

Abstract
Hepatitis B virus (HBV) infection is widely distributed in both human and ape populations throughout the world and is a major cause of human morbidity and mortality. HBV variants are currently classified into the human genotypes A to H and species-associated chimpanzee and gibbon/orangutan groups. To examine the role of recombination in the evolution of HBV, large-scale data retrieval and automated phylogenetic analysis (TreeOrder scanning) were carried out on all available published complete genome sequences of HBV. We detected a total of 24 phylogenetically independent potential recombinants (different genotype combinations or distinct breakpoints), eight of which were previously undescribed. Instances of intergenotype recombination were observed in all human and ape HBV variants, including evidence for a novel gibbon/genotype C recombinant among HBV variants from Vietnam. By recording sequence positions in trees generated from sequential fragments across the genome, violations of phylogeny between trees also provided evidence for frequent intragenotype recombination between members of genotypes A, D, F/H, and gibbon variants but not in B, C, or the Asian B/C recombinant group. In many cases, favored positions for both inter- and intragenotype recombination matched positions of phylogenetic reorganization between the human and ape genotypes, such as the end of the surface gene and the core gene, where sequence relationships between genotypes changed in the TreeOrder scan. These findings provide evidence for the occurrence of past, extensive recombination events in the evolutionary history of the currently classified genotypes of HBV and potentially in changes in its global epidemiology and associations with human disease.
Infection with hepatitis B virus (HBV) is globally distributed, infecting approximately one-third of the world's human population. It is responsible for substantial proportion of liver disease that kills over one million people through uncompensated liver disease or hepatocellular carcinoma each year (43). Populations in South and East Asia, sub-Saharan Africa, and Central and South America show particularly high frequencies of HBV infection that may be maintained through mother-to-child perinatal transmission (13) or horizontal transmission in childhood (8, 41).
HBV variants infecting humans show genetic and antigenic heterogeneity and are currently classified into seven or eight genotypes that differ from each other in nucleotide sequence by 10 to 13% (29-31, 33). While genotypes A, D, and possibly G have global distributions, genotypes B and C are found predominantly in East and Southeast Asia, genotype E is found in West Africa, and genotypes F and H are found among various population groups, including indigenous peoples in Central and South America. Active and resolved HBV infections are also found at high frequencies in chimpanzees (17, 17, 24, 42, 45) and Southeast Asian apes (12, 28, 46, 49) with species-associated HBV variants distinct from those found in human populations. Finally, an HBV variant was recovered from a captive woolly monkey (21), whose sequence is highly divergent from all other human and ape sequences described to date.
It is increasingly accepted that, in addition to the currently classified genotypes of HBV, recombination between genotypes generates novel variants that contribute to the genetic diversity of HBV. Recombination in human HBV was first detected in sequential samples from infected individuals and among integrated sequences from cases of hepatocellular carcinoma (11, 44). However, recombination between different genotypes was also suspected from incongruities between phylogenetic trees constructed from different regions of the HBV genome (30) and was formally demonstrated through the identification of specific breakpoints in certain published sequences, such as HBVDNA (4), proposed to be a recombinant between genotypes A and D. Both the existence of well-defined breakpoints and the exchange of large sections of sequence between genotypes indicate that recombination, rather than phenotypically selected accelerated evolution and/or convergence (e.g., such as mediated by the immune system [22]), most plausibly accounts for the existence of phylogenetic incongruities between HBV variants in different parts of the genome.
Since then, using similar analytical methods, there have been several published descriptions of intergenotype recombinants among newly obtained or existing variants of HBV. Among these, recombinant forms between A and D (5, 26, 34), B and C (5, 26, 26, 27, 30, 40, 52), and C and D (7, 48) have been identified. However, in many cases, recombinants may be separate instances of phylogenetically related viruses that have become widely distributed geographically. For example, a recombinant generated through the exchange of the core gene of genotype C into a genotype B variant, often described as genotype Ba (with Bj designating nonrecombinant genotype B), has become prevalent in mainland East Asia and independently recorded in several surveys (5, 26, 40).
In this study, we have sought to identify independent intergenotype recombinants of HBV by large-scale screening and phylogenetic analysis of complete genome sequences available in public sequence databases. Using a series of novel phylogenetic and bioinformatic analysis methods, we have mapped recombination hot spots to positions of phylogeny rearrangements within currently classified HBV genotypes. These findings, combined with the new evidence for extensive recombination between members of the same genotype (intragenotype recombination), provide new data on the central role of recombination in the past evolution of HBV and in the generation of its current genetic diversity.
MATERIALS AND METHODS
Nucleotide sequences.
The study sequences comprised all available complete genome sequences of HBV available initially in October 2004 from GenBank, EMBL, and DDBJ. Sequences were screened to exclude patents, artificial mutants, and identical entries. The remaining sequences were aligned manually in the Simmonic sequence editor package (available from http://www.polio.vir.gla.ac.uk). From the initial download, identical sequences or sequences showing less than 1% sequence divergence from each other were removed, leaving a total of 526. Using genotyping information in the original sources, or through analysis of the hepatitis B surface antigen gene, these sequences comprised 79 genotype A, 80 genotype B, 161 genotype C, 56 genotype D, 6 genotype E, 23 genotype F, 8 genotype G, and 7 genotype H sequences. A further 13 and 30 complete HBV genome sequences from chimpanzees and gibbons/orangutans, respectively, were added to the study data set, along with the divergent HBV variant (NC_001896) from the woolly monkey (used as an outgroup in most subsequent phylogenetic analyses).
Sequence selection.
To ensure as fair a weighting as possible in the analysis of sequence groups and recombination, several complete genome sequences of the overrepresented genotypes A to C were removed by similarity scanning. This ensured that the sequences used for analysis were both representative and maximally informative phylogenetically. After recombinant sequences were removed, target numbers of approximately 40 to 60 sequences from each of the three genotypes were obtained by excluding sequences with similarity thresholds of below 1.5% over aligned complete genome sequences for genotype A and B sequences (leaving 43 and 52, respectively) and 2.5% for genotype C (leaving 64). Genotype B sequences were further divided by exclusion of B/C recombinants (Ba genotype [26]) to leave 24 nonrecombinant variants (corresponding to Bj). For other genotypes (including those from primates), all available sequences were used, although genotypes unavoidably differed from each other in their intragenotype variability. These sequences comprised set A and were used for group scanning analysis. Consensus sequences from each group (50%) were used for SIMPLOT.
After the initial analysis, a second download of sequences published from October 2004 until June 2005 was carried out, and using the selection criteria described above to eliminate closely related sequences, the following sequences were combined to form set B: genotype A, 45; genotype B (nonrecombinant), 25; genotype B/C recombinants (recB), 51; genotype C, 70; genotype D, 59; genotype E, 13; genotype F, 25; genotype G, 9; and genotype H, 9 sequences. No further nonhuman primate-derived sequences were available. A full listing of sequences in sets A and B and recombinant sequences identified in the study is available from the authors.
Assignment of recombinant forms of HBV.
HBV variants were assigned as confirmed intergenotype recombinants where there was published evidence for their sequences having been obtained from two or more clones demonstrating identical recombination breakpoints or from variants with identical breakpoints described in separate studies or from different HBV-infected study subjects in the same study. The remaining sequences were assigned as provisional recombinants. For these variants, there was insufficient published or unpublished data to formally rule out the possibility that the complete genome sequences were not composites of fragments amplified from samples of individuals with mixed genotype infections.
RESULTS
Phylogenetic relationships between HBV genotypes.
To visualize phylogenetic relationships in different parts of the HBV genome, a new method was developed to record positions of each sequence in a series of phylogenetic trees generated by sets of overlapping fragments across the genome. Sequence fragments of 250 bases, incrementing in 25-base steps were used (125 trees in total) (Fig. 1). Boot-strapped phylogenetic trees were constructed using the programs SEQBOOT, DNADIST, NEIGHBOR, and CONSENSE in the PHYLIP version 3.62 package (10). The sequence data set comprised a total of 348 human and ape HBV sequences (set B) from which all recombinant sequences had been excluded with the exception of B/C recombinant sequences (recB) (Fig. 1, bright green).
FIG. 1.

TreeOrder Scan of nonrecombinant HBV and recB sequences, showing positions of individual sequences (y axis) in phylogenetic trees generated from sequential 250-base sequence fragments, incrementing by 25 bases (midpoints indicated in x axis). Changes in sequence orders resulting from changes in phylogeny at the 70% bootstrap level are shown. Sequences are color coded by genotype, as indicated by labels in left and right margins: genotype A, red; B, light green; recB (rB), green; C, yellow; D, blue; E, purple; F, pink; G, pale yellow; H, brown; chimpanzee (Ch), gray; gibbon (Gi), white; orangutan (Or, turquoise; woolly monkey (outgroup on line 1), black. The recombinant sequence, AY161141, is shown as a dotted line. The boundaries of sequence groupings with 70% or greater bootstrap support are indicated with black horizontal lines; for clarity, only clades with six or more members were demarcated. All sequence positions were numbered relative to the HBVADW2 reference sequence (length, 3,221 bp). For comparison, the TreeOrder Scan was aligned with a scale genome diagram of HBV (upper panel) and with positions of breakpoints in recombinant HBV variants (triangles in lower panel). Each independent breakpoint is indicated as a square, localized to one of 61 separate 250-base sequence fragments spanning the genome, with segment 62 representing the junction between the beginning and end of the alignment. Genotype transitions are shown in the upper-left and lower-right quadrants of the square and are color coded as for the TreeOrder Scan; unfilled triangles represent unknown genotypes; gibbon sequences are in black.
The vertical ordering of sequences in each tree was determined by the following criteria. (i) Each tree was permuted to match as closely as possible the sequence order of the tree generated from the immediately preceding fragment. (ii) Allowable permutations comprised branch rotations and positional reordering of sequences or groups of sequences, provided this occurred within groupings below the specified bootstrap threshold value (70% for the analyses shown in Fig. 1). (iii) Each tree was rooted using the woolly monkey HBV sequence (NC_001896), whose position was predefined at position 1.
The ordering of sequences in sequential trees according to the above rules was automated in the program TreeOrder Scan in the Simmonic 2005 version 1.4 Sequence Editor package (which contains full information on required files and output formats). The output of the program comprises a numeric array that records the position of each sequence (columns) for each tree (rows). Each line in the graph (Fig. 1), representing the position of each sequence across the genome, was color coded by genotype (see left and right columns). Because the algorithm attempts to match sequence order between fragments, alterations in the tree order of individual sequences and of whole genotypes between fragments indicate changes in the phylogenetic relationships of clades supported by bootstrap values, in this case, of 70% or greater. For sequences with constant phylogenetic relationships across the genome, each sequence would follow a horizontal line across Fig. 1. In contrast, phylogenetic relationships between many genotypes changed frequently in different parts of the genome.
First, there were differences between sequence fragments in the degree of phylogenetic differentiation between genotypes, with clear delineation of genotypes in some cases in bootstrap-supported clades (delineated by black dividing lines). In other cases, such as between positions 1600 and 1850 (corresponding to the region of the genome encoding the X gene), sequences of genotypes C, A, E, D, and B were phylogenetically undifferentiated at the 70% bootstrap threshold level.
Second, phylogenetic relationships between genotypes changed frequently, leading to several forced alterations in branching order. For example, genotype D became an outgroup for all genotypes except genotypes F and H between positions 780 (end of the S gene) and 980. Genotype G moved to a similar outlier position from 980 to 2750. Genotype B sequences split at position 1950 (start of core gene), with the majority (recB) grouping with genotype C sequences and rejoining the others at around position 2500 (end of core gene). A similar change in phylogenetic relationships was observed between genotypes D and E. These genotypes were phylogenetically distinct over most of the genome, but genotype E fell within the genotype D clade in the core gene (positions 1950 to 2500). Genotypes F and H did not invariably form separate clades (350 to 500). Finally, sequences from orangutans are interspersed with gibbon sequences throughout the genome except for the pre-S1 and pre-S2 regions where they were distinct.
Overall, the major phylogenetic discontinuities detected in the TreeOrder Scan were at positions 750, 900, 1600, 1950, 2500, 2650, and 2750. Most of these occurred at, or close to, gene boundaries and frequently occurred independently in different genotypes. Particularly frequent changes in phylogenetic relationships were found at each end of the core and surface genes. Since the grouping of many genotype B sequences in the genotype C clade in the core gene has been previously interpreted as evidence for intergenotype recombination, this process plausibly accounts for other changes in tree order evident in the scan.
Screening for HBV recombination.
To further investigate the potential of recombination in the phylogenetic discontinuities observed between HBV genotypes, a complete catalogue of recombination breakpoints among previously published HBV sequences was compiled. All sequences available in the GenBank database in June 2005 were screened using the program TreeOrder Scan incorporating the previously shown data set of nonrecombinant sequences as controls (set B; n = 347). The TreeOrder Scan provides a rapid method to detect intergenotype recombination among individual sequences. To demonstrate this, a previously undescribed recombinant sequence AY161141 was included in the data set (Fig. 1). This sequence fell within the genotype A clade (red) throughout the genome except for the core gene, where it grouped with genotype D (blue). This method was applied using several sets of downloaded sequences for rapid, large-scale initial screening of all published HBV sequences for recombination.
Sequences identified as recombinant or potentially recombinant because of changes in branching order relative to the control groups were further analyzed using SIMPLOT, version 2.5 (23), and a newly developed program called Group Scan available in the Simmonic Sequence Editor package. The latter program records how deeply embedded a test sequence lies within each of the clades formed by standard phylogenetic analysis of preassigned groups (nonrecombinant HBV genotypes in set A, excluding recB sequences), from which it calculates a grouping score (Fig. 2). The method for scoring is related to that used by the Association Index, a method developed previously to quantify the degree of segregation of different predefined groups (6, 47).
FIG. 2.

(A) Comparison of Group Scanning and SIMPLOT analysis of the recombinant sequence AY161141. In each case, sequence fragments of 250 bases incrementing by 50 bases, 100 bootstrap replicates, were compared with sequence groups (Group Scan) or 50% consensus sequences of the eight human genotypes and four primate genotypes (color coded as described in the legend of Fig. 1). For the group scanning analysis, only values of >0.5 indicate grouping within a genotype group. (B) Group Scan analysis of examples of newly discovered recombinant HBV variants (listed in Table 1) and of novel recombinant sites in a previously analyzed sequence, HBVDNA. (C) Group Scan and SIMPLOT analysis of the sequence AB048704. Because the recombinant region in the S gene was relatively phylogenetically uninformative, longer sequence fragments of 400 bases incrementing by 15 bases (Group Scan) or 20 bases (SIMPLOT) were analyzed. Sequences are color coded as described in the legend of Fig. 1, except that the gibbon HBV variants are shown in black. The closely related variant, AB048705, produced comparable results (data not shown).
For the Group Scan, the grouping score of a test sequence in a phylogeny is computed by reference to the assigned groups of its nearest neighbors in unrooted trees. Grouping scores (G) for a predefined group, a, can be represented by the following formula:
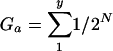 |
1 |
where N is the number of nodes separating the test sequence from each member of the group, and y is the total number of sequences in group a. By definition, the total of grouping scores of each preassigned group, Ga to Gn, adds up to 1. For robustness, each score is the mean of a predefined number of bootstrap replicate trees (100 in the analyses shown) generated from permuted data sets where either the nucleotide sequence data (using SEQBOOT in the PHYLIP package) or the population (selection of a random two-thirds of sequences from each preassigned group) is resampled. To localize sites of potential recombination, consecutive fragments of predefined length and incremental steps between fragments were analyzed.
For each fragment (midpoint plotted on x axis), the depth of grouping in each genotype was plotted (y axis) with values ranging from close to 0 (no grouping) to close to 1 (deeply buried within a clade formed by sequences of a genotype). Changes in genotype grouping, potentially arising from recombination, are characterized by abrupt changes in grouping values of the sequence, as shown for AY161141 (Fig. 2A). Concordant with its changing position on the TreeOrder Scan (Fig. 1), this sequence fell within the genotype A clade between positions 2400 to 1650 (values of >0.5; i.e., mean values for the 50 bootstrap replicated trees lay within the outgroup value) and within the genotype D clade from 1700 to 2400. The underlying methodology of the Group Scan is different from SIMPLOT, which records the bootstrap value of a test sequence with artificially generated consensus sequences of each genotype. However, the output from both programs is frequently comparable, with evidence for alternate groupings of the AY161141 sequence with the genotype A (consensus) sequence between positions 2400 to 1600 and with genotype D between 1800 to 2400 (Fig. 2A), as found in the Group Scan.
A detailed comparison of the two methodologies will be presented elsewhere; for the purposes of the current study, both methods were used to scan recombinant and potential recombinant sequences in data set A of HBV complete genome sequences (Table 1). Only breakpoints identified by both methods were included in the analysis, since inconsistencies between methods (such as the apparent recombination site between genotypes A and G at position 700 in the SIMPLOT analysis of AY161141) (Fig. 2A) may represent artifacts, in this case arising from a more general change in phylogenetic relationships between genotypes at the end of the surface gene. However, in general, the methods produced concordant results for almost all recombination sites.
TABLE 1.
Compilation of recombinant HBV sequences
| Sequencea | Genotypes | Breakpoint | Reference(s)e |
|---|---|---|---|
| AF297621 | A, D | 4 | 34 |
| HBVDNA | D, A | 4 | 4, 26, 5 |
| HBVAYWCI | D, A | 4 | 26, 5 |
| HBVAYWE | D, A | 2 | 26, 5 |
| AF297619 | D, A | 4 | 34 |
| AF297620 | D, A | 4 | 34 |
| AY236161b | D, A | 2 | Unpublished (U1) |
| AY161141, AY161146 | A, D | 2 | Unpublished (U2) |
| AY161149 | A, D | 6 | Unpublished (U2) |
| AF418681 | D, A | 2 | Unpublished (U3) |
| AB194949 | A, E | 2 | 20 |
| AB056516 | A, G | 2 | 18 |
| AY057947 | C, A | 2 | 7 |
| RecB (Ba) | B, C | 2 | 5, 26, 30, 40 |
| HPBADRM | C, B | 2 | 26, 27 |
| AB031265 | C, B | 2 | 52 |
| AF233236 | C, B | 2 | Unpublished (U4) |
| AY057948, AY817511 | D, C | 2 | 7, 48 |
| AF461043, AY817515, et al. | D, C | 2 | Unpublished (U5), 48 |
| AF241408, AF241409 | Uc, C | 2 | 16 |
| AB214516 | F, C | 2 | Unpublished (U6) |
| AB048704, AB048705 | C, Gibbond | 2 | 38f |
| AF498266 | C, Chimp | 2 | 25, 45 |
| AB046525 | Chimp, U | 2 | 42 |
Accession numbers of sequences reported as recombinant previously and/or found to be recombinant in this study. Each line contains variants with separate, phylogenetically independent breakpoints.
HBV sequences provisionally assigned as intergenotype recombinants shown in italics (see Materials and Methods).
U, unknown genotype.
Assignment was based on the Group Scan and separate phylogenetic analysis (Fig. 3).
Published sources of recombinant sequence or description of recombination or attributions for unpublished (U) sequences are as follows: G. Raimondo, T. Pollicino, and G. Raffa for U1; C. Vaishali, S. K. Acharya, and S. K. Panda for U2; V. Chaudhuri, S. K. Acharya, and S. K. Panda for U3; T. Cheng, M. Chen, J. Zhang, and N. S. Xia for U4; W. J. Huang, H. Y. Zhang, Y. C. Wang, J. X. Lin, W. J. Gu, X. Wu, and H. Y. Lan for U5; H. Tran and K. Abe for U6.
Not reported as recombinant in original publication.
The screening carried out on GenBank sequences independently identified all recombinant sequences described to date in the HBV literature (Table 1). However, the analysis in this study identified a series of previously undescribed recombinant forms, including several A/D recombinants, a C/B, a D/C, and an F/C recombinant from Bolivia (Fig. 2B). For the sequence HBVDNA, reported as a D/A recombinant (4), there was evidence from both Group Scanning and SIMPLOT for additional recombination points between 1300 and 1450 (Fig. 2B). For the sequences, HPBADW1 and HBVP4PCXX, which have previously been described as recombinant (4, 5, 26), neither group scanning nor SIMPLOT identified clear breakpoints, and these sequences have therefore been omitted from subsequent analysis.
Finally, the Group Scan identified AB048704 and AB048705 from Vietnamese HBV patients as potential gibbon/genotype C recombinants (Fig. 2C and 3) (the only published analysis of them was carried out before the gibbon-derived sequences were available). Between positions 300 and 1000, both sequences either formed outgroups of or fell within the gibbon clade (grouping values of ≥0.5). The grouping of AB048704 and AB048705 with gibbon-derived variants was confirmed by separate phylogenetic analysis, where both sequences (along with those from orangutans) clearly fell within the gibbon sequence clade (Fig. 3). Because this region of the S gene is relatively phylogenetically uninformative, SIMPLOT failed to record high bootstrap scores in the recombinant region, although the analysis confirmed a change in grouping from genotype C to gibbon and back at the same breakpoints identified by the Group Scan (Fig. 2C).
FIG. 3.

Phylogenetic analysis of positions 251 to 650 (corresponding to the putative recombinant region in the S gene) (Fig. 2C) of sequences AB048704 and AB048705 (Rec), all available gibbon- and orangutan-derived sequences, and nonrecombinant human- and chimpanzee-derived HBV variants (set A). Since sequences from each genotype were monophyletic, only the most recent common ancestor (MRCA) is shown for non-gibbon genotypes. The tree was constructed by neighbor-joining using Jukes-Cantor corrected distances in the MEGA2 package (19), using 200 bootstrap replicates (values of 70% or greater shown).
For analysis of breakpoint positions, it was apparent that many recombinant HBV variants were phylogenetically related and contained identical recombination sites (such as among the widespread B/C recombinant form in East Asia and the newly discovered D/C recombinants, such as AY057948 and AY817511 from Tibet and China [7, 48]). Therefore, only HBV variants that showed clearly different breakpoints and/or genotype composition were included in subsequent analysis to ensure that the recombination events were phylogenetically independent and not multiply scored. Breakpoints were localized to the segment (from a total of 62) where grouping or bootstrap values crossed the threshold 0.5 value for the two sequences. The positions of the breakpoints were aligned with the HBV genome diagram and with the TreeOrder Scan of nonrecombinant sequences (Fig. 1).
Overall, the distribution of recombination breakpoints was nonrandom, with certain apparently favored sites (such as fragment 14 [positions 650 to 900]), where several different recombination events between different genotypes occurred (between genotype A and D variants, as well as between D and C). Favored recombination sites frequently localized to gene boundaries, such as the end of the S gene (fragment 14), the precore/C gene start, the end of C gene, and the pre-S1/pre-S2 junction. Elsewhere, however, breakpoints were more randomly distributed, such as between fragments 19 and 32 (the 3′ end of the P gene and the X gene).
Recombination within HBV genotypes.
Apart from the changes in branching order of different genotypes and the multiple examples of phylogenetically independent recombinant events (previous sections), further evidence for recombination was provided by changes in tree order between consecutive fragments among variants conventionally regarded as nonrecombinant. For example, following the tree position of individual genotype A (red) and D (blue) sequences across the genome (Fig. 1) reveals several sites where positions change in response to changes in phylogenetic relationships between members of the same genotype.
To investigate whether these violations of intragenotype phylogenies between fragments arose more frequently than would be expected by chance, frequencies of sequence order changes within genotypes were compared with those of artificially generated, nonrecombinant control data sets of sequences corresponding exactly in sequence divergences between native sequences. A total of 50 independently replicated data sets of the original 311 native sequences in set A were created using a maximum likelihood method implemented in the program EVOLVER in the PAML package (51), with likelihood-derived parameters for the model, rate variation between sites, frequencies of invariant sites, and sequence changes in different model categories. Separate analysis was also carried out on separate subsets of certain HBV genotypes that contained more than 20 representative sequences (A, B, recombinant B/C [recB], C, D, F/H combined, and Gibbon). Phylogeny violations in native and control data sets were calculated as the mean number of tree order changes observed on pairwise comparison of each sequence fragment, normalized by the number of sequences, using a range of bootstrap threshold values (Table 2).
TABLE 2.
Phylogeny violations within HBV genotypes
| Group | No. of sequencesa | Divergence (%)b | No. of violations/sequencec
|
Z scoree | Pe | Total no. of violationsf | ||
|---|---|---|---|---|---|---|---|---|
| Native | Control | SDd | ||||||
| A | 39 | 4.1 | 0.364 | 0.190 | ±0.038 | 4.65 | 4 × 10−6 | 40 |
| B | 24 | 2.7 | 0.152 | 0.109 | ±0.039 | 1.03 | NS | 6 |
| RecB | 47 | 3.0 | 0.116 | 0.118 | ±0.029 | −0.07 | NS | −1 |
| C | 64 | 4.0 | 0.148 | 0.147 | ±0.031 | 0.05 | NS | 1 |
| D | 52 | 2.9 | 0.235 | 0.149 | ±0.031 | 2.78 | 0.005 | 26 |
| F/H | 30 | 3.3 | 0.314 | 0.216 | ±0.042 | 2.33 | 0.02 | 17 |
| Gibbon | 28 | 6.7 | 0.380 | 0.180 | ±0.042 | 4.42 | 1 × 10−5 | 30 |
| All | 310 | 3.4 | 0.206 | 0.143 | ±0.013 | 4.91 | 9 × 10−7 | 110 |
Number of sequences compared within genotype.
Mean, uncorrected pairwise distance between sequences within genotype.
Number of phylogeny violations per sequence per 1,000 bases using a 60% bootstrap value. For controls, mean number of 50 nonrecombinant control data sets is shown. For this analysis, fragments were 400 bp in length, overlapping by 200 bases (16 fragments per genome).
Sampled standard deviation of 50 nonrecombinant control data sets.
Data sets with significantly greater frequencies of phylogeny violations than expected by chance (P < 0.05) are indicated in bold. NS, not significant.
Total net (number of native − number of control) number of phylogeny violations within each sequence group.
For all meaningful bootstrap threshold values (i.e., equal to or greater than 50%), higher frequencies of phylogeny violations were observed in native genotype A, D, F/H, and gibbon sequences compared with mean values for control data sets (Table 2). To estimate the statistical significance of these differences, the positions of native scores in the distribution of those from control sequences was determined and expressed as a Z score. Values of greater than 2 represent frequencies of phylogeny violations outside the 95th percentile for the control values (two-tailed test). Sequences within genotypes A, D, F/H, and gibbon-derived HBV variants showed significantly greater frequencies of phylogeny violations than would be expected by chance (Z scores ranging from 3.4 to 7.7), while no significantly greater tree order changes were found in genotype B, recB, or C (Z scores of −0.1 to 0.94).
The number of phylogeny violations attributable to recombination was calculated by subtracting the control from the native value (Table 2, column 9); this indicated substantial number of separate recombination events in genotype A, D, and gibbon sequences, and none or close to none in genotypes B, recB, and C. Frequencies of phylogeny violations were frequently, but not invariably, greater among the more divergent genotypes (measured as mean intragenotype pairwise distance) (Table 2, column 3). Although carried out as a separate analysis, the number of phylogeny violations in the whole data set of 312 sequences of all genotypes (n = 110) approximated to the total number of events from the individual genotypes analyzed (n = 119). As expected, repeating the analysis using higher bootstrap threshold values recorded progressively fewer recombination events (for the whole data set: 62 at 70%, 52 at 75%, 44 at 80%, and 28 at 90%) but retained disparities in phylogeny violations between native and control data sets for each genotype (Z scores for whole data set of 4.91 at 60%, 3.44 at 70%, 3.15 at 75%, 2.6 at 80%, and 2.43 at 90%).
The locations of phylogeny violations in gibbon-derived HBV sequences were determined by plotting frequencies for individual pairwise comparisons of fragments in the form of a half-diagonal phylogenetic compatibility matrix (Fig. 4). This analysis reveals several regions of low values (blue) where phylogenies were compatible, interrupted by often sharp boundaries (arrows) splitting sequences into a series of noncompatible blocks. In two of the three indicated positions, these corresponded to favored sites of recombination among intergenotype recombinant HBV variants (Fig. 1, lower panel).
FIG. 4.

Phylogenetic compatibility matrix of gibbon-derived HBV sequences, showing frequencies of phylogeny violations for each pairwise comparison of sequence fragments. Frequencies are color coded to indicate number of phylogeny violation per sequence (see key). Phylogenetically compatible regions are shown as deep blue. Arrows indicates the main sites where changes in phylogeny of the gibbon sequences lead to regions of incompatibility between fragments. For this analysis, sequence fragments of 400 bases, incrementing by 25 bases across the genome, were compared (117 fragments total), using a bootstrap value of 70% above which phylogeny violations were computed.
DISCUSSION
Intergenotype recombination.
This study provides comprehensive information on the occurrence and composition of HBV intergenotype recombinants. A total of 24 phylogenetically independent recombinant forms of HBV were found, involving all human genotypes as well as both chimpanzee and gibbon variants. From the available data on their geographical distributions and the finding of the same recombinants from different published sources, it is apparent that recombinant forms are capable of spread in human populations and of developing their own specific distributions and epidemiologies. This is particularly clearly demonstrated for the B/C recombinant, which now accounts for the majority of genotype B sequences in mainland Asia (39). Another example is the D/C recombinant, found in a separate survey in Tibet (AY057948 [7]) and China (AY817511 [48]). While the current study is as up to date as possible, clearly more extensive sampling and sequence analysis are likely to reveal further examples of recombinant forms of HBV as a wider range of human populations are surveyed in the future.
Apart from documenting individual examples of intergenotype recombinants, this study provides evidence that the evolution of what are conventionally regarded as “nonrecombinant” genotypes of HBV has been accompanied by a similar series of recombination events. This process is manifested most clearly in the frequent changes in phylogenetic relationships between genotypes across the genome in the TreeOrder Scan at positions that frequently coincide with breakpoints in recombinant viruses (end of S gene, start of pre-S2, and both ends of the C gene) (Fig. 1). For example, genotype G shows regions where it is highly divergent in sequence from genotypes A to E and primate viruses although in the S gene, genotype G is approximately equidistant from genotypes A to E; in a third region within the small S fragment, it becomes interspersed with genotype A (250 to 350). Genotype E has a similarly variable relationship with genotype D (interspersed in the core gene and distinct elsewhere), as do genotypes F and H (positions 350 to 500) and gibbon and orangutan variants, which are only phylogenetically distinct from each other in the pre-S1/S2 region.
Part of the difficulty with conceptualizing the recombination process is that, in some cases, one or other of the ancestral HBV variants involved in a recombination event may be missing from the data set. In the simple case, the widely distributed recombinant virus, recB, has readily identifiable parents as the genotype B and C nonrecombinant forms (5, 26), both of which still cocirculate in Asia with recB (40). However, a potentially analogous recombination process may have occurred between genotypes D and E, although in this case, the putative ancestor genotype E sequence with a phylogenetically distinct core gene sequence has become rare or extinct and therefore not identifiable as a recombination parent. That both genotype E and recombinant B/C sequences are each monophyletic in the core region (although contained within the genotype D and C clades) provides further evidence for their clonal origin from discrete recombination events in the past. It is similarly likely that genotype G is a recombinant virus between an ancestral virus with S gene sequences comparable in divergence to those between genotypes A to E and a much more divergent HBV variant that contributed the rest of the genome (18, 36). An abrupt change in phylogenetic relationships in indeed observed in the TreeOrder Scan, where genotype G sequences (pale yellow) migrated to an outlier position between positions 950 and 2750 (Fig. 1). This observation is consistent with previously observed differences in pairwise distances or phylogenetic trees of different genes of genotype G and other HBV variants (18, 36). An analogous process of core gene replacement with a much more divergent HBV variant has been shown to have occurred in the chimpanzee HBV variant, AF498266 (Table 2) (25, 45).
Intragenotype recombination.
Using novel analysis methods based on recording frequencies of phylogeny violations between members of the same genotype, we have been able to infer frequent recombination within genotypes A, D, and F/H as well as among gibbon variants recovered from a variety of species in Southeast Asia (Table 2; Fig. 3). Without any knowledge of what the recombination partners might be within the same genotype, it was nevertheless possible to show that their tree order changed more frequently in native sequences compared to controls that were matched for their sequence diversity but were nonrecombinant in phylogeny. The frequencies of recombination events varied by genotype, with frequent occurrences in genotypes A, D,F/H, and gibbon sequences but few if any in genotypes B, recB, and C (Table 2, column 9). These observations provide further evidence for large-scale recombination in the evolutionary history of several HBV genotypes.
Interestingly, the recB (B/C) recombinants (which were monophyletic in the core region; see above) showed no evidence for intragenotype recombination, in common with the parental genotype C sequences (Table 2). The period over which their current sequence diversity arose (mean pairwise distances, 3% and 4%) indicates that the timescale for intragenotype recombination can be extremely slow. For this calculation, the rate of sequence change of HBV of 1.5 × 10−5 to 5 × 10−5 substitutions per site per year observed in hepatitis B e antigen carriers (14, 32) is most likely to approximate to the rate found among carriers infected through perinatal transmission in Asia, where genotypes B, C, and recB predominate (2, 13). The current sequence diversity within recB (3%) therefore corresponds to a period of 300 to 800 years of separate evolution without fixation of detectable recombinant forms of this genotype. Mother-to-child transmission and the establishment of a persistent carrier state resistant to adult reinfection may indeed be an epidemiological factor that reduces opportunities for recombination among these Asian genotypes (B, recB, and C). The higher frequencies observed in genotypes A and D may, in contrast, reflect greater epidemiological opportunities for recombination associated with horizontal transmission patterns later in life found in sub-Saharan Africa (41, 50) and in Western countries where these genotypes predominate. Recombination may also be favored over longer timescales over which some genotypes may have circulated, such as the more divergent gibbon HBV sequences (Table 2).
Recombination and the origin of HBV genotypes.
The time scales of HBV recombination implied by this analysis are clearly compatible with the existence of geographically widespread individual recombinants (such as the B/C variant in Asia and genotype E in central Africa). Over this extended timescale, individual recombinant forms of HBV may spread and replace their ancestors and therefore obscure the sequence of recombination events that led to their origin (such as hypothesized for genotype E). The evidence in this study for frequent intragenotype recombination, combined with past multiple recombination events with often divergent HBV variants, as shown by the TreeScan analysis, indicates that many of the currently classified genotypes of HBV are themselves mosaics. The extraordinary difficulty in developing a plausible timescale in the evolutionary history of HBV (3, 9) and understanding the relationship between human and nonhuman HBV variants may be, at least in part, accounted for by the complexity of the recombination events in their evolution (3).
While changes in the circulating pool of HBV variants are clearly subject to stochastic events and complex epidemiological factors (such as transmission route), their spread may be enhanced by the evolution of changes in frequencies of persistence and e antigen expression (37, 39, 40) generated though recombination that enhance transmissibility. In the future, observed differences in treatment responsiveness (1), combined with the growing evidence for frequent coinfection with different genotypes (15), may provide a further driving force for recombination to generate resistant viruses. Recombination potentially may therefore play a large, underestimated role in both the origins of currently known genotypes of HBV found in human and nonhuman primate populations and in the future evolution of their pathogenicity and transmissibility.
Finally, the evidence for recombination between human and ape HBV variants, such as the chimpanzee/genotype C recombinant (25, 45), and the evidence from group scanning for a gibbon/genotype C recombinant among HBV variants in Vietnam (Fig. 2C and 3) have important implications for eradication and control measures for HBV. Their occurrence demonstrates that human and nonhuman-associated HBV variants can indeed share hosts in nature. The existence of widespread, endemic infection in nonhuman primates and their potential for cross-species transmission into humans will clearly hamper efforts toward the ultimate goal of eradication of HBV infection in geographical regions where humans and apes share habitats (35).
Acknowledgments
The authors are grateful to Dan Haydon, Department of Zoology, University of Glasgow, for generating the control data set used in the intragenotype recombination analysis (Table 2) and for useful discussion of the results.
REFERENCES
- 1.Akuta, N., and H. Kumada. 2005. Influence of hepatitis B virus genotypes on the response to antiviral therapies. J. Antimicrob. Chemother. 55:139-142. [DOI] [PubMed] [Google Scholar]
- 2.Andre, F. 2000. Hepatitis B epidemiology in Asia, the Middle East and Africa. Vaccine 18(Suppl. 1):S20-S22. [DOI] [PubMed] [Google Scholar]
- 3.Bollyky, P. L., and E. C. Holmes. 1999. Reconstructing the complex evolutionary history of hepatitis B virus. J. Mol. Evol. 49:130-141. [DOI] [PubMed] [Google Scholar]
- 4.Bollyky, P. L., A. Rambaut, P. H. Harvey, and E. C. Holmes. 1996. Recombination between sequences of hepatitis B virus from different genotypes. J. Mol. Evol. 42:97-102. [DOI] [PubMed] [Google Scholar]
- 5.Bowyer, S. M., and J. G. Sim. 2000. Relationships within and between genotypes of hepatitis B virus at points across the genome: footprints of recombination in certain isolates. J. Gen. Virol. 81:379-392. [DOI] [PubMed] [Google Scholar]
- 6.Cochrane, A., B. Searle, A. Hardie, R. Robertson, T. Delahooke, S. Cameron, R. S. Tedder, G. M. Dusheiko, L. De, X., and P. Simmonds. 2002. A genetic analysis of hepatitis C virus transmission between injection drug users. J. Infect. Dis. 186:1212-1221. [DOI] [PubMed] [Google Scholar]
- 7.Cui, C., J. Shi, L. Hui, H. Xi, Zhuoma, Quni, Tsedan, and G. Hu. 2002. The dominant hepatitis B virus genotype identified in Tibet is a C/D hybrid. J. Gen. Virol. 83:2773-2777. [DOI] [PubMed] [Google Scholar]
- 8.Dumpis, U., E. C. Holmes, M. Mendy, A. Hill, M. Thursz, A. Hall, H. Whittle, and P. Karayiannis. 2001. Transmission of hepatitis B virus infection in Gambian families revealed by phylogenetic analysis. J. Hepatol. 35:99-104. [DOI] [PubMed] [Google Scholar]
- 9.Fares, M. A., and E. C. Holmes. 2002. A revised evolutionary history of hepatitis B virus (HBV). J. Mol. Evol. 54:807-814. [DOI] [PubMed] [Google Scholar]
- 10.Felsenstein, J. 1993. PHYLIP inference package version 3.5. Department of Genetics, University of Washington, Seattle.
- 11.Georgi-Geisberger, P., H. Berns, I. F. Loncarevic, Z. Y. Yu, Z. Y. Tang, H. Zentgraf, and C. H. Schroder. 1992. Mutations on free and integrated hepatitis B virus DNA in a hepatocellular carcinoma: footprints of homologous recombination. Oncology 49:386-395. [DOI] [PubMed] [Google Scholar]
- 12.Grethe, S., J. O. Heckel, W. Rietschel, and F. T. Hufert. 2000. Molecular epidemiology of hepatitis B virus variants in nonhuman primates. J. Virol. 74:5377-5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gust, I. D. 1996. Epidemiology of hepatitis B infection in the Western Pacific and South East Asia. Gut 38(Suppl. 2):S18-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannoun, C., P. Horal, and M. Lindh. 2000. Long-term mutation rates in the hepatitis B virus genome. J. Gen. Virol. 81:75-83. [DOI] [PubMed] [Google Scholar]
- 15.Hannoun, C., K. Krogsgaard, P. Horal, and M. Lindh. 2002. Genotype mixtures of hepatitis B virus in patients treated with interferon. J. Infect. Dis. 186:752-759. [DOI] [PubMed] [Google Scholar]
- 16.Hannoun, C., H. Norder, and M. Lindh. 2000. An aberrant genotype revealed in recombinant hepatitis B virus strains from Vietnam. J. Gen. Virol. 81:2267-2272. [DOI] [PubMed] [Google Scholar]
- 17.Hu, X., H. S. Margolis, R. H. Purcell, J. Ebert, and B. H. Robertson. 2000. Identification of hepatitis B virus indigenous to chimpanzees. Proc. Natl. Acad. Sci. USA 97:1661-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato, H., E. Orito, R. G. Gish, F. Sugauchi, S. Suzuki, R. Ueda, Y. Miyakawa, and M. Mizokami. 2002. Characteristics of hepatitis B virus isolates of genotype G and their phylogenetic differences from the other six genotypes (A through F). J. Virol. 76:6131-6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei. 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244-1245. [DOI] [PubMed] [Google Scholar]
- 20.Kurbanov, F., Y. Tanaka, K. Fujiwara, F. Sugauchi, D. Mbanya, L. Zekeng, N. Ndembi, C. Ngansop, L. Kaptue, T. Miura, E. Ido, M. Hayami, H. Ichimura, and M. Mizokami. 2005. A new subtype (subgenotype) Ac (A3) of hepatitis B virus and recombination between genotypes A and E in Cameroon. J. Gen. Virol. 86:2047-2056. [DOI] [PubMed] [Google Scholar]
- 21.Lanford, R. E., D. Chavez, K. M. Brasky, R. B. Burns III, and R. Rico-Hesse. 1998. Isolation of a hepadnavirus from the woolly monkey, a New World primate. Proc. Natl. Acad. Sci. USA 95:5757-5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. St. John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282-289. [DOI] [PubMed] [Google Scholar]
- 23.Lole, K. S., R. C. Bollinger, R. S. Paranjape, D. Gadkari, S. S. Kulkarni, N. G. Novak, R. Ingersoll, H. W. Sheppard, and S. C. Ray. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald, D. M., E. C. Holmes, J. C. Lewis, and P. Simmonds. 2000. Detection of hepatitis B virus infection in wild-born chimpanzees (Pan troglodytes verus): phylogenetic relationships with human and other primate genotypes. J. Virol. 74:4253-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magiorkinis, E. N., G. N. Magiorkinis, D. N. Paraskevis, and A. E. Hatzakis. 2005. Re-analysis of a human hepatitis B virus (HBV) isolate from an East African wild born Pan troglodytes schweinfurthii: evidence for interspecies recombination between HBV infecting chimpanzee and human. Gene 349:165-171. [DOI] [PubMed] [Google Scholar]
- 26.Morozov, V., M. Pisareva, and M. Groudinin. 2000. Homologous recombination between different genotypes of hepatitis B virus. Gene 260:55-65. [DOI] [PubMed] [Google Scholar]
- 27.Mukaide, M., T. Kumazawa, A. Hoshi, R. Kawaguchi, and K. Hikiji. 1992. The complete nucleotide sequence of hepatitis B virus, subtype adr (SRADR) and phylogenetic analysis. Nucleic Acids Res. 20:6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noppornpanth, S., B. L. Haagmans, P. Bhattarakosol, P. Ratanakorn, H. G. Niesters, A. D. Osterhaus, and Y. Poovorawan. 2003. Molecular epidemiology of gibbon hepatitis B virus transmission. J. Gen. Virol. 84:147-155. [DOI] [PubMed] [Google Scholar]
- 29.Norder, H., A. M. Courouce, and L. O. Magnius. 1993. Complete nucleotide sequences of six hepatitis B viral genomes encoding the surface antigen subtypes ayw4, adw4q−, and adrq− and their phylogenetic classification. Arch. Virol. Suppl. 8:189-199. [DOI] [PubMed] [Google Scholar]
- 30.Norder, H., A. M. Courouce, and L. O. Magnius. 1994. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology 198:489-503. [DOI] [PubMed] [Google Scholar]
- 31.Norder, H., B. Hammas, S. Lofdahl, A. M. Courouce, and L. O. Magnius. 1992. Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of the corresponding hepatitis B virus strains. J. Gen. Virol. 73:1201-1208. [DOI] [PubMed] [Google Scholar]
- 32.Okamoto, H., M. Imai, M. Kametani, T. Nakamura, and M. Mayumi. 1987. Genomic heterogeneity of hepatitis B virus in a 54-year-old woman who contracted the infection through materno-fetal transmission. Jpn. J. Exp. Med. 57:231-236. [PubMed] [Google Scholar]
- 33.Okamoto, H., F. Tsuda, H. Sakugawa, R. I. Sastrosoewignjo, M. Imai, Y. Miyakawa, and M. Mayumi. 1988. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J. Gen. Virol. 69:2575-2583. [DOI] [PubMed] [Google Scholar]
- 34.Owiredu, W. K., A. Kramvis, and M. C. Kew. 2001. Hepatitis B virus DNA in serum of healthy black African adults positive for hepatitis B surface antibody alone: possible association with recombination between genotypes A and D. J. Med. Virol. 64:441-454. [DOI] [PubMed] [Google Scholar]
- 35.Sall, A. A., S. Starkman, J. M. Reynes, S. Lay, T. Nhim, M. Hunt, N. Marx, and P. Simmonds. 2005. Frequent infection of Hylobates pileatus (pileated gibbon) with species-associated variants of hepatitis B virus in Cambodia. J. Gen. Virol. 86:333-337. [DOI] [PubMed] [Google Scholar]
- 36.Stuyver, L., S. De Gendt, C. Van Geyt, F. Zoulim, M. Fried, R. F. Schinazi, and R. Rossau. 2000. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J. Gen. Virol. 81:67-74. [DOI] [PubMed] [Google Scholar]
- 37.Sugauchi, F., H. Kumada, S. A. Acharya, S. M. Shrestha, M. T. Gamutan, M. Khan, R. G. Gish, Y. Tanaka, T. Kato, E. Orito, R. Ueda, Y. Miyakawa, and M. Mizokami. 2004. Epidemiological and sequence differences between two subtypes (Ae and Aa) of hepatitis B virus genotype A. J. Gen. Virol. 85:811-820. [DOI] [PubMed] [Google Scholar]
- 38.Sugauchi, F., M. Mizokami, E. Orito, T. Ohno, H. Kato, S. Suzuki, Y. Kimura, R. Ueda, L. A. Butterworth, and W. G. Cooksley. 2001. A novel variant genotype C of hepatitis B virus identified in isolates from Australian Aborigines: complete genome sequence and phylogenetic relatedness. J. Gen. Virol. 82:883-892. [DOI] [PubMed] [Google Scholar]
- 39.Sugauchi, F., E. Orito, T. Ichida, H. Kato, H. Sakugawa, S. Kakumu, T. Ishida, A. Chutaputti, C. L. Lai, R. G. Gish, R. Ueda, Y. Miyakawa, and M. Mizokami. 2003. Epidemiologic and virologic characteristics of hepatitis B virus genotype B having the recombination with genotype C. Gastroenterology 124:925-932. [DOI] [PubMed] [Google Scholar]
- 40.Sugauchi, F., E. Orito, T. Ichida, H. Kato, H. Sakugawa, S. Kakumu, T. Ishida, A. Chutaputti, C. L. Lai, R. Ueda, Y. Miyakawa, and M. Mizokami. 2002. Hepatitis B virus of genotype B with or without recombination with genotype C over the precore region plus the core gene. J. Virol. 76:5985-5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabor, E., A. C. Bayley, J. Cairns, L. Pelleu, and R. J. Gerety. 1985. Horizontal transmission of hepatitis B virus among children and adults in five rural villages in Zambia. J. Med. Virol. 15:113-120. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi, K., B. Brotman, S. Usuda, S. Mishiro, and A. M. Prince. 2000. Full-genome sequence analyses of hepatitis B virus (HBV) strains recovered from chimpanzees infected in the wild: implications for an origin of HBV. Virology 267:58-64. [DOI] [PubMed] [Google Scholar]
- 43.Thomas, H. C., and M. R. Jacyna. 1993. Hepatitis B virus: pathogenesis and treatment of chronic infection. 185-207.
- 44.Tran, A., D. Kremsdorf, F. Capel, C. Housset, C. Dauguet, M. A. Petit, and C. Brechot. 1991. Emergence of and takeover by hepatitis B virus (HBV) with rearrangements in the pre-S/S and pre-C/C genes during chronic HBV infection. J. Virol. 65:3566-3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vartanian, J. P., P. Pineau, M. Henry, W. D. Hamilton, M. N. Muller, R. W. Wrangham, and S. Wain-Hobson. 2002. Identification of a hepatitis B virus genome in wild chimpanzees (Pan troglodytes schweinfurthi) from East Africa indicates a wide geographical dispersion among equatorial African primates. J. Virol. 76:11155-11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verschoor, E. J., K. S. Warren, S. Langenhuijzen, Heriyanto, R. A. Swan, and J. L. Heeney. 2001. Analysis of two genomic variants of orang-utan hepadnavirus and their relationship to other primate hepatitis B-like viruses. J. Gen. Virol. 82:893-897. [DOI] [PubMed] [Google Scholar]
- 47.Wang, T. H., Y. K. Donaldson, R. P. Brettle, J. E. Bell, and P. Simmonds. 2001. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 75:11686-11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang, Z., Z. Liu, G. Zeng, S. Wen, Y. Qi, S. Ma, N. V. Naoumov, and J. Hou. 2005. A new intertype recombinant between genotypes C and D of hepatitis B virus identified in China. J. Gen. Virol. 86:985-990. [DOI] [PubMed] [Google Scholar]
- 49.Warren, K. S., J. L. Heeney, R. A. Swan, Heriyanto, and E. J. Verschoor.1999. A new group of hepadnaviruses naturally infecting orangutans (Pongo pygmaeus). J. Virol. 73:7860-7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whittle, H. C., A. K. Bradley, K. McLauchlan, A. B. Ajdukiewicz, C. R. Howard, A. J. Zuckerman, and I. A. McGregor. 1983. Hepatitis B virus infection in two Gambian villages. Lancet 1:1203-1206. [DOI] [PubMed] [Google Scholar]
- 51.Yang, Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555-556. [DOI] [PubMed]
- 52.Yuasa, R., K. Takahashi, B. V. Dien, N. H. Binh, T. Morishita, K. Sato, N. Yamamoto, S. Isomura, K. Yoshioka, T. Ishikawa, S. Mishiro, and S. Kakumu. 2000. Properties of hepatitis B virus genome recovered from Vietnamese patients with fulminant hepatitis in comparison with those of acute hepatitis. J. Med. Virol. 61:23-28. [PubMed] [Google Scholar]