

Abstract
A considerable body of evidence now shows that PrP (prion protein) binds metal ions with high affinity and it has been claimed that the binding of copper (II) ions to PrP confers SOD (superoxide dismutase) activity. In turn, it has been suggested that PrP is a synaptic dismutase and that loss of this function, as a result of the conversion of PrPC into PrPSc, results in pathology and hence morbidity associated with prion disease. However, contrary to previous reports, in the present study we have found that PrP exhibits no detectable dismutase activity above baseline levels measured for copper (II) ions in water when assayed using a reliable procedure with a detection limit of at least 2 units of activity/mg of protein. This was true when the assay was performed with either PrP refolded from a denatured state in the presence of copper, as in previous studies, or native PrP loaded with copper. Thus if PrP has any role in oxidative stress, it must be indirect as a regulator of protective cellular responses.
Keywords: copper (II) ion, oxidative stress, neurodegenerative disorder, prion protein (PrP), superoxide dismutase (SOD) activity
Abbreviations: BSE, bovine spongiform encephalopathy; CJD, Creutzfeldt–Jacob disease; PrP, prion protein; PrPC, normal cellular PrP; PrPSc, abnormal disease-specific conformation of PrP; SOD, superoxide dismutase; VC, background control rate of dismutase activity; VS, sample assay activity; XO, xanthine oxidase
INTRODUCTION
Prion diseases are a group of related, fatal neurodegenerative disorders that include CJD (Creutzfeldt–Jacob disease), GSS (Gerstmann–Straussler–Scheinker disease), FFI (fatal familial insomnia) and Kuru in humans, scrapie in sheep and BSE (bovine spongiform encephalopathy) in cattle. Previously called transmissible spongiform encephalopathies, the human prion diseases are unique in that they may have inherited, transmissible or idiopathic origins [1].
The nature of the transmissible agent has been hotly debated since the first suggestions that the infectious agent may not contain nucleic acids [2,3]. International interest in prion disease has grown rapidly since the protein-only hypothesis of prion replication, first proposed by Griffith [4], was brought to the attention of a new generation by Prusiner [5]. Renewed interest was accelerated further by concerns over public health following the revelation that exposure to BSE leads to human disease in the form of vCJD (variant CJD) [6–8].
The protein-only hypothesis argues that the infectious agent, the prion, is composed of a conformational isomer of a normal host-encoded protein that is able to convert other isoforms into itself in an auto-catalytic manner. Thus infection, replication and the inevitable onset of disease occur without the transmission of a nucleic acid genome.
A wealth of experimental evidence now shows that the central feature of prion diseases is indeed the post-translational conversion of a normal host-encoded GPI (glycosylphosphatidylinositol)-anchored sialoglycoprotein, PrPC (where PrP is prion protein) [9], into an abnormal form, designated PrPSc. This transition appears to involve only conformational changes as amino acid sequencing and systematic study of known post-translational modifications have failed to identify any differences between PrPC and PrPSc [10]. The structure of PrPC has been elucidated and consists of a principally α-helical globular C-terminal domain and an unstructured N-terminal region [11,12]. The N-terminal domain contains several copies of an octapeptide motif that has been demonstrated to play a role in the chelation of divalent metal ions [13,14], which may, in turn, be of relevance to the physiological role of PrP.
During disease progression PrP is accumulated as PrPSc, which is distinct from PrPC, as it is composed largely of β-sheet elements aggregated into detergent-insoluble assemblies that are partially resistant to proteolysis [15,16].
The precise molecular nature of the infectious agent and the events that lead to neuronal cell death remain unclear. Various hypotheses have been proposed to explain the mechanism of spongiform change and neuronal cell loss, based upon either loss of functional PrPC or a toxic gain of function by PrPSc. These have included direct neurotoxic effects from a region of PrP encompassing residues 106–126 [17], normally unstructured in PrPC, to increased oxidative stress in neurons as a result of PrPC depletion, which has been proposed to function as an antioxidant molecule [18]. It has also been suggested that PrPC may play a role in regulating apoptosis with disturbance of normal levels of PrPC during infection leading to neuronal cell death [19,20].
A number of diverse lines of evidence strongly suggest that PrPC exists as a metalloprotein in vivo [13,21], and this is supported by the observation that recombinant PrP has a high affinity for divalent metal ions [14]. It has been demonstrated that different PrPSc types, characteristic of clinically distinct subtypes of sporadic CJD, can be interconverted in vitro by altering the metal ion occupancy [22]. PrP has been proposed to function as a copper transport protein for internalization of copper (II) ions [23], and it has been claimed that the levels of copper in the brains of PrP0/0 mice lacking the Prn-p gene are lower than in wild-type mice [24], although this has not been replicated by other workers [25]. Copper binding has also been reported to stabilize interactions between PrP and glycosoaminoglycans [26] and that PrP can activate plasminogen in a copper-dependent manner [27]. Most significantly, it has been reported that recombinant PrP possesses copper-dependent SOD (superoxide dismutase) activity [18], which has led to the suggestion that prion disease pathology is a direct result of metal imbalance and compromised antioxidant function in neurons as a result of the depletion of PrPC by conversion into PrPSc [28]. Controversy has been heightened further by the report that PrPC does not influence or possess intrinsic SOD activity in vivo [29].
In the present study, we examine the dismutase activity of recombinant human PrP when treated in a variety of conditions, including those reported previously, using two separate assay systems.
MATERIALS AND METHODS
Recombinant PrP production
Recombinant human PrP encompassing residues 23–231 (PrP23−231) and a truncated form lacking the octapeptide repeat region which contains residues 91–231 (PrP91−231) were prepared by a modification of the method described previously [30]. To ensure the proteins were free of any contaminating metal ions before use, they were refolded in the presence of 50 mM EDTA and dialysed extensively against 10 mM Tris/10 mM sodium acetate (pH 8.0). Reduced forms of the protein lacking the native disulphide bond were produced in a similar manner with refolding carried out in the presence of DTT (dithiothreitol) as described previously [30]. To replicate the observation that PrP can exhibit SOD-1 mimetic activity, PrP was refolded in the presence of 5 mM CuSO4, followed by extensive dialysis to remove free copper as described previously [31]. Protein concentration was determined by UV absorption using a calculated molar absorption coefficient of 19893 M−1·cm−1 at 280 nm. For proteins and peptides that received additions of CuSO4, this was added to a stoichiometry of either 1:1 or 10:1.
Assay for SOD activity using the tetracyclic catechol assay
The assay used to analyse SOD and SOD mimetic activity is based upon a SOD-mediated increase in the rate of auto-oxidation of the tetracyclic catechol, 5,6,6a,11b tetrahydro-3,9,10-trihydroxybenzo[c]fluorene. Hydrolysis results in the generation of a chromophore with a wavelength of maximal absorbance at 525 nm [32]. A proprietary assay kit was used according to the instructions of the manufacturer (BIOXYTECH® SOD-525; OXIS Health Products).
Reactions were performed in the buffer supplied by the manufacturer at pH 8.8 in a total volume of 1 ml and were initiated by the addition of enzyme or PrP. A525 was recorded for a period of 100 s at 37 °C, and reaction rate was determined over a linear range typically between 20 and 40 s from initiation.
Determination of activity
A calculation interval between 20 and 40 s was chosen as the best pseudo-linear range about the inflection point over which the increase in activity could be best determined. A background rate for spontaneous dismutase activity was established by averaging four ‘blank’ control reactions and was designated VC.
For each unique sample, the rate of dismutase activity was expressed as a ratio of VS/VC (where VS is the sample assay activity) and was determined by averaging four separate reactions. SOD activity was calculated from VS/VC using the following equation:
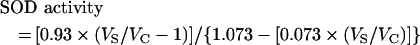 |
(1) |
Defined as the lowest concentration that can be distinguished from zero at the 95% confidence level, the lower limit of detection is 0.1 SOD unit/ml of assay volume or a VS/VC ratio of 1.106689.
Assay for SOD activity by measuring the inhibition of XO (xanthine oxidase)
The second assay exploits a reaction in which a water-soluble tetrazolium salt [1,2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2H-tetrazolium monosodium salt] is reduced by the superoxide anion. This reaction produces a water-soluble formazan dye with a characteristic absorption spectrum, the concentration of which can be measured spectrophotometrically. The superoxide anion that feeds this reaction is generated by XO. In the presence of SOD-1, the superoxide anion is removed and, hence, the rate of formation of the formazan chromophore is reduced. In this system this rate is inversely proportional to the dismutase activity.
The SOD Assay Kit (Dojindo Molecular Technologies) was used for the experiments described in the present study, and the assay was performed according to the manufacturer's protocol.
RESULTS
Tetracyclic catechol assay
In order to validate the sensitivity limit of the assay, we quantified two levels of SOD-1 activity using a proprietary source of Cu/Zn SOD-1 (BIOXYTECH®). The background level of dismutasae activity was recorded in four replicate assays (Figure 1, curve C), followed by a further four replicate assays in the presence of 7 units/ml SOD-1. The addition of SOD-1 gave the expected acceleration of dismutase activity shown in Figure 1 (curve A). A 10-fold dilution of the enzyme was assayed at a final concentration of 0.7 unit/ml SOD-1 activity and the dismutase reaction was recorded as in Figure 1 (curve B).
Figure 1. Examples of SOD assay reactions.

Assays were performed in quadruplicate as described in the Materials and method section. The reaction course for 7 units/ml (A) and 0.7 unit/ml (B) SOD-1 activity are shown. (C) The control reaction to define a background level of dismutase activity. For each condition, the linear part of the reaction taken between 20 and 40 s after initiation was used to calculate SOD-1 activity.
Quantification of the SOD-1 activity assayed according to the calculation described above gave 8 units/ml and 0.9 unit/ml for the respective reactions.
The purity of recombinant PrP was verified as >95% by SDS/PAGE (results not shown), and the concentration was determined as detailed in the Materials and methods section following the removal of aggregated material by centrifugation. In the case of refolding in the presence of 5 mM CuSO4, there was considerable loss of PrP due to aggregation. All assays were performed four times and the results are shown in Figure 2. Activity is expressed as VS/VC. The detection limit of the assay is shown as a broken line, and all the conditions assayed fall below this limit of detection with no activity detected for any protein or peptide condition other than for authentic SOD-1.
Figure 2. Levels of SOD activity as determined for various proteins and peptides.

Activity is expressed as a ratio of Vs/Vc. The threshold for activity is defined as described in the Materials and methods section and is shown as a broken line. The samples assayed were: 7 units/ml SOD-1 (30 nM), 0.7 unit/ml SOD-1 (3 nM), 0.3 μM PrP23−231 refolded in the presence of EDTA, 0.3 μM PrP23−231 refolded in the presence of 5 mM CuSO4 and 0.3 μM PrP23−231 in the presence of stoichiometric CuSO4. Similar experiments were performed on PrP91−231, the truncated form of PrP lacking the octapeptide repeat region. A synthetic peptide encompassing residues 52–98 (PrP52−98) was assayed at 0.3 μM without and with the addition of stoichiometric amounts of CuSO4 (Cu2+). Caeruloplasmin was assayed at 3 μM as a control for potential low levels of non-specific activity both without and with the addition of stoichiometric amounts of CuSO4. Additionally, reduced PrP91−231 and a 10-fold higher concentration (3 μM) of PrP23−231 refolded in the presence of 5 mM CuSO4 were assayed, but displayed no measurable activity.
XO assay
In order to compare our data directly with those reported previously [18], we used the same assay method to assess the SOD activity of a variety of samples, including recombinant PrP refolded in presence of 5 mM copper (II) ions, as described by Wong et al. [31] (Figure 3). As a positive control, the activity of SOD-1 was determined at a range of enzyme concentrations and our data were found to be commensurate with the standard curve determined by the manufacturers, i.e. the non-linearity at low- and highenzyme activities is an intrinsic property of the inhibition assay. However, a cupro-protein, known to be devoid of dismutase activity (caeruloplasmin) and included as a negative control, had significant activity in the assay. All of the copper-treated PrP samples, either in the folded or refolded forms, gave activities that were the equivalent of 0.5–1.0 units/ml SOD-1 activity.
Figure 3. Levels of SOD activity as determined by XO inhibition.

The level of SOD activity is proportional to the inhibition of formazan formation. The samples assayed were: 5, 1.0 and 0.5 units/ml SOD-1; 0.3 μM and 3 μM caeruloplasmin; PrP91−231 at 0.3 and 3 μM with added stoichiometric amounts of CuSO4 (Cu2+); PrP23−231 at concentrations of 0.3 and 3 μM with added stoichiometric amounts of CuSO4; and PrP23−231 at concentrations of 0.3 and 3 μM with CuSO4 added at a 10-fold molar excess. Also, 0.3 and 3 μM PrP23−231 refolded in the presence of 5 mM CuSO4, as described previously [18], were included. U, units.
DISCUSSION
One of the major questions that remains unanswered in prion biology concerns the function of PrPC. The report that PrP possesses dismutase activity [18] appears to answer this question and provides plausible hypotheses to explain the neuropathology associated with these diseases and, in turn, offers avenues for the development of effective therapeutic strategies.
A recent report [29] has challenged several observations related to this postulated function. In this study [29], the authors crossed transgenic mice expressing various levels of PrPC, including PrP-null mice, with mice expressing various levels of SOD-1, again including SOD-1 knockout animals. They found no variation in the levels of detectable SOD-1 or SOD-2 activity in any of the tissues analysed when PrP gene dosage varied. In contrast, Sod1 gene dosage correlated precisely with detectable SOD-1 levels.
We were able to demonstrate that our tetracyclic catechol assay was sensitive to at least 0.7 unit/ml SOD-1, which corresponded to a protein concentration of only 3 nM. Assays with recombinant PrP, peptides and caeruloplasmin were performed at 300 nM and 3 μM, all of which failed to display any detectable activity. Given the detection limit of the assay used, we can determine that PrP has<0.1% of the activity of an authentic SOD-1 enzyme, which leads to the conclusion that PrP does not display activity in vitro.
The discrepancy between our findings and those reported previously are difficult to reconcile. A potential explanation is the behaviour of the XO assay used by Brown et al. [18], which has been reported previously to be susceptible to error [29]. In this regard, it is interesting to note that our measurements of SOD activity using this assay also have inconsistencies which we did not encounter using the tetracyclic catechol assay.
In particular, 3 μM caeruloplasmin produced almost a 50% inhibition of formazan production. This inhibition was detected in a chromatographically pure preparation of caeruloplasmin that is known not to possess dismutase activity and equates to approx. 1 unit/ml of authentic SOD-1. By comparison, the highest apparent dismutase activity of PrP was less than 1 unit/ml at a protein concentration of 3 μM. By consideration of the specific activity of SOD-1 at 3 units/μg, PrP displays<0.02 unit/μg or<2% of the level of authentic SOD-1.
Another possible explanation for the discrepancy between our results and the results of previous work is the method used for expression and purification of PrP. Wong et al. [31] describe the use of PrP fused to a polyhistidine tag which is not removed by proteolytic cleavage during purification. The fusing of recombinant proteins to polyhistidine tags is a common strategy to aid purification, but usually the fusion peptide is removed before the expressed protein is used for study. The retention of the polyhistidine tag will undoubtedly result in high levels of copper ions being chelated by the fusion protein and could explain the SOD-1 activity described by this group [31].
Although the bacterially expressed PrP we studied does not display significant activity, it could be argued that the post-translational modifications to PrP that occur in mammalian cells may lead to the acquisition of SOD-1 activity. However, the study by Hutter et al. [29] demonstrates elegantly that this is not the case and that it is unlikely that PrP participates in any indirect modulation of SOD-1 activity. Thus we must conclude that PrP does not act as a SOD-1 mimetic enzyme in vitro or in vivo.
Acknowledgments
We thank Ray Young for his assistance in the preparation of the Figures for this manuscript. This work was supported solely by the Medical Research Council.
References
- 1.Prusiner S. B. Prions. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alper T., Haig D. A., Clarke M. C. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966;22:278–284. doi: 10.1016/0006-291x(66)90478-5. [DOI] [PubMed] [Google Scholar]
- 3.Alper T., Cramp W. A., Haig D. A., Clarke M. C. Does the agent of scrapie replicate without nucleic acid? Nature (London) 1967;214:764–766. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 4.Griffith J. S. Self replication and scrapie. Nature (London) 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner S. B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 6.Collinge J., Sidle K. C. L., Meads J., Ironside J., Hill A. F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature (London) 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 7.Hill A. F., Desbruslais M., Joiner S., Sidle K. C. L., Gowland I., Collinge J. The same prion strain causes vCJD and BSE. Nature (London) 1997;389:448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 8.Bruce M. E., Will R. G., Ironside J. W., McConnell I., Drummond D., Suttie A., McCardle L., Chree A., Hope J., Birkett C., et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature (London) 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 9.Stahl N., Borchelt D. R., Hsiao K., Prusiner S. B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 10.Stahl N., Baldwin M. A., Teplow D. B., Hood L., Gibson B. W., Burlingame A. L., Prusiner S. B. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 11.Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., Wuthrich K. NMR structure of the mouse prion protein domain PrP (121–231) Nature (London) 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 12.Riek R., Hornemann S., Wider G., Glockshuber R., Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231) FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 13.Brown D. R., Qin K., Herms J. W., Madlung A., Manson J., Strome R., Fraser P. E., Kruck T., von Bohlen A., Schulz-Schaeffer W., et al. The cellular prion protein binds copper in vivo. Nature (London) 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 14.Jackson G. S., Murray I., Hosszu L. L. P., Gibbs N., Waltho J. P., Clarke A. R., Collinge J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8531–8535. doi: 10.1073/pnas.151038498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolton D. C., McKinley M. P., Prusiner S. B. Molecular characteristics of the major scrapie prion protein. Biochemistry. 1984;23:5898–5906. doi: 10.1021/bi00320a002. [DOI] [PubMed] [Google Scholar]
- 16.Bolton D. C., McKinley M. P., Prusiner S. B. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 17.Forloni G., Angeretti N., Chiesa R., Monzani E., Salmona M., Bugiani O., Tagliavini F. Neurotoxicity of a prion protein fragment. Nature (London) 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 18.Brown D. R., Wong B. S., Hafiz F., Clive C., Haswell S. J., Jones I. M. Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 1999;344:1–5. [PMC free article] [PubMed] [Google Scholar]
- 19.Kurschner C., Morgan J. I. The cellular prion protein (PrP) selectively binds to Bcl-2 in the yeast two-hybrid system. Brain Res. Mol. Brain Res. 1995;30:165–168. doi: 10.1016/0169-328x(95)00013-i. [DOI] [PubMed] [Google Scholar]
- 20.Kurschner C., Morgan J. I. Analysis of interaction sites in homo- and heteromeric complexes containing Bcl-2 family members and the cellular prion protein. Mol. Brain Res. 1996;37:249–258. doi: 10.1016/0169-328x(95)00323-k. [DOI] [PubMed] [Google Scholar]
- 21.Stockel J., Safar J., Wallace A. C., Cohen F. E., Prusiner S. B. Prion protein selectively binds copper(II) ions. Biochemistry. 1998;37:7185–7193. doi: 10.1021/bi972827k. [DOI] [PubMed] [Google Scholar]
- 22.Wadsworth J. D. F., Hill A. F., Joiner S., Jackson G. S., Clarke A. R., Collinge J. Strain-specific prion-protein conformation determined by metal ions. Nat. Cell Biol. 1999;1:55–59. doi: 10.1038/9030. [DOI] [PubMed] [Google Scholar]
- 23.Pauly P. C., Harris D. A. Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- 24.Brown D. R., Schulz-Schaeffer W. J., Schmidt B., Kretzschmar H. A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 1997;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- 25.Waggoner D. J., Drisaldi B., Bartnikas T. B., Casareno R. L. B., Prohaska J. R., Gitlin J. D., Harris D. A. Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J. Biol. Chem. 2000;275:7455–7458. doi: 10.1074/jbc.275.11.7455. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Iglesias R., Pajares M. A., Ocal C., Carlos E. J., Oesch B., Gasset M. Prion protein interaction with glycosaminoglycan occurs with the formation of oligomeric complexes stabilized by Cu(II) bridges. J. Mol. Biol. 2002;319:527–540. doi: 10.1016/S0022-2836(02)00341-8. [DOI] [PubMed] [Google Scholar]
- 27.Ellis V., Daniels M., Misra R., Brown D. R. Plasminogen activation is stimulated by prion protein and regulated in a copper-dependent manner. Biochemistry. 2002;41:6891–6896. doi: 10.1021/bi025676g. [DOI] [PubMed] [Google Scholar]
- 28.Wong B. S., Brown D. R., Pan T., Whiteman M., Liu T., Bu X., Li R., Gambetti P., Olesik J., Rubenstein R., Sy M. S. Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. J. Neurochem. 2001;79:689–698. doi: 10.1046/j.1471-4159.2001.00625.x. [DOI] [PubMed] [Google Scholar]
- 29.Hutter G., Heppner F. L., Aguzzi A. No superoxide dismutase activity of cellular prion protein in vivo. Biol. Chem. 2003;384:1279–1285. doi: 10.1515/BC.2003.142. [DOI] [PubMed] [Google Scholar]
- 30.Jackson G. S., Hill A. F., Joseph C., Hosszu L. L. P., Clarke A. R., Collinge J. Multiple folding pathways for heterologously expressed human prion protein. Biochim. Biophys. Acta. 1999;1431:1–13. doi: 10.1016/s0167-4838(99)00038-2. [DOI] [PubMed] [Google Scholar]
- 31.Wong B. S., Wang H., Brown D. R., Jones I. M. Selective oxidation of methionine residues in prion proteins. Biochem. Biophys. Res. Commun. 1999;259:352–355. doi: 10.1006/bbrc.1999.0802. [DOI] [PubMed] [Google Scholar]
- 32.Nebot C., Moutet M., Huet P., Xu J. Z., Yadan J. C., Chaudiere J. Spectrophotometric assay of superoxide dismutase activity based on the activated autoxidation of a tetracyclic catechol. Anal. Biochem. 1993;214:442–451. doi: 10.1006/abio.1993.1521. [DOI] [PubMed] [Google Scholar]