more vigorously by doing in vitro helicase assays with a mutated NS3 that cannot be methylated on arginine 467 and therefore should not be inhibited by preincubation with PRMT1. However, the mutation of arginine 467 in the NS3 helicase domain leads to a complete loss of helicase activity (16). Because PP2Ac can inhibit PRMT1, as shown above, the upregulation of PP2Ac by HCV ultimately may result in enhanced helicase activity of NS3.
If PP2A enhances NS3 helicase activity, then inhibition of PP2A should have consequences for HCV replication. We therefore used HuH-7.5 cells harboring a subgenomic HCV replicon (3, 4) and the PP2A inhibitor OA to test this hypothesis. OA was added to the cells at a final concentration of 25 nM. This concentration was used because it significantly inhibited the phosphatase activity of PP2A with only minimal toxicity (data not shown). Treatment of replicon cells with OA indeed inhibited the replication of the HCV replicon (Fig. 5, lane 3). This inhibition was not as strong as that seen after treatment with IFN-α or with the HCV polymerase inhibitor 2′-C-methyladenosine (Fig. 5, lanes 4 and 5) but was still highly significant.
FIG. 5.

HuH-7.5 cells harboring a subgenomic HCV replicon were left untreated or were treated for 18 h with 5 μl/ml dimethyl sulfoxide (DMSO), 25 nM OA, 2 μM 2′-C-methyladenosine (2′C-Me-A), or 1,000 U/ml human IFN-α2a as indicated. Eight μg of total cellular RNA per lane was analyzed by 1.2% denaturing agarose gel electrophoresis, which was followed by Northern blotting using HCV- and GAPDH-specific probes. (A) Representative Northern blot. (B) The Northern blot shown in panel A and two additional blots from a representative experiment performed in triplicate were quantified by phosphorimaging. The mean intensity (± standard deviation) of each HCV-specific band relative to that of each GAPDH-specific band is shown.
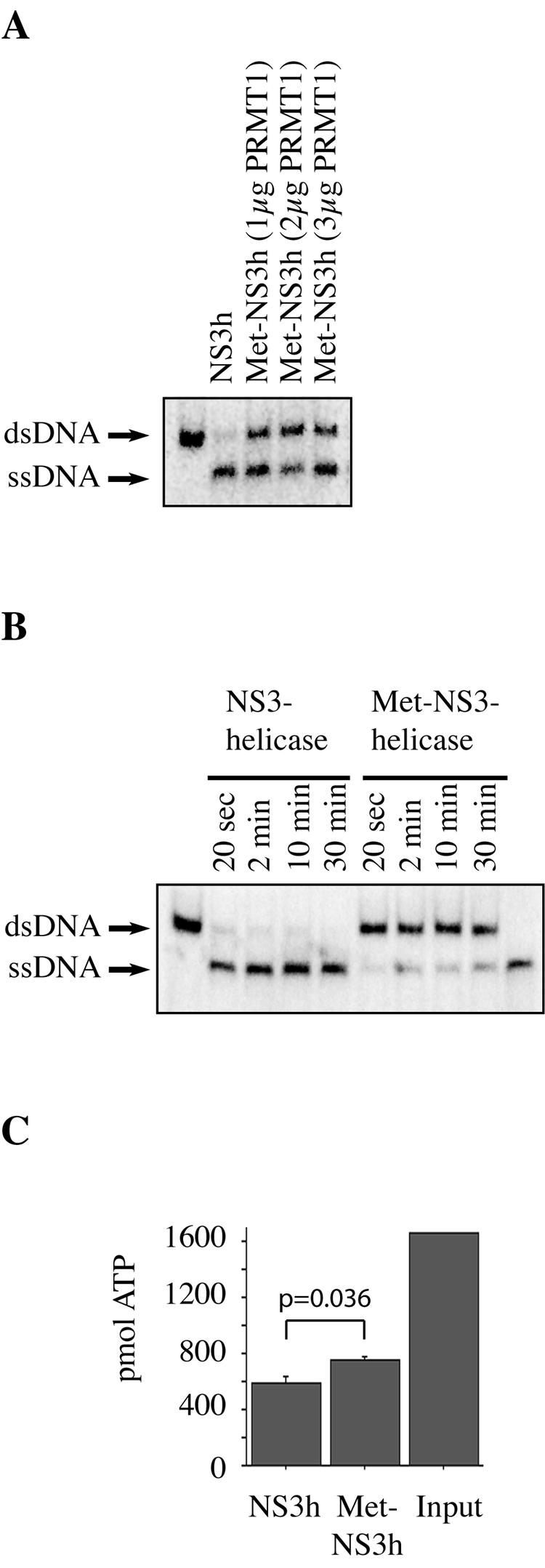
To confirm these results, PP2Ac expression was inhibited in HuH-7.5 cells by use of siRNA (Fig. 6). Again, a reduction of replicon RNA to 70% of the initial value found for HuH-7.5 cells was observed.
FIG. 6.
Suppression of PP2Ac expression by siRNA treatment of HuH-7.5 cells inhibits the replication of the HCV replicon. HuH-7.5 cells (two plates each) were left untransfected or were transfected with a nonsilencing or PP2Ac-silencing oligonucleotide, as indicated. (A) The expression of PP2Ac mRNA was quantified with real-time RT-PCR. (B) The expressions of PP2Ac and of PRMT1 proteins were analyzed by Western blotting. (C) The expression of HCV replicon RNA was quantified with real-time RT-PCR. In panels A and C, the expression levels of PP2Ac and HCV replicon found in one of the untransfected cells was set to 100% (reference sample), and the expression levels found in the other five samples are shown relative to that of the reference sample. Below panels A and C are the measured CT and ΔCT (dcT) values.
DISCUSSION
Given the limited coding capacity of a 9.6-kb RNA genome, HCV has an astonishing ability to evade the powerful immune system of the host and to establish a persistent infection over decades. In its interference with host defense mechanisms, HCV therefore probably targets a limited number of key enzymes of the cells in which it replicates. Here we provide evidence that PP2A is such a key enzyme. We have found that HCV infection or expression of HCV proteins induces the expression of PP2Ac in liver biopsy samples from patients with chronic hepatitis C (10), in liver cells of HCV transgenic mice (10), and in UHCV-57.3 and UHCV-32 cells. PP2A is a widely expressed serine/threonine phosphatase involved in a wide range of cellular processes (15, 27, 34). It can be expected that the induction of such an important phosphatase will have a number of effects on the host cell. Here we have concentrated our efforts on the study of the effect of PP2A on PRMT1, an important arginine methyltransferase. During previous studies, we found that HCV interferes with IFN-α-induced signaling by inhibiting the methylation of STAT1, an important signal transducer of IFN-α. Since STAT1 methylation is catalyzed by PRMT1, we hypothesized that PP2A directly or indirectly inhibits the enzymatic activity of PRMT1. We show here with purified proteins that PP2Ac binds directly to PRMT1 and, furthermore, that PP2Ac binding inhibits the methyltransferase activity of PRMT1 in vitro. Therefore, as a first and important consequence of PP2Ac induction by HCV, STAT1 methylation is reduced. Unmethylated STAT1 is bound by its inhibitor PIAS1, and IFN-α-induced induction of target genes important for cellular defense against HCV is impaired.
Surprisingly, the same mechanism of interference with the host cell is exploited twice by HCV. It has been shown recently that NS3, an HCV protein with protease and helicase activity, can be modified by arginine methylation through the cellular enzyme PRMT1 (31). The functional relevance of this modification was not known, but mutational analysis of arginine residues in the helicase domain provided strong evidence that these arginines are important for the enzymatic activity (16). For example, the mutation of arginine residue 467 of NS3 helicase (arginine 1493 in the polyprotein) to lysine disrupts the enzymatic activity completely (16). Here we show that methylation of NS3 reduces its helicase activity but does not completely abrogate it. Given the direct interaction of PP2Ac with PRMT1, it was not surprising that we did find an inhibition of NS3 methylation by PP2Ac. We conclude that, as a second important consequence of PP2Ac induction by HCV, NS3 helicase activity is increased.
The characterization of PP2A as a key target of HCV interference with the host cell identifies PP2A as a potential target of treatment strategies against the virus. In a proof-of-concept experiment, we treated replicon cells with OA, a reasonably specific inhibitor of PP2A (27), and found a significant inhibition of replication. The same degree of inhibition of the replicon was found after decreasing the expression of PP2Ac by siRNA. In our model (Fig. 7), inhibition of PP2A by OA results in an enhanced enzymatic activity of PRMT1. NS3 helicase is one of the substrates of PRMT1 and becomes methylated on critical arginine residues. This methylation reduces the helicase activity of NS3, leading to a decrease of viral (or replicon) replication. There are two caveats that we would like to mention. First, compared to IFN-α or the HCV polymerase inhibitor, OA is a relatively weak inhibitor of replication. After 18 h of OA treatment, a 30% reduction of the amount of replicon RNA in the cell was observed. This is clearly less than the 70% inhibition observed after treatment with IFN-α or the HCV polymerase inhibitor. However, in the long course of a natural HCV infection, even relatively small increases or decreases of viral replication activity can make a big difference on the virus-host interaction. Second, we would also like to stress that, given the numerous other functions of PP2A in cell homeostasis, we cannot exclude that inhibition of the HCV replicon by OA or by siRNA inhibition of PP2Ac expression involves mechanisms other than those described here. However, our in vitro data obtained with purified proteins show that these proteins can and do interact, and it should be worthwhile to further study interventions targeted to disrupt some of the HCV-induced changes of PP2Ac expression or of PRMT1 activity.
FIG. 7.

Enhanced HCV replication is a consequence of both the increase of NS3 helicase activity and the inhibition of IFN-α-induced Jak-STAT signaling.
Acknowledgments
We thank Elke Bieck for technical assistance, Charles M. Rice for replicon constructs and Huh-7.5 cells, Steven S. Carroll for 2′-C-methyladenosine, Pamela Silver for pGEX-HRMT1L2 (human PRMT1), and David Frick for NS3 helicase expression vectors.
The work was supported by grants 3200-063838 and 3200B0-103958 from the Swiss National Science Foundation (to M.H.H.), by the Oncosuisse grant OCS-01475-02-2004 (to M.H.H.), by grants 01 KI 9951 from the Bundesministerium für Bildung und Forschung and DFG no. 799/1-3 from the Deutsche Forschungsgemeinschaft (to D.M.), and by the Stiftung für Gastroenterologische Forschung Basel (to F.H.T.D.).
REFERENCES
- 1.Baharians, Z., and A. H. Schonthal. 1998. Autoregulation of protein phosphatase type 2A expression. J. Biol. Chem. 273:19019-19024. [DOI] [PubMed] [Google Scholar]
- 2.Biron, C. A., and G. C. Sen. 2001. Interferons and other cytokines, p. 321-351. In D. M. Knipe and P. M. Howley (ed.), Fundamental virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 3.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 4.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blindenbacher, A., F. H. Duong, L. Hunziker, S. T. Stutvoet, X. Wang, L. Terracciano, D. Moradpour, H. E. Blum, T. Alonzi, M. Tripodi, N. La Monica, and M. H. Heim. 2003. Expression of hepatitis c virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology 124:1465-1475. [DOI] [PubMed] [Google Scholar]
- 6.Carroll, S. S., J. E. Tomassini, M. Bosserman, K. Getty, M. W. Stahlhut, A. B. Eldrup, B. Bhat, D. Hall, A. L. Simcoe, R. LaFemina, C. A. Rutkowski, B. Wolanski, Z. Yang, G. Migliaccio, R. De Francesco, L. C. Kuo, M. MacCoss, and D. B. Olsen. 2003. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J. Biol. Chem. 278:11979-11984. [DOI] [PubMed] [Google Scholar]
- 7.Darnell, J. E., Jr., I. M. Kerr, and G. R. Stark. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415-1421. [DOI] [PubMed] [Google Scholar]
- 8.Darnell, J. E., Jr. 1997. STATs and gene regulation. Science 277:1630-1635. [DOI] [PubMed] [Google Scholar]
- 9.Di Bisceglie, A. M. 2000. Natural history of hepatitis C: its impact on clinical management. Hepatology 31:1014-1018. [DOI] [PubMed] [Google Scholar]
- 10.Duong, F. H., M. Filipowicz, M. Tripodi, N. La Monica, and M. H. Heim. 2004. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology 126:263-277. [DOI] [PubMed] [Google Scholar]
- 11.Durbin, J. E., R. Hackenmiller, M. C. Simon, and D. E. Levy. 1996. Targeted disruption of the mouse Stat1 results in compromised innate immunity to viral disease. Cell 84:443-450. [DOI] [PubMed] [Google Scholar]
- 12.Fort, P., L. Marty, M. Piechaczyk, S. el Sabrouty, C. Dani, P. Jeanteur, and J. M. Blanchard. 1985. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 13:1431-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frick, D. N., R. S. Rypma, A. M. Lam, and B. Gu. 2004. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J. Biol. Chem. 279:1269-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heim, M. H., D. Moradpour, and H. E. Blum. 1999. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J. Virol. 73:8469-8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janssens, V., and J. Goris. 2001. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353:417-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim, D. W., J. Kim, Y. Gwack, J. H. Han, and J. Choe. 1997. Mutational analysis of the hepatitis C virus RNA helicase. J. Virol. 71:9400-9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiyosawa, K., T. Sodeyama, E. Tanaka, Y. Gibo, K. Yoshizawa, Y. Nakano, S. Furuta, Y. Akahane, K. Nishioka, R. H. Purcell, and H. J. Alter. 1990. Interrelationship of blood transfusion, non-A, non-B hepatitis and hepatocellular carcinoma: analysis by detection of antibody to hepatitis C virus. Hepatology 12:671-675. [DOI] [PubMed] [Google Scholar]
- 18.Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570-574. [DOI] [PubMed] [Google Scholar]
- 19.Koonin, E. V. 1991. Similarities in RNA helicases. Nature 352:290. [DOI] [PubMed] [Google Scholar]
- 20.Krebs, D. L., and D. J. Hilton. 2001. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19:378-387. [DOI] [PubMed] [Google Scholar]
- 21.Kwong, A. D., J. L. Kim, and C. Lin. 2000. Structure and function of hepatitis C virus NS3 helicase. Curr. Top. Microbiol. Immunol. 242:171-196. [DOI] [PubMed] [Google Scholar]
- 22.Lam, A. M., R. S. Rypma, and D. N. Frick. 2004. Enhanced nucleic acid binding to ATP-bound hepatitis C virus NS3 helicase at low pH activates RNA unwinding. Nucleic Acids Res. 32:4060-4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauer, G. M., and B. D. Walker. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41-52. [DOI] [PubMed] [Google Scholar]
- 24.Linder, P., P. F. Lasko, M. Ashburner, P. Leroy, P. J. Nielsen, K. Nishi, J. Schnier, and P. P. Slonimski. 1989. Birth of the D-E-A-D box. Nature 337:121-122. [DOI] [PubMed] [Google Scholar]
- 25.Liu, B., J. Liao, X. Rao, S. A. Kushner, C. D. Chung, D. D. Chang, and K. Shuai. 1998. Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA 95:10626-10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meraz, M. A., J. M. White, K. C. F. Sheehan, E. A. Bach, S. J. Rodig, A. S. Dighe, D. H. Kaplan, J. K. Riley, A. C. Greenlund, D. Campbell, K. Carvermoore, R. N. Dubois, R. Clark, M. Aguet, and R. D. Schreiber. 1996. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the Jak-Stat signaling pathway. Cell 84:431-442. [DOI] [PubMed] [Google Scholar]
- 27.Millward, T. A., S. Zolnierowicz, and B. A. Hemmings. 1999. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 24:186-191. [DOI] [PubMed] [Google Scholar]
- 28.Moradpour, D., P. Kary, C. M. Rice, and H. E. Blum. 1998. Continuous human cell lines inducibly expressing hepatitis C virus structural and nonstructural proteins. Hepatology 28:192-201. [DOI] [PubMed] [Google Scholar]
- 29.Moradpour, D., B. Wolk, A. Cerny, M. H. Heim, and H. E. Blum. 2001. Hepatitis C: a concise review. Minerva Med. 92:329-339. [PubMed] [Google Scholar]
- 30.Mowen, K. A., J. Tang, W. Zhu, B. T. Schurter, K. Shuai, H. R. Herschman, and M. David. 2001. Arginine methylation of stat1 modulates ifnalpha/beta-induced transcription. Cell 104:731-741. [DOI] [PubMed] [Google Scholar]
- 31.Rho, J., S. Choi, Y. R. Seong, J. Choi, and D. S. Im. 2001. The arginine-1493 residue in QRRGRTGR1493G motif IV of the hepatitis C virus NS3 helicase domain is essential for NS3 protein methylation by the protein arginine methyltransferase 1. J. Virol. 75:8031-8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt-Mende, J., E. Bieck, T. Hugle, F. Penin, C. M. Rice, H. E. Blum, and D. Moradpour. 2001. Determinants for membrane association of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 276:44052-44063. [DOI] [PubMed] [Google Scholar]
- 33.Shuai, K. 2000. Modulation of STAT signaling by STAT-interacting proteins. Oncogene 19:2638-2644. [DOI] [PubMed] [Google Scholar]
- 34.Sontag, E. 2001. Protein phosphatase 2A: the Trojan horse of cellular signaling. Cell Signal 13:7-16. [DOI] [PubMed] [Google Scholar]
- 35.ten Hoeve, J., M. de Jesus Ibarra-Sanchez, Y. Fu, W. Zhu, M. Tremblay, M. David, and K. Shuai. 2002. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 22:5662-5668. [DOI] [PMC free article] [PubMed] [Google Scholar]