

Abstract
Kaposi's sarcoma is an angioproliferative disseminated tumor of endothelial cells linked to infection with Kaposi's sarcoma-associated herpesvirus (KSHV). AP-1 transcription factors are involved in diverse biological processes, including infection and replication of viruses, cell growth, oncogenesis, angiogenesis, and invasion of cancer cells. Here we show that KSHV activates AP-1 during primary infection. The activation of AP-1 at the early stage of KSHV infection is mainly mediated by virus entry events. Concurrently, KSHV infection strongly activates MEK, JNK, and to a lesser extent, p38 mitogen-activated protein kinase (MAPK) pathways. Specific inhibitors or dominant negative constructs of MEK and JNK completely abolish AP-1 activation by KSHV, while those of p38 reduce it by half. Furthermore, individual MAPK pathways differentially regulate KSHV activation of AP-1 components. KSHV activation of AP-1 leads to the transcriptional induction of interleukin 6 (IL-6), which is inhibited by inhibitors or dominant negative constructs of MAPK pathways. Together, these results demonstrate that KSHV induces AP-1 and IL-6 during primary infection by modulating multiple MAPK pathways. Because of the diverse roles of IL-6, AP-1, and MAPK pathways in viral infection and tumor induction and promotion, these results have important implications in the pathogenesis of KSHV-induced malignancies.
Viral infection causes a deregulation of host cellular pathways, some of which reflect cellular responses to the infection while others are the results of viral manipulation of cellular environments (39, 49, 60). A common strategy that a virus uses to facilitate its infection and replication is to exploit the altered cellular pathways. For example, the modulation of mitogen-activated protein kinase (MAPK) pathways is essential for infection and replication of hepatitis B virus, Epstein-Barr virus (EBV), and vaccinia virus (15, 26, 70), while modulation of the NF-κB pathway facilitates infection and replication of EBV, herpes simplex virus type 1, and influenza virus (25, 46, 59). The deregulated cellular pathways could also contribute to the pathogenesis of diseases induced by viral infections (49).
Activator protein 1 (AP-1) complexes are commonly activated during viral infection (39, 49, 60). AP-1 complexes are ubiquitous heterodimeric transcriptional factors of Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, FosB, Fra-1, and Fra-2) subfamilies. Through binding to either a 12-O-tetradecanoylphorbol-13-acetate (TPA) response element or a cyclic AMP response element, AP-1 regulates a wide variety of cellular processes, such as cell growth, differentiation, death, and survival (56). Generally, AP-1 complexes function as positive regulators of cell proliferation by regulating the expression of essential cell cycle proteins such as cyclin D1, p53, p21, p19, and p16, yet differential effects are also present among individual members (56). The inhibition of jun and fos gene expression by antisense RNAs reduced the proliferation rate of mouse fibroblasts, while the introduction of specific antibodies against Fos and Jun proteins into fibroblasts resulted in similar effects (57). A double knockout of c-fos and fosB genes reduced mouse size (10). AP-1 is also important for cellular transformation. Both c-jun and c-fos themselves are cellular retroviral oncogenes (4). Some AP-1 members such as the Jun proteins can assist activated Ras in cellular transformation (61). The overexpression of c-fos caused osteosarcomas and chondrosarcomas in transgenic mice (51). In contrast, the expression of a dominant negative (DN) c-jun transgene in mice inhibited TPA-induced tumor promotion (69). Besides its role in cell growth, cellular transformation, and induction of tumor formation, AP-1 is also involved in other aspects of tumor progression, such as invasion and angiogenesis, by regulating the expression of matrix metalloproteinases (MMPs) and angiogenic factors (14, 64).
Kaposi's sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus associated with Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD) (20). KS is a vascular spindle tumor of proliferating endothelial cells commonly found in patients with AIDS (21). The early stage of KS is mostly seen on the skin of the lower extremities, but the advanced stage of KS is highly disseminated and often involved with visceral organs. A unique feature of KS tumors is that they are highly angiogenic and regularly contain infiltrated inflammatory cells (21). Thus, KS tumors frequently secrete angiogenic and inflammatory cytokines. Among these cytokines, interleukin 6 (IL-6) is overexpressed in KS tumors as well as in PEL and MCD, contributing to the pathogenesis of these malignancies (5, 37, 41). IL-6 is an autocrine growth factor for KS cells (19, 31, 41, 66) and a growth and survival factor for PEL cells (5). IL-6 is also important for the development of MCD in mice (54). While a recent microarray analysis has shown that KSHV infection induces the expression of IL-6, the mechanism of induction remains unclear (12, 45).
Considering the known biologic functions of AP-1 and the features of KS tumors, one can envisage the likely roles of AP-1 in various facets of KS pathogenesis, including malignant cellular proliferation, angiogenesis, induction of inflammatory cytokines, and dissemination of tumor cells. The expression of IL-6 is also regulated by AP-1 (16). Furthermore, the consensus AP-1-binding element is present in the promoter regions of several KSHV lytic replication genes, including the RTA (Orf50), RAP (K8), and MTA (Orf57) genes (11, 62), as well as in lytic origins of DNA replication (oriLyt) (6). Thus, KSHV lytic replication and the expression of KSHV genes are likely dependent on the activation of AP-1.
AP-1 activity can be regulated at multiple levels, including transcription, posttranslational modifications, protein turnover, and interactions with various cellular proteins. MAPK pathways, which have important roles in a broad spectrum of cellular functions, including cell proliferation, differentiation, survival, death, and gene expression, mediate AP-1 activation (35, 56, 65). MAPK pathways relay extracellular stimuli into the nucleus through a series of phosphorylation events that activate the upstream trimeric G proteins and downstream MAPK kinase kinases (MAPKKKs), MAPK kinases (MAPKKs), and MAPKs (32). There are three groups of MAPKs in mammalian cells: the extracellular signal-related kinases (ERK1 and -2), the c-Jun NH2-terminal kinases (JNK1, -2, and -3), and the p38 kinases (p38α, p38β, p38γ, and p38δ) (13). Members of the ERK pathway, after translocation into the nucleus, directly phosphorylate c-Fos and ternary complex factors, which further bind and activate the fos promoter (30, 42). The JNK members phosphorylate c-Jun to activate AP-1 (35). The p38 pathway activates AP-1 by phosphorylating the transcriptional factors ATF2, MEF2C, and ternary complex factors (28).
KSHV entry into cells relies on interactions with cellular receptors. Integrin α3β1 was initially identified as a KSHV receptor; however, recent studies indicated that other cellular membrane proteins could also be involved in KSHV entry (55). Interactions of glycoprotein B with integrin α3β1 result in acute activation of the ERK1/2 pathway (44). Therefore, it is reasonable to postulate that KSHV regulates AP-1 through activation of the ERK1/2 pathway, and possibly other MAPK pathways, during the early stage of infection. Two KSHV latent genes, encoding LANA (Orf73) and vFLIP (Orf-K13), have been shown to activate AP-1 as well as induce IL-6 expression (1, 2). Thus, LANA and vFLIP are likely to modulate AP-1 during latent KSHV infection. vFLIP and several other KSHV genes, including the vGPCR (Orf74), vPK (Orf36), and LAMP (K15) genes, also activate MAPK pathways (2, 7, 9, 27). Both the vGPCR and vPK genes are lytic cycle genes and therefore can contribute to the activation of AP-1 during KSHV lytic replication. While the LAMP gene has been considered a latent gene, it is also induced by TPA (20). Thus, its gene class remains undefined. Regardless of their gene classes, given that these genes are capable of activating MAPK pathways and AP-1, they are likely to induce the expression of IL-6. Furthermore, a transcriptional activator, RTA (Orf50), itself has also been shown to activate the expression of IL-6 (17). Using an efficient KSHV infection model of primary human umbilical vein endothelial cells (HUVEC) (24), we have found that KSHV activates AP-1 during the early stage of infection, which is mainly mediated by the virus entry events rather than the expression of KSHV genes. This is the first time that AP-1 activation by KSHV has been observed in the context of viral infection. Furthermore, we have found that multiple MAPK pathways mediate the activation of AP-1 by KSHV, which results in a rapid increase in IL-6 expression in KSHV-infected cells and the secretion of IL-6 into the extracellular environment. These results underscore some of the important molecular events of virus-cell interactions during the early stage of KSHV infection.
MATERIALS AND METHODS
Plasmids.
The AP-1 reporter pAP-1-luc was purchased from Stratagene (La Jolla, CA). pFC-MEKK, expressing constitutively active MEKK, was also purchased from Stratagene. pGL-3-IL-6, an IL-6 promoter reporter, was obtained by PCR cloning the 1,175-bp IL-6 promoter sequence upstream of the transcriptional start site into the SacI/HindIII sites of the pGL-3 vector (Promega, Madison, WI). The following plasmids were described before: pcDNA3-p38/AF is a DN construct of p38 (29), HA-JNK[APF] is a DN construct of JNK (18), pCEP4L-HA-ERK1K71R is a DN construct of ERK1 (23), A-Fos is a DN construct of c-Fos (47), and pcDNA3 Flag-c-Fos expresses an active form of c-Fos (43).
Cell culture.
Human embryonic kidney 293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 μg/ml gentamicin, and 2 mM l-glutamine. Primary HUVEC were purchased commercially and cultured in EBM2 endothelial cell growth medium (Clonetics, Walkersville, MD).
Virus preparation and infection.
Concentrated virus was prepared as previously described (24, 71). Fresh virus preparations with titers of about 2 × 106 green fluorescent protein (GFP)-expressing cells/ml were used in the experiments. HUVEC or 293 cells were infected with KSHV as previously described (24). For all experiments, we infected the cells at a multiplicity of infection (MOI) of 2, i.e., 2 × 106 GFP-expressing cells for every 106 cells, which resulted in a 70 to 80% infection rate based on GFP expression at 2 days postinfection. For UV light treatment, virus preparations were exposed to a UV source for 5 min, which reduced the virus infectivity from 70 to 80% to <1%. To inhibit a MAPK pathway, one of the following specific inhibitors, all of which were purchased from Calbiochem (Oakland, CA), was added to the culture 1 h prior to infection: U0126 (10 μM), an inhibitor of MEKs; SB203580 (50 μM), an inhibitor of p38; and JNK inhibitor II (50 μM), a JNK inhibitor.
Western blot analysis.
Protein preparations from HUVEC or 293 cells infected with KSHV were separated in sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes as previously described (24). The membranes were incubated first with antibodies specific for total and phosphorylated forms of JNK, ERK1/2, and p38 (Santa Cruz, Santa Cruz, CA) and then with a goat anti-rabbit-horseradish peroxidase (HRP) conjugate (Sigma, St. Louis, MO). A mouse antibody to α-tubulin (Sigma) was used to monitor sample loading. Specific signals were revealed with chemiluminescence substrates and recorded on films.
EMSA.
Nuclear extracts were prepared from mock- or KSHV-infected HUVEC or 293 cells as previously described (63). Annealed double-stranded consensus AP-1 oligonucleotides (5′GGGTTATGAGTCAGTTGC3′) were labeled with [γ-32P]ATP. Oligonucleotides for the consensus AP-1 binding site in the IL-6 promoter (5′CCAAGTGCTGAGTCACTAATA3′) were also used for electrophoretic mobility shift assays (EMSAs). For gel shift assays, 4 μg of nuclear extract was incubated for 20 min at room temperature with 5 × 105 cpm of the labeled probe in 20 μl of binding buffer containing 10 mM Tris-HCl at pH 7.6, 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 5% glycerol, 1 μg/μl bovine serum albumin, and 2 μg of poly(dI-dC). For antibody supershift assays, 2 μg of polyclonal antibodies to c-Fos or c-Jun (Santa Cruz) was added, and the reactions were continued for 15 min. Samples were separated by 6% nondenaturing polyacrylamide gel electrophoresis. Competition assays were carried out in the same manner, except that the above reaction mixture was preincubated with excess cold probe for 10 min at 4°C before the addition of the labeled probe.
AP-1 ELISA.
An enzyme-linked immunosorbent assay (ELISA)-based TransAM AP-1 kit was used to quantify activated AP-1 components (Active Motif, Carlsbad, CA). Briefly, nuclear extracts were prepared in 50 μl of lysis buffer with the cocktail of protease inhibitors provided in the kit. Nuclear extracts diluted in the binding buffer were added at 5 μg per well to 96-well plates previously coated with a TPA response element oligonucleotide (5′-TGAGTCA-3′) and incubated for 1 h. For specificity control, an excess amount (20 pmol) of mutant or wild-type probe was added to the reaction in a competition assay. The plates were washed three times with washing buffer and incubated with rabbit antibodies to AP-1 members for 1 h. The plates were again washed and then incubated with a goat anti-rabbit-HRP conjugate for 1 h. After repeating the washes, the plates were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) HRP substrate, and the results were recorded with an ELISA plate reader at 450 nm, using 655 nm as a reference wavelength.
IL-6 ELISA.
Secreted IL-6 in the culture medium was assayed with a commercial IL-6 ELISA kit (RayBiotech, Norcross, GA). Supernatants were incubated at room temperature for 2.5 h in 96-well plates previously coated with antibodies to IL-6. The plates were washed and incubated with biotinylated antibodies to IL-6 for 1 h at room temperature. The plates were again washed and incubated with a streptavidin-HRP conjugate solution for 45 min at room temperature. After extensive washes, the plates were developed with TMB substrate.
Reporter assay.
Reporter assays were carried out as previously described (63). HUVEC or 293 cells in six-well plates were transfected with the pAP-1-luc or pGL-3-IL-6 reporter plasmid together with other plasmids. For HUVEC, transfection was carried out using the CytoPure-huv transfection reagent (Qbiogene, Irvine, CA). For 293 cells, transfection was carried out using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). To monitor the transfection efficiency, we performed cotransfection with plasmid pEGFP-C1 expressing GFP (Clontech, Palo Alto, CA). We achieved about 30% and 90% transfection efficiencies with HUVEC and 293 cells, respectively, based on the expression of GFP at 2 days posttransfection. To further calibrate the transfection efficiency, samples were normalized by cotransfection with the reporter plasmid pSV-β-galactosidase (Promega). At 36 h posttransfection, cells were collected in 200 μl of lysis buffer (Promega). Aliquots of 20 μl were tested in a luciferase assay system (Promega), and the results were recorded with a Veritus microplate luminometer (Turner Biosystems, Sunnyvale, CA). All reporter assays were carried out three to five times in triplicate. Results calculated as averages with standard deviations from one representative experiment are presented.
Reverse transcription-real-time quantitative PCR (RT-qPCR).
Total RNA (5 μg) was reverse transcribed in 40 μl to obtain first-strand cDNAs, using the Superscript III first-strand synthesis system (Invitrogen). A control without reverse transcriptase was conducted in parallel. The primers for IL-6 were 5′-TGGTCTTTTGGAGTTTGAGGTA-3′ (IL-6-401F) and 5′-AGGTTTCTGACCAGAAGAAGGA-3′ (IL-6-746R), which amplify a product of 325 bp. The primers for glyceraldehye-3-phosphate dehydrogenase (GAPDH) were 5′-ACAGTCAGCCGCATCTTCTT-3′ (GAPDH-F) and 5′-ACGACCAAATCCGTTGACTC-3′ (GAPDH-R), which amplify a product of 94 bp. Real-time PCR was carried out in a DNA Engine Opticon 2 continuous fluorescence detector (Bio-Rad) as described previously (67). Each sample was examined in triplicate.
RESULTS
KSHV activates AP-1 reporter during primary infection.
We used an AP-1 reporter, pAP-1-luc, containing tandem copies of AP-1 binding sites [(TGACTAA)7] upstream of a luciferase reporter gene, to examine KSHV activation of AP-1 during primary infection. Since AP-1 complexes have strong transactivation activity, if activated, they will bind to the consensus sites and drive the transcription of the downstream reporter gene. We assayed the luciferase activities in 293 cells that were infected with KSHV for 6 h after transfection with pAP-1-luc plasmid DNA for 30 h. Compared with mock-infected cells, KSHV-infected cells have a 7.5-fold higher luciferase activity (Fig. 1A, panel a). As expected, treatment with TPA increased the luciferase activity (Fig. 1A, panel a), since TPA is known to activate AP-1 (3). Cotransfection with pFC-MEKK expressing constitutively active MEKK increased the luciferase activity 6.2-fold compared with that in cells cotransfected with control plasmid DNA alone (Fig. 1A, panel b), since active MEKK can activate the AP-1 complexes through the MEK/ERK MAPK pathway (56). We then determined whether KSHV activates AP-1 in a more biologically relevant primary cell type. As shown in Fig. 1B, KSHV infection increased luciferase activity 6.2-fold in HUVEC. These results indicated that KSHV infection enhanced the transactivation activity of AP-1 complexes. We next determined the time kinetics of KSHV-induced AP-1 activation. pAP-1-luc-transfected cells were harvested at different time points after KSHV infection and assayed for luciferase activity. The luciferase activities in both HUVEC and 293 cells started to increase as early as 2 h postinfection (hpi), peaked at 6 hpi, and then decreased (Fig. 1C and D). These results indicated that KSHV activation of AP-1 occurs during the early stage of virus infection.
FIG. 1.

KSHV activates the AP-1 reporter during primary infection. (A and B) 293 cells (A) or HUVEC (B) transfected with the pAP-1-luc reporter plasmid for 30 h were either mock or KSHV infected for 6 h and then harvested for luciferase assays. Cells treated with TPA at 0.1 mM for 0.5 h (a) or cotransfected with pMEK1 (b) were used as positive controls. (C and D) Time kinetics of KSHV activation of the AP-1 reporter in 293 cells (C) and HUVEC (D). Cells were transfected with the pAP-1-luc reporter and assayed for luciferase activity at 36 h posttransfection. Prior to harvest, the cells were infected with KSHV for 0, 2, 4, 6, 10, and 16 h. KSHV infection experiments were carried out at an MOI of 2. The experiments were independently carried out three times, with three repeats each. Results presented are averages with standard deviations from one representative experiment.
KSHV induces AP-1 binding to the consensus element during primary infection.
To rule out nonspecific binding of the AP-1 consensus cis element by other proteins that could also result in the increase in reporter activity, we carried out EMSAs to directly detect the binding of AP-1 complexes to the consensus element. We incubated 32P-labeled consensus oligonucleotides with nuclear extracts prepared from 293 cells infected with KSHV for 6 h (Fig. 2A). Compared with mock-infected cells, an increase in the shifted band corresponding to the AP-1-DNA complexes was observed in KSHV-infected cells. The addition of unlabeled cold probes significantly reduced the intensity of the AP-1 band, confirming the specificity of the protein-DNA interaction (Fig. 2A). Similar results were also observed with KSHV-infected HUVEC (Fig. 2B). To further verify that the observed band shifts were caused by the AP-1 complexes, specific antibodies to c-Fos and c-Jun were added to the reactions. Both antibodies caused supershifts of the AP-1 bands in 293 cells and HUVEC (Fig. 2A and B), again confirming the specific binding of AP-1 complexes to the probe. These results also indicated that both c-Jun and c-Fos were parts of the KSHV-activated AP-1 complexes.
FIG. 2.

KSHV infection induces binding of AP-1 complexes to consensus elements. (A and B) Nuclear extracts from 293 cells (A) or HUVEC (B), either mock infected (lanes 2) or infected with KSHV (lanes 3 to 9) for 6 h, were subjected to EMSA using a 32P-labeled AP-1 consensus oligonucleotide probe. The labeled probe alone is shown in lanes 1. For the competition assay, a 100× excess amount of unlabeled probe was added to the reaction mixture (lanes 4). The addition of 2 or 1 μg of antibodies to c-Fos (lanes 5 and 8) or c-Jun (lanes 6 and 9), respectively, supershifted the AP-1-probe bands, while the addition of 2 μg control immunoglobulin G did not supershift or change the intensity of the AP-1-probe band (lanes 7). (C and D) Nuclear extracts from 293 cells (C) or HUVEC (D) infected with KSHV for 0, 2, 4, 6, 10, and 16 h were examined by EMSA to determine the time kinetics of KSHV-induced binding of AP-1 complexes. All KSHV infection experiments were carried out at an MOI of 2. The experiments were independently carried out five (A and B) or two (C and D) times. The results presented are from one representative experiment.
Next, we examined the time kinetics of KSHV-induced AP-1 binding to the consensus element in both 293 cells (Fig. 2C) and HUVEC (Fig. 2D). In concordance with the reporter assay results, the induction of AP-1 binding to the consensus element during KSHV infection occurred as early as 2 hpi, peaked at 6 hpi, and decreased at the subsequent time points tested. These results confirmed the early activation of AP-1 by KSHV during infection.
KSHV differentially activates AP-1 family members during primary infection.
AP-1 components are differentially regulated in response to stimuli, resulting in diverse biological effects on the cells. We determined which of the AP-1 members were activated during KSHV primary infection by an ELISA in which an AP-1 consensus oligonucleotide was immobilized on 96-well plates to capture different AP-1 components. The addition of specific antibodies to individual phosphorylated forms of AP-1 members quantitatively detects their presence. As shown in Fig. 3, compared with mock-infected cells, KSHV-infected cells had increased activated forms of members of both Fos and Jun families, including c-Fos, FosB, c-Jun, JunB, and JunD, at 2.8-, 3.4-, 2.3-, 1.6-, and 1.5-fold, respectively, while both Fra-1 and Fra-2 remained unchanged. Within the Fos family, both c-Fos and FosB are known to have positive effects on cell proliferation (56). In the Jun family, c-Jun is a strong promoting factor for cell proliferation (33), whereas JunB generally behaves as a negative regulator (50). The effect of JunD on cellular proliferation remains promiscuous, and it is less potent than c-Jun when it exerts a positive effect (56). The observed stronger activation of c-Fos, FosB, and c-Jun than that of other AP-1 components points to a positive effect on cell proliferation exerted by KSHV during primary infection.
FIG. 3.

KSHV differential activation of AP-1 family members during primary infection. Nuclear extracts from 293 cells either mock infected (empty bars) or infected with KSHV at an MOI of 2 (solid bars) for 6 h were incubated with AP-1 consensus oligonucleotides immobilized in a 96-well plate. Active forms of AP-1 components were detected with the respective specific antibodies. The fold activation of individual AP-1 components was calculated, using the mock-infected group as the baseline (onefold). The experiments were independently carried out two times, with three repeats each. The results presented are averages with standard deviations from one representative experiment.
KSHV activation of AP-1 is mainly mediated by virus entry events.
The observation of KSHV activation of AP-1 at times as early as 2 hpi indicates that virus entry is involved in this process. Nevertheless, a small number of KSHV genes are expressed at low levels at this early stage of infection (68), thus possibly contributing to AP-1 activation. To distinguish the role of virus entry from viral gene expression in the early activation of AP-1 by KSHV, we carried out AP-1 reporter assays with cells infected by virions or UV-irradiated virions. A large proportion of the KSHV-activated AP-1 reporter activity was retained after UV treatment of the virions (74%) (Fig. 4A), as confirmed by EMSA (Fig. 4B). These results indicate that KSHV activation of AP-1 during the early stage of infection is mainly mediated by virus entry events.
FIG. 4.
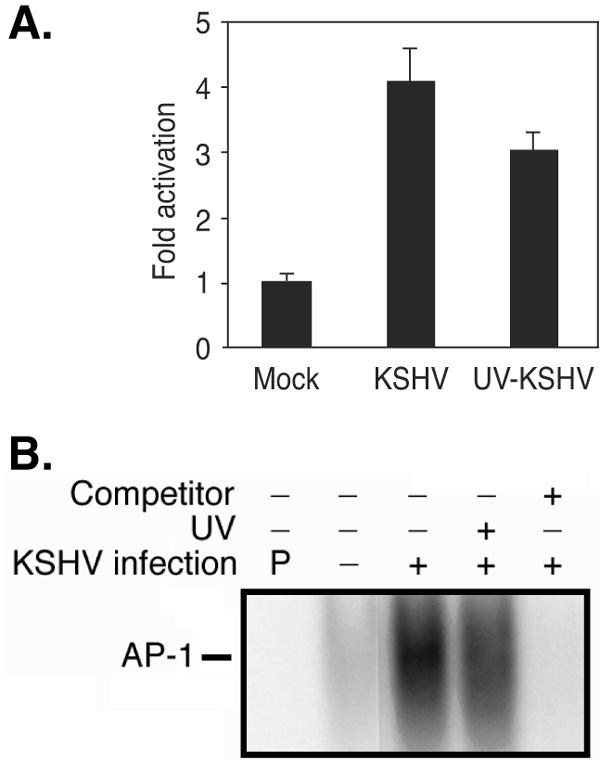
KSHV activation of AP-1 is mediated by virus entry events. (A) UV irradiation only minimally reduced KSHV activation of AP-1, as measured by an AP-1 reporter assay. 293 cells were transfected with the pAP-1-luc reporter and assayed for luciferase activity at 36 h posttransfection. Six hours prior to harvest, the cells were mock infected or infected with KSHV or UV-irradiated KSHV. The majority of KSHV-activated reporter activity (74%) was retained after exposing the virions to UV irradiation. The experiments were independently carried out three times, with three repeats each. The results presented are averages with standard deviations from one representative experiment. (B) UV irradiation of KSHV virions only minimally reduced KSHV activation of AP-1, as measured by EMSA. Cells were mock infected or infected with KSHV or UV-irradiated KSHV for 6 h. The experiments were carried out two times. The results presented are from one representative experiment. All KSHV infection experiments were carried out at an MOI of 2. UV irradiation reduced the virus infectivity from 70 to 80% to <1%.
KSHV activates MAPK pathways during primary infection.
A previous report has shown that KSHV activates the ERK pathway immediately following infection (44). We wished to determine whether KSHV also activates the JNK and p38 pathways during primary infection. HUVEC were infected with KSHV, collected at different hpi, and analyzed for phosphorylated forms of JNK, ERK1/2, and p38. Concurring with the previous report, we observed an activation of ERK1/2 from 0.25 to 1 hpi (Fig. 5). In addition, we observed an activation of JNK from 0.25 to 1 hpi, and to a lesser extent, of p38 at 0.5 hpi. The detected activation of p38 was modest, at about twofold. The phosphorylated forms of JNK and ERK1/2 dropped between 1 and 2 hpi and reached a normal level afterward (Fig. 5). We loaded all the samples in similar amounts, as shown by their comparable intensities of α-tubulin staining. The total amounts of JNK, ERK1/2, and p38 remained unchanged after KSHV infection (Fig. 5), indicating that the activation of these proteins was likely done through posttranslational modifications.
FIG. 5.

KSHV activates multiple MAPK pathways during primary infection. Proteins prepared from HUVEC that were mock infected or infected with KSHV at an MOI of 2 for 0.25, 0.5, 1, 2, 4, 8, or 16 h were resolved in a sodium dodecyl sulfate-polyacrylamide gel and transferred onto a nitrocellulose membrane. Phosphorylated or total ERK1/2, JNK, and p38 proteins were detected with the respective specific antibodies. Proteins from HUVEC treated with TPA for 0.5 h were used as positive controls for phosphorylated ERK1/2. Antibodies to α-tubulin were used to normalize sample loading. The experiments were independently carried out two times. The results presented are from one representative experiment.
AP-1 activation during primary KSHV infection is mediated by multiple MAPK pathways.
The above data showed that KSHV activation of MAPK pathways preceded that of AP-1. To determine whether MAPK pathways mediated KSHV activation of AP-1, we performed the AP-1 reporter assay with 293 cells, with or without the presence of inhibitors of MAPK pathways. As shown in Fig. 6A, inhibitors of MEK and JNK completely abolished KSHV induction of AP-1 reporter activity, while the inhibitor of p38 reduced it by half. In EMSAs, the addition of all three MAPK inhibitors to cells during KSHV primary infection reduced the intensities of the KSHV-induced AP-1-DNA bands in both 293 cells and HUVEC. Stronger effects were observed with inhibitors of MEK and JNK, and a less robust effect was seen with the inhibitor of p38 (Fig. 6B). Western blotting confirmed that the specific inhibitors blocked the activation of their respective MAPK pathways in HUVEC (Fig. 6C). We further applied biological methods to inhibit the MAPK pathways and examined the effects on KSHV activation of AP-1. We cotransfected 293 cells with DN constructs of the JNK, ERK, and p38 pathways in the AP-1 reporter assay. The results were similar to those obtained with chemical inhibitors (Fig. 6D). DN constructs of MEK and JNK completely abolished KSHV induction of the AP-1 reporter activity, while that of p38 reduced it by half. These results clearly indicated that multiple MAPK pathways mediated KSHV activation of AP-1.
FIG. 6.

Inhibition of MAPK pathways prevents KSHV activation of AP-1 during primary infection. (A) Inhibitors of MAPK pathways inhibited the activation of the AP-1 reporter by KSHV infection. Cells (293 cells) transfected with pAP-1-luc reporter plasmid DNA for 30 h were either mock infected or infected with KSHV for 6 h with or without the presence of specific inhibitors of ERK1/2, p38, and JNK, lysed, and assayed for luciferase activity. (B) Inhibitors of MAPK pathways inhibited KSHV-induced binding of AP-1 complexes to consensus elements. Nuclear extracts prepared from 293 cells (upper panel) and HUVEC (lower panel) either mock infected or infected with KSHV for 6 h, with or without the presence of specific inhibitors of JNK, ERK1/2, and p38, were subjected to EMSA using a 32P-labeled AP-1 consensus oligonucleotide probe. P, free probe. A 100-fold excess of cold probe was used for the competition assay. (C) Western blotting was carried out with specific antibodies to detect the phosphorylated forms of ERK1/2 (first panel), JNK (second panel), and p38 (third panel) in HUVEC that were mock infected, KSHV infected, or KSHV infected in the presence of the respective specific MAPK inhibitors. An anti-α-tubulin antibody was used to normalize sample loading (fourth panel). (D) DN constructs of MAPK pathways inhibited activation of the AP-1 reporter by KSHV infection. Cells (293 cells) transfected with pAP-1-luc reporter plasmid DNA alone or together with DN constructs of MEK, p38, and JNK for 30 h were either mock infected or infected with KSHV for 6 h, lysed, and assayed for luciferase activity. (E) Inhibitors of MAPK pathways differentially inhibited the activation of AP-1 components by KSHV infection. Cells (293 cells) that were mock infected or infected with KSHV for 6 h, with or without specific inhibitors of ERK1/2, p38, and JNK, were lysed and assayed for the active forms of AP-1 components by ELISA, as described in the legend to Fig. 3. All KSHV infection experiments were carried out at an MOI of 2. For the reporter assay (A and D), the experiments were independently carried out three times, with three repeats each. The results presented are averages with standard deviations from one representative experiment. For EMSA (B) and Western blotting (C), the experiments were repeated two times. The results presented are from one representative experiment. For ELISA (E), the experiments were independently carried out two times, with three repeats each. The results presented are averages with standard deviations from one representative experiment.
The above results have shown that inhibition of just one of the MAPK pathways leads to a reduction in KSHV activation of AP-1, indicating that the activation of all three MAPK pathways is necessary for the maximum KSHV activation of AP-1 during primary infection. Importantly, inhibition of either the ERK or JNK pathway by a chemical inhibitor or DN construct in a reporter assay or EMSA resulted in more reduction of KSHV activation of the AP-1 pathway, while inhibition of the p38 pathway had fewer effects. These data are consistent with the strong activation of ERK1/2 and JNK and the less robust activation of p38 detected during primary KSHV infection (Fig. 5).
Multiple MAPK pathways differentially activate AP-1 components during primary KSHV infection.
We further investigated which AP-1 component(s) was prevented from activation during primary KSHV infection by MAPK inhibitors. In concordance with previous results (Fig. 3), neither KSHV infection nor inhibitors of MAPK pathways had any effects on the activation of Fra-1 and Fra-2 (Fig. 6E). In contrast, all three inhibitors of MAPK pathways displayed differential inhibitory effects on the activation of other AP-1 components (Fig. 6E). Inhibitors of both the ERK and JNK pathways totally abolished the activation of Jun members c-Jun, JunB, and JunD and prevented 51% and 46% of c-Fos activation and 80% and 16% of FosB activation, respectively. These results indicated that members of the AP-1 complex had differential roles in KSHV activation of AP-1. The Jun members of the AP-1 complex, particularly c-Jun, which was activated the most, were likely to have a major role in KSHV activation of AP-1 and to account for the total abolishment of AP-1 activity by inhibitors of the ERK and JNK pathways observed in the reporter assay (Fig. 6A). The inhibitor of p38 had no effect on the activation of any AP-1 components except for a complete abolishment of JunB activation. These results are consistent with those obtained with the reporter assay and EMSA and indicate that both MEK and JNK MAPK pathways are essential for the activation of AP-1 during primary KSHV infection, while the p38 pathway is involved to a lesser extent. Both the MEK and JNK pathways contribute to AP-1 activation: the MEK pathway mediates c-Jun, JunB, and JunD, and to a lesser extent, c-Fos and FosB, and the JNK pathway mediates c-Jun, JunB, JunD, and FosB, and to a lesser extent, c-Fos. The p38 pathway contributes to AP-1 activation by mediating JunB.
KSHV induces IL-6 transcriptional expression during primary infection.
Since the above results clearly demonstrated KSHV activation of AP-1 during primary infection, we wished to further investigate its biological significance. Previous studies using microarrays have shown that KSHV infection induces IL-6 expression (12, 45); however, the mechanism of induction remains unclear. Figure 7A shows that KSHV-infected HUVEC secreted IL-6 at 10 times higher levels than those in mock-infected cells at 6 hpi. To determine whether the rapid induction of IL-6 by KSHV was mediated at the transcriptional level, we examined the expression of the IL-6 transcript. As shown in Fig. 7B, KSHV infection rapidly induced the expression of the IL-6 transcript. We further examined KSHV activation of the IL-6 promoter in a reporter assay (Fig. 7C). In concordance with the results of transcript expression, KSHV infection rapidly increased the luciferase activity of the IL-6 reporter, which peaked at 6 hpi. Thus, KSHV induction of IL-6 expression is at least partly due to direct transcriptional activation of the IL-6 promoter.
FIG. 7.

KSHV induces IL-6 expression through AP-1 activation and is mediated by multiple MAPK pathways. (A) KSHV infection induces IL-6 secretion into the culture medium. The culture medium from mock- or KSHV-infected HUVEC was collected at 6 hpi and assayed for IL-6 by ELISA. (B) KSHV infection induces expression of the IL-6 transcript. Total RNA isolated from HUVEC infected with KSHV for different time points was assayed for the abundance of IL-6 transcripts by RT-qPCR. The relative abundances of IL-6 transcripts were calculated by setting mock-infected cells (0 h) as onefold (a). Transcripts of human GAPDH were used to calibrate loading. Products from RT-qPCR were also examined on a gel to confirm the results (b). (C) KSHV infection activated the IL-6 promoter reporter. Cells (293 cells) were transfected with IL-6 reporter plasmid DNA and assayed for luciferase activity at 36 h posttransfection. Prior to harvest, the cells were infected with KSHV for 0, 2, 4, 6, 10, and 16 h. (D) KSHV infection induces binding of AP-1 complexes to the consensus element in the IL-6 promoter. Cells (293 cells) were infected with KSHV for 6 h. The experimental conditions are described in the legend to Fig. 2. (E) DN c-Fos (A-Fos) inhibited induction of the IL-6 promoter by KSHV infection. Cells (293 cells) cotransfected with IL-6 reporter plasmid DNA together with A-Fos or a vector control for 30 h were either mock infected or infected with KSHV for 6 h, lysed, and assayed for luciferase activity (a). As controls, the IL-6 reporter was cotransfected with a vector control or an active form of c-Fos, with or without A-Fos (b). (F) Inhibitors of MAPK pathways prevent activation of the IL-6 reporter during KSHV primary infection. Cells (293 cells) transfected with IL-6 reporter plasmid DNA for 30 h were either mock infected or infected with KSHV for 6 h, with or without specific inhibitors of ERK1/2, p38, and JNK, lysed, and assayed for luciferase activity. The inhibitors were added 1 h prior to KSHV infection. (G) Inhibition of MAPK pathways with DN constructs reduces activation of the IL-6 reporter during primary KSHV infection. Cells (293 cells) transfected with IL-6 reporter plasmid DNA alone or together with DN constructs of MEK, p38, and JNK for 30 h were either mock infected or infected with KSHV for 6 h, lysed, and assayed for luciferase activity. All KSHV infection experiments were carried out at an MOI of 2. For the IL-6 ELISA (A), the experiments were independently carried out two times, with three repeats each. The results are averages with standard deviations from one representative experiment. For IL-6 RT-qPCR (B), the experiments were independently carried out two times, with three repeats each. The results are from one set of samples from one representative experiment. For the reporter assay (C, E, F, and G), the experiments were independently carried out three times, with three repeats each. The results presented are averages with standard deviations from one representative experiment. For EMSA (D), the experiments were repeated two times. The results presented are from one representative experiment.
Induction of IL-6 by KSHV infection is dependent on AP-1 activation and is mediated by multiple MAPK pathways.
To determine whether AP-1 activation during primary KSHV infection was essential to the induction of IL-6, we first carried out EMSA using an AP-1 consensus-binding site in the IL-6 promoter as a probe (Fig. 7D). KSHV infection drastically increased the intensity of the AP-1-DNA complexes. As expected, the addition of unlabeled cold probes abolished the intensity of the AP-1 band, while the addition of specific antibodies to c-Fos and c-Jun caused supershifts of the AP-1 bands. These results indicated that the KSHV-activated AP-1 complexes indeed bound to the IL-6 promoter. To further confirm the essential role of AP-1 activation in KSHV induction of IL-6 during primary infection, we infected 293 cells in the presence of a DN construct of c-Fos (A-Fos). The addition of A-Fos prevented 68% of the KSHV induction of the IL-6 reporter (Fig. 7E, panel a). As expected, overexpression of c-Fos increased the IL-6 reporter activity 1.6-fold, and this was abolished by A-Fos (Fig. 7E, panel b), indicating the specificity of the DN construct. To determine whether the activation of MAPK pathways during KSHV primary infection also mediated the induction of IL-6, we applied inhibitors of MAPK pathways in the IL-6 reporter assay (Fig. 7F). All three inhibitors reduced KSHV induction of the IL-6 reporter during primary infection. The inhibitors of MEK and JNK totally abolished KSHV induction of the IL-6 reporter, while that of p38 reduced it to a lesser extent. Similar results were observed when the IL-6 reporter was cotransfected with DN constructs of MAPK pathways (Fig. 7G). Together, the above results clearly demonstrated that KSHV induces IL-6 during primary infection, at least partly by activating AP-1, which is mediated by multiple MAPK pathways.
DISCUSSION
Like infections with other viruses, KSHV infection is a complicated biological process that modulates diverse cellular pathways. In this study, we have shown that KSHV activates AP-1 complexes during primary infection, leading to their increased binding to consensus elements and the induction of reporter gene expression. We have also demonstrated that KSHV activation of AP-1 is mediated by multiple MAPK pathways, including the ERK, JNK, and p38 signal transduction cascades. In addition, MAPK pathways differentially activate individual AP-1 components during KSHV primary infection, and maximum AP-1 activation requires the participation of all three MAPK cascades. The activation of AP-1 and multiple MAPK pathways could have important roles in different aspects of virus-cell interactions during KSHV primary infection, including the facilitation of KSHV infection and replication and the induction and promotion of KSHV-related malignancies.
AP-1 complexes are potent transcription complexes that regulate the expression of a large variety of cellular genes (56). Previous studies have shown that the expression of IL-6 is regulated by various transcriptional factors, including NF-κB, NF-IL-6, C/EBP, AP-1, Sp-1, and IRF-1 (16, 36, 38, 53, 58). We have shown that KSHV activation of AP-1 leads to the induction of IL-6, which is also mediated by multiple MAPK cascades. Several KSHV genes have been shown to directly upregulate the expression of IL-6 (1, 2, 17); however, our results indicate that the induction of IL-6 during primary KSHV infection is largely due to the activation of MAPK pathways and AP-1 mediated by virus entry events rather than the expression and effects of individual KSHV genes (Fig. 4). Nevertheless, it remains to be determined which other transcriptional factors are activated and involved in KSHV induction of IL-6 during primary infection. IL-6 is overexpressed in KS tumors, PEL, and MCD and is implicated in the pathogenesis of these KSHV-induced malignancies (5, 19, 31, 37, 41, 54, 66). Thus, our studies have identified a molecular mechanism by which KSHV modulates cellular signal pathways to induce IL-6 during the early stage of infection. The rapid production of IL-6 can stimulate cellular proliferation and prevent infected cells from apoptosis resulting from KSHV infection.
KSHV activation of AP-1 can lead directly to the promotion of cell proliferation and cell cycle progression by regulating key cell cycle proteins. In fact, AP-1 has been shown to regulate several cell cycle proteins, such as cyclin D1, p53, p21, p19, and p16 (56). However, individual AP-1 members can exert different effects on a given targeted gene, resulting in diverse or even opposite outcomes (56). For examples, c-Jun and Fos proteins are generally positive regulators (10, 33), while JunB has a negative effect (50). Thus, the effect of AP-1 activation on cell proliferation could be temporally and spatially restricted. In this study, we have observed that the activation of AP-1 by KSHV occurs primarily through members that have positive effects on cell proliferation, including c-Jun (2.4-fold), c-Fos (2.8-fold), and FosB (3.5-fold). Other AP-1 members, including JunB (1.6-fold), JunD (1.4-fold), Fra-1 (no change), and Fra-2 (no change), were not activated or only weakly activated. In concordance with these results, we have observed KSHV stimulation of cell proliferation during the early stage of infection by promoting cell exit from G0/G1 phase and entry into S phase (24; unpublished data).
AP-1 activation is involved in cellular transformation (56). Members of both the Jun and Fos families cooperate with other oncogenes to enhance their cellular transformation (40, 61). While extensive epidemiological studies have demonstrated an essential role of KSHV infection in the development of KS, the elucidation of the molecular mechanisms underlying KSHV-induced tumorigenesis remains elusive (20). Some early studies have described KS as a cytokine-driven hyperplasia; however, recent evidence indicates that KS lesions are clonal or multiclonal, manifest a neoplastic transforming nature, and have various genomic abnormalities (21). KSHV infection of human primary microvascular endothelial cells can indeed lead to cellular transformation (22). It is likely that KSHV activation of AP-1 and individual AP-1 components could contribute to KSHV-induced malignant transformation. We have previously shown that KSHV infection of primary HUVEC causes chromosome instability, which could predispose the infected cells to cellular transformation (48). In fact, activation of the MEK MAPK pathway is implicated in uncontrolled centrosome duplication, leading to the formation of aberrant mitotic spindles and chromosomal abnormalities (52).
KS is a highly angiogenic and disseminated tumor (21). AP-1 activation can contribute to the angiogenesis and cell invasion of KS tumors. Besides IL-6, the expression of several other proliferative and angiogenic factors, such as vascular endothelial growth factor and IL-8, and of MMPs that are important for cell invasion is also mediated by AP-1 and MAPK pathways (8, 14, 34, 64).
AP-1 activation is likely to facilitate KSHV infection and replication. The promoter regions of several key KSHV lytic replication genes, encoding RTA, RAP, and MTA, contain the AP-1 consensus element (11, 62). RTA, a viral immediate-early gene, alone is sufficient to drive KSHV into lytic replication and activates the expression of KSHV lytic cycle genes. Furthermore, KSHV oriLyts contain AP-1 consensus sites (6), indicating that the activation of AP-1 is required for the functions of oriLyts. Thus, the activation of AP-1 is likely important for KSHV lytic replication and the expression of viral lytic cycle genes. We have previously reported that efficient KSHV infection of HUVEC is productive at the early stage of infection (24). Obviously, the enhanced survival effect exerted by activated AP-1 and induced IL-6 can indirectly sustain KSHV productive replication and possibly facilitate the establishment of a latent infection during primary infection. It remains to be determined whether AP-1 activation is directly required for productive KSHV infection during primary infection. A sustaining productive primary infection cycle in the host could contribute to the rapid progression of KS by not only producing large quantities of virions for infecting new cells but also contributing to other aspects of KS pathogenesis, including the production of IL-6 and other inflammatory cytokines and the promotion of cellular transformation, cell invasion, and angiogenesis.
Considering the early activation of AP-1 (at 2 hpi) and the MAPK pathways (at <1 hpi) during KSHV infection, it is likely that it occurred during virus binding or entry events. Indeed, UV irradiation of the virions only minimally reduced KSHV activation of AP-1 activities (Fig. 4). A number of KSHV genes that are known to activate MAPK pathways, including those for vGPCR, vPK, LAMP, and vFLIP (2, 7, 9, 27), are expressed at very low levels during this period (68) and could contribute, to a lesser extent, to KSHV activation of AP-1 at the early stage of infection. Previous studies have demonstrated that KSHV infection activates the ERK pathway early in infection (44). In this study, we also observed early activation of the ERK pathway during primary KSHV infection. In addition, we have demonstrated a strong activation of JNK and weak activation of p38 pathways. It is likely that the early activation of JNK and p38 pathways is also mediated through interactions of KSHV glycoproteins with their cellular receptors.
Figure 8 shows a working model illustrating the molecular events underlying the complicated virus-cell interactions during primary KSHV infection. In this study, we have clearly demonstrated KSHV activation of multiple MAPK pathways and AP-1 during primary infection. We have also shown KSHV induction of IL-6, which is mediated by MAPK pathways and AP-1. Future studies will further determine to what extent the activated AP-1 could facilitate KSHV infection and replication and contribute to KSHV-induced malignancies.
FIG. 8.

Working model illustrating the molecular basis of virus-cell interactions during primary KSHV infection. The binding of KSHV virions to cellular receptors activates multiple MAPK pathways, which further activate AP-1 and other transcriptional factors, resulting in the induction of IL-6 and other cytokines as well as cellular phenotypic alterations. The activation of cellular signaling pathways and transcriptional factors also facilitates KSHV infection and replication, which further modulate host cells to promote malignant cellular proliferation.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (CA096512 and DE017333), and by an American Cancer Society Research Scholar grant (RSG-04-195), and by an Outstanding Abroad Young Scientist award (30328001) from the National Science Foundation of China to S.-J. Gao.
We thank Jiahua Han (Scripps Institute), Lin Mantell (New York University School of Medicine), Roger Davis (University of Massachusetts Medical School), Melanie Cobb (University of Texas Southwest Medical Center), Charles Vinson (National Institutes of Health) and John Blenis (Harvard Medical School) for kindly providing reagents. We thank Senlin Li and members of Gao's laboratory for technical assistance and helpful comments.
REFERENCES
- 1.An, J., A. K. Lichtenstein, G. Brent, and M. B. Rettig. 2002. The Kaposi's sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood 99:649-654. [DOI] [PubMed] [Google Scholar]
- 2.An, J., Y. Sun, R. Sun, and M. B. Rettig. 2003. Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways. Oncogene 22:3371-3385. [DOI] [PubMed] [Google Scholar]
- 3.Angel, P., M. Imagawa, R. Chiu, B. Stein, R. J. Imbra, H. J. Rahmsdorf, C. Jonat, P. Herrlich, and M. Karin. 1987. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 49:729-739. [DOI] [PubMed] [Google Scholar]
- 4.Angel, P., and M. Karin. 1991. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072:129-157. [DOI] [PubMed] [Google Scholar]
- 5.Asou, H., J. W. Said, R. Yang, R. Munker, D. J. Park, N. Kamada, and H. P. Koeffler. 1998. Mechanisms of growth control of Kaposi's sarcoma-associated herpes virus-associated primary effusion lymphoma cells. Blood 91:2475-2481. [PubMed] [Google Scholar]
- 6.AuCoin, D. P., K. S. Colletti, S. A. Cei, I. Papouskova, M. Tarrant, and G. S. Pari. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318:542-555. [DOI] [PubMed] [Google Scholar]
- 7.Bais, C., B. Santomasso, O. Coso, L. Arvanitakis, E. G. Raaka, J. S. Gutkind, A. S. Asch, E. Cesarman, and E. A. Mesri. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86-89. [DOI] [PubMed] [Google Scholar]
- 8.Bobrovnikova-Marjon, E. V., P. L. Marjon, O. Barbash, D. L. Vander Jagt, and S. F. Abcouwer. 2004. Expression of angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 is highly responsive to ambient glutamine availability: role of nuclear factor-kappaB and activating protein-1. Cancer Res. 64:4858-4869. [DOI] [PubMed] [Google Scholar]
- 9.Brinkmann, M. M., M. Glenn, L. Rainbow, A. Kieser, C. Henke-Gendo, and T. F. Schulz. 2003. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77:9346-9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown, J. R., E. Nigh, R. J. Lee, H. Ye, M. A. Thompson, F. Saudou, R. G. Pestell, and M. E. Greenberg. 1998. Fos family members induce cell cycle entry by activating cyclin D1. Mol. Cell. Biol. 18:5609-5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byun, H., Y. Gwack, S. Hwang, and J. Choe. 2002. Kaposi's sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol. Cell 14:185-191. [PubMed] [Google Scholar]
- 12.Carroll, P. A., E. Brazeau, and M. Lagunoff. 2004. Kaposi's sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 328:7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang, L., and M. Karin. 2001. Mammalian MAP kinase signal cascades. Nature 410:27-40. [DOI] [PubMed] [Google Scholar]
- 14.Crowe, D. L., and T. N. Brown. 1999. Transcriptional inhibition of matrix metalloproteinase 9 (MMP-9) activity by a c-Fos/estrogen receptor fusion protein is mediated by the proximal AP-1 site of the MMP-9 promoter and correlates with reduced tumor cell invasion. Neoplasia 1:368-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Magalhaes, J. C., A. A. Andrade, P. N. Silva, L. P. Sousa, C. Ropert, P. C. Ferreira, E. G. Kroon, R. T. Gazzinelli, and C. A. Bonjardim. 2001. A mitogenic signal triggered at an early stage of vaccinia virus infection: implication of MEK/ERK and protein kinase A in virus multiplication. J. Biol. Chem. 276:38353-38360. [DOI] [PubMed] [Google Scholar]
- 16.Dendorfer, U., P. Oettgen, and T. A. Libermann. 1994. Multiple regulatory elements in the interleukin-6 gene mediate induction by prostaglandins, cyclic AMP, and lipopolysaccharide. Mol. Cell. Biol. 14:4443-4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng, H., J. T. Chu, M. B. Rettig, O. Martinez-Maza, and R. Sun. 2002. Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100:1919-1921. [DOI] [PubMed] [Google Scholar]
- 18.Derijard, B., M. Hibi, I. H. Wu, T. Barrett, B. Su, T. Deng, M. Karin, and R. J. Davis. 1994. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76:1025-1037. [DOI] [PubMed] [Google Scholar]
- 19.De Wit, R., M. H. Raasveld, R. J. ten Berge, P. A. van der Wouw, P. J. Bakker, and C. H. Veenhof. 1991. Interleukin-6 concentrations in the serum of patients with AIDS-associated Kaposi's sarcoma during treatment with interferon-alpha. J. Intern. Med. 229:539-542. [DOI] [PubMed] [Google Scholar]
- 20.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ensoli, B., C. Sgadari, G. Barillari, M. C. Sirianni, M. Sturzl, and P. Monini. 2001. Biology of Kaposi's sarcoma. Eur. J. Cancer 37:1251-1269. [DOI] [PubMed] [Google Scholar]
- 22.Flore, O., S. Rafii, S. Ely, J. J. O'Leary, E. M. Hyjek, and E. Cesarman. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588-592. [DOI] [PubMed] [Google Scholar]
- 23.Frost, J. A., T. D. Geppert, M. H. Cobb, and J. R. Feramisco. 1994. A requirement for extracellular signal-regulated kinase (ERK) function in the activation of AP-1 by Ha-Ras, phorbol 12-myristate 13-acetate, and serum. Proc. Natl. Acad. Sci. USA 91:3844-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao, S. J., J. H. Deng, and F. C. Zhou. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738-9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao, X., K. Ikuta, M. Tajima, and T. Sairenji. 2001. 12-O-Tetradecanoylphorbol-13-acetate induces Epstein-Barr virus reactivation via NF-kappaB and AP-1 as regulated by protein kinase C and mitogen-activated protein kinase. Virology 286:91-99. [DOI] [PubMed] [Google Scholar]
- 26.Gao, X., H. Wang, and T. Sairenji. 2004. Inhibition of Epstein-Barr virus (EBV) reactivation by short interfering RNAs targeting p38 mitogen-activated protein kinase or c-Myc in EBV-positive epithelial cells. J. Virol. 78:11798-11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamza, M., R. Reyes, Y. Izumiya, R. Wisdom, H.-J. Kung, and P. Luciw. 2004. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 279:38325-38330. [DOI] [PubMed] [Google Scholar]
- 28.Han, J., Y. Jiang, Z. Li, V. Kravchenko, and R. Ulevitch. 1997. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature 386:296-299. [DOI] [PubMed] [Google Scholar]
- 29.Han, J., B. Richter, Z. Li, V. Kravchenko, and R. J. Ulevitch. 1995. Molecular cloning of human p38 MAP kinase. Biochim. Biophys. Acta 1265:224-227. [DOI] [PubMed] [Google Scholar]
- 30.Hill, C., J. Wynne, and R. Treisman. 1994. Serum-regulated transcription by serum response factors (Srf): a novel role for the DNA binding domain. EMBO J. 13:5421-5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang, Y. Q., J. J. Li, K. S. Kim, A. Nicolaides, W. G. Zhang, J. Le, B. J. Poiesz, and A. E. Friedman-Kien. 1993. HIV-1 infection and modulation of cytokine and growth factor expression in Kaposi's sarcoma-derived cells in vitro. AIDS 7:317-322. [DOI] [PubMed] [Google Scholar]
- 32.Johnson, G., and R. Lapadat. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinase. Science 298:1911-1912. [DOI] [PubMed] [Google Scholar]
- 33.Johnson, R., B. Van Lingen, and V. Papaioannou. 1993. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 7:1309-1317. [DOI] [PubMed] [Google Scholar]
- 34.Jones, M. K., K. Tsugawa, A. S. Tarnawski, and D. Baatar. 2004. Dual actions of nitric oxide on angiogenesis: possible roles of PKC, ERK, and AP-1. Biochem. Biophys. Res. Commun. 318:520-528. [DOI] [PubMed] [Google Scholar]
- 35.Karin, M. 1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270:16483-16486. [DOI] [PubMed] [Google Scholar]
- 36.Kick, G., G. Messer, A. Goetz, G. Plewig, and P. Kind. 1995. Photodynamic therapy induces expression of interleukin 6 by activation of AP-1 but not NF-kappa B DNA binding. Cancer Res. 55:2373-2379. [PubMed] [Google Scholar]
- 37.Leger, R. M., M. Peuchmaur, O. Devergne, J. Audouin, M. Raphael, D. J. Van, P. Galanaud, J. Diebold, and D. Emilie. 1991. Interleukin-6 gene expression in Castleman's disease. Blood 78:2923-2930. [PubMed] [Google Scholar]
- 38.Libermann, T. A., and D. Baltimore. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 10:2327-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ludwig, S., O. Planz, S. Pleschka, and T. Wolff. 2003. Influenza-virus-induced signaling cascades: targets for antiviral therapy? Trends Mol. Med. 9:46-52. [DOI] [PubMed] [Google Scholar]
- 40.Mechta, F., D. Lallemand, C. M. Pfarr, and M. Yaniv. 1997. Transformation by ras modifies AP1 composition and activity. Oncogene 14:837-847. [DOI] [PubMed] [Google Scholar]
- 41.Miles, S. A., A. R. Rezai, J. F. Salazar-Gonzalez, M. Vander Meyden, R. H. Stevens, D. M. Logan, R. T. Mitsuyasu, T. Taga, T. Hirano, T. Kishimoto, et al. 1990. AIDS Kaposi's sarcoma-derived cells produce and respond to interleukin 6. Proc. Natl. Acad. Sci. USA 87:4068-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monje, P., M. J. Marinissen, and J. S. Gutkind. 2003. Phosphorylation of the carboxyl-terminal transactivation domain of c-Fos by extracellular signal-regulated kinase mediates the transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol. Cell. Biol. 23:7030-7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy, L. O., S. Smith, R. H. Chen, D. C. Fingar, and J. Blenis. 2002. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 4:556-564. [DOI] [PubMed] [Google Scholar]
- 44.Naranatt, P. P., S. M. Akula, C. A. Zien, H. H. Krishnan, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-ζ-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naranatt, P. P., H. H. Krishnan, S. R. Svojanovsky, C. Bloomer, S. Mathur, and B. Chandran. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72-84. [DOI] [PubMed] [Google Scholar]
- 46.Nimmerjahn, F., D. Dudziak, U. Dirmeier, G. Hobom, A. Riedel, M. Schlee, L. M. Staudt, A. Rosenwald, U. Behrends, G. W. Bornkamm, and J. Mautner. 2004. Active NF-kappaB signaling is a prerequisite for influenza virus infection. J. Gen. Virol. 85:2347-2356. [DOI] [PubMed] [Google Scholar]
- 47.Olive, M., D. Krylov, D. R. Echlin, K. Gardner, E. Taparowsky, and C. Vinson. 1997. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J. Biol. Chem. 272:18586-18594. [DOI] [PubMed] [Google Scholar]
- 48.Pan, H., F. Zhou, and S. J. Gao. 2004. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 64:4064-4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panteva, M., H. Korkaya, and S. Jameel. 2003. Hepatitis viruses and the MAPK pathway: is this a survival strategy? Virus Res. 92:131-140. [DOI] [PubMed] [Google Scholar]
- 50.Passegue, E., and E. Wagner. 2000. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 19:2969-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruther, U., D. Komitoski, R. Muller, and F. Wagner. 1987. Deregulated c-fos expression interferes with normal bone development in transgenic mice. Nature 325:412-416. [DOI] [PubMed] [Google Scholar]
- 52.Saavedra, H. I., K. Fukasawa, C. W. Conn, and P. J. Stambrook. 1999. MAPK mediates RAS-induced chromosome instability. J. Biol. Chem. 274:38083-38090. [DOI] [PubMed] [Google Scholar]
- 53.Sanceau, J., T. Kaisho, T. Hirano, and J. Wietzerbin. 1995. Triggering of the human interleukin-6 gene by interferon-gamma and tumor necrosis factor-alpha in monocytic cells involves cooperation between interferon regulatory factor-1, NF kappa B, and Sp1 transcription factors. J. Biol. Chem. 270:27920-27931. [DOI] [PubMed] [Google Scholar]
- 54.Screpanti, I., P. Musiani, D. Bellavia, M. Cappelletti, F. B. Aiello, M. Maroder, L. Frati, A. Modesti, A. Gulino, and V. Poli. 1996. Inactivation of the IL-6 gene prevents development of multicentric Castleman's disease in C/EBP beta-deficient mice. J. Exp. Med. 184:1561-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma-Walia, N., P. P. Naranatt, H. H. Krishnan, L. Zeng, and B. Chandran. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-Rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaulian, E., and M. Karin. 2002. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4:E131-E136. [DOI] [PubMed] [Google Scholar]
- 57.Shaulian, E., and M. Karin. 2001. AP-1 in cell proliferation and survival. Oncogene 20:2390-2400. [DOI] [PubMed] [Google Scholar]
- 58.Shimizu, H., K. Mitomo, T. Watanabe, S. Okamoto, and K. Yamamoto. 1990. Involvement of an NF-kappa B-like transcription factor in the activation of the interleukin-6 gene by inflammatory lymphokines. Mol. Cell. Biol. 10:561-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taddeo, B., W. Zhang, F. Lakeman, and B. Roizman. 2004. Cells lacking NF-kappaB or in which NF-kappaB is not activated vary with respect to ability to sustain herpes simplex virus 1 replication and are not susceptible to apoptosis induced by a replication-incompetent mutant virus. J. Virol. 78:11615-11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tyler, K. L., P. Clarke, R. L. DeBiasi, D. Kominsky, and G. J. Poggioli. 2001. Reoviruses and the host cell. Trends Microbiol. 9:560-564. [DOI] [PubMed] [Google Scholar]
- 61.Vandel, L., N. Montreau, E. Vial, C. M. Pfarr, B. Binetruy, and M. Castellazzi. 1996. Stepwise transformation of rat embryo fibroblasts: c-Jun, JunB, or JunD can cooperate with Ras for focus formation, but a c-Jun-containing heterodimer is required for immortalization. Mol. Cell. Biol. 16:1881-1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, S., F. Wu, H. Chen, M. Shamay, Q. Zheng, and G. Hayward. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang, X. P., Y. J. Zhang, J. H. Deng, H. Y. Pan, F. C. Zhou, and S. J. Gao. 2002. Transcriptional regulation of Kaposi's sarcoma-associated herpesvirus-encoded oncogene viral interferon regulatory factor by a novel transcriptional silencer, Tis. J. Biol. Chem. 277:12023-12031. [DOI] [PubMed] [Google Scholar]
- 64.Westermarck, J., and V. Kahari. 1999. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 13:781-792. [PubMed] [Google Scholar]
- 65.Whitmarsh, A., and R. Davis. 1996. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 74:589-607. [DOI] [PubMed] [Google Scholar]
- 66.Yang, J., M. K. Hagan, and M. K. Offermann. 1994. Induction of IL-6 gene expression in Kaposi's sarcoma cells. J. Immunol. 152:943-955. [PubMed] [Google Scholar]
- 67.Ye, F. C., F. C. Zhou, S. M. Yoo, J. P. Xie, P. J. Browning, and S. J. Gao. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121-11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoo, S. M., F. C. Zhou, F. C. Ye, H. Y. Pan, and S.-J. Gao. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology, in press. [DOI] [PMC free article] [PubMed]
- 69.Young, M., J.-J. Li, M. Rincon, R. Flavell, B. Sathyanarayana, R. Hunziker, and N. Colburn. 1999. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. USA 96:9827-9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng, Y., J. Li, D. L. Johnson, and J. H. Ou. 2003. Regulation of hepatitis B virus replication by the Ras-mitogen-activated protein kinase signaling pathway. J. Virol. 77:7707-7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]