

Abstract
Electrical excitability in neurons depends on the expression and activity of voltage-gated sodium channels in the neuronal plasma membrane. The ion-conducting α-subunit of the channel is associated with auxiliary β-subunits of which there are four known types. In the present study, we describe the first detailed structure/function analysis of the β3-subunit. We correlate the effect of point mutations and deletions in β3 with the functional properties of the sodium channel and its membrane-targeting behaviour. We show that the extracellular domain influences sodium channel gating properties, but is not required for the delivery of β3 to the plasma membrane when expressed with the α-subunit. In contrast, the intracellular domain is essential for correct subunit targeting. Our results reveal the crucial importance of the Cys21–Cys96 disulphide bond in maintaining the functionally correct β3 structure and establish a role for a second putative disulphide bond (Cys2–Cys24) in modulating channel inactivation kinetics. Surprisingly, our results imply that the wild-type β3 molecule can traverse the secretory pathway independently of the α-subunit.
Keywords: auxiliary β-subunit, channel-gating kinetics, extracellular domain, intracellular targeting, site-directed mutagenesis, sodium channel
Abbreviations: CHO, Chinese-hamster ovary; ECD, extracellular domain; ΔECD, ECD-deletion mutant; ICD, intracellular domain; ΔICD, ICD-deletion mutant; EGFP, enhanced green fluorescent protein; ER, endoplasmic reticulum; Ig, immunoglobulin; NGF, nerve growth factor; TMD, transmembrane domain
INTRODUCTION
Voltage-gated sodium channels play a fundamental role in all electrically excitable cells and tissues. They respond to an increase in the membrane potential by transiently enhancing the sodium permeability of the plasma membrane and, in neuronal axons, they help to propagate the action potential [1,2]. Sodium channels are major targets for anticonvulsants and local anaesthetics [3] and mutations in sodium channel genes have been implicated in a variety of pathologies including epilepsy, cardiac arrhythmias such as long QT syndrome and Brugada syndrome, and some muscle myotonias [4–6].
The sodium channel contains a central 260 kDa α-subunit that forms the ion pore and dictates the channel specificity. There are at least ten distinct α-subunits that vary in their tissue distribution and kinetic properties [1]. Sodium channels also contain two functional classes of auxiliary subunits, of which β1 and β2 are the originally identified members [7,8]. A third auxiliary subunit, β3, is structurally most similar to β1, and a fourth subunit β4 is homologous with β2 [9,10]. The β3- and β1-subunits share only approx. 50% amino acid sequence identity and show clearly distinct expression patterns both within and between tissues and during embryonic and postnatal development [9,11]. This suggests that despite their overall sequence similarity, β1 and β3 play distinct roles. The β3-subunit increases the fraction of channels operating in the fast-gating mode and thus enhances the activation and inactivation gating kinetics of the channel [9]. Under some conditions, the β3-subunit increases the expression of the sodium channel at the plasma membrane. Thus it may help either in the assembly and trafficking of the channel to the surface or in channel retention at the plasma membrane [12]. However, unlike β1 [13–15], there have so far been no published studies that correlate structure and function within the β3-subunit. In the present study, we use site-directed mutagenesis to identify distinct domains on the β3-subunit that modify the electrophysiological properties of the sodium channel. In addition, we examine how mutations in β3 affect the intracellular location of the subunit and the extent to which the mistargeting of the mutations correlates with their electrophysiological phenotypes.
MATERIALS AND METHODS
Generation of EGFP (enhanced green fluorescent protein) fusion constructs
PCR was used to amplify the open reading frame of β3 from full-length cDNA. The 3′-primer was designed to exclude the TAA stop codon and to include an AgeI site. The PCR product was subcloned into the pGEM T Easy vector (Promega), which was then cut with SacII and AgeI to generate a SacII–AgeI fragment containing the β3-subunit. The fragment was ligated in-frame into the SacII–AgeI site of the vector pEGFP-N1 (Clontech). The ATG start codon for EGFP was deleted using the QuikChange XL site-directed mutagenesis (Stratagene), to ensure that EGFP transcription did not occur separately from β3. The final fusion construct encodes the β3 signal sequence with the full protein sequence of β3 (amino acids 1–191) [9] together with EGFP connected via the amino acid sequence EPVAT as a linker region (Figure 1). This construct was used as the template for all subsequent mutations. ΔECD (ECD-deletion mutant, where ECD stands for extracellular domain) and ΔICD (ICD-deletion mutant, where ICD stands for intracellular domain) were made by removing the relevant regions in the β3-subunit using PCR. The ΔECD mutation is a deletion of the extracellular region of β3 (amino acids 1–135) with the putative signal sequence left intact. The ΔICD mutation is a deletion of the intracellular region of β3 (amino acids 157–191). The cysteine mutants C24A and C96A were made by site-directed mutagenesis using the Stratagene kit as above. All constructs were verified by sequencing of both sense and anti-sense strands. Plasmids were amplified in Escherichia coli strain DH5α and purified with a Qiagen Plasmid Maxi kit.
Figure 1. Summary of the mutant constructs.

(A) Diagramatic representation of the EGFP-tagged β3 constructs showing the relative positions of the ECD, TMD and ICD. The ΔECD mutant had amino acids 1–135 removed. The ΔICD mutant had amino acids 158–191 removed. Point mutations C24A and C96A in the ECD are indicated. (B) Modelling of the V-type Ig fold of the ECD. The representation is based on the known structure of myelin P0, a protein showing approx. 26% amino acid sequence identity with the ECD of β3 [9] and for which an accurate structure is known [25]. The model shows the proposed location and disulphide bonds for Cys2–Cys24 and Cys21–Cys96.
Cell culture
PC12 cells were obtained from the A.T.C.C. (Manassas, VA, U.S.A.). In the present study, we used two separate CHO (Chinese-hamster ovary) cell lines: CHO-K1 cells (obtained from the European Collection of Cell Cultures, Porton Down, Salisbury, Wilts., U.K.) and CHO-K1 cells stably expressing the cardiac α-subunit Nav1.5 sodium channel (hereafter referred to as CHO-K1/1.5 cells) [16]. All cells were grown at 37 °C in a humidified atmosphere with 5% CO2. The PC12 cells were cultured in Kaighn's modification of Ham's F12 medium (Gibco, Paisley, U.K.), supplemented with 2 mM L-glutamine, 1.5 g/l sodium bicarbonate, 15% (v/v) horse serum and 2.5% (v/v) foetal bovine serum. The CHO cell lines were grown in DMEM (Dulbecco's modified Eagle's medium)/F12 mixture (Invitrogen) supplemented with 10% foetal bovine serum, penicillin (100 units/ml) and streptomycin (100 μg/ml). Media for the CHO-K1/1.5 cells were supplemented with G418 (500 μg/ml; Sigma).
Immunocytochemistry
Cells were seeded on to borosilicate glass coverslips (BDH) coated with poly(L-lysine) (Sigma) and grown to confluency before being transiently transfected with 1 μg of DNA using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's instructions. After 24 h, cells were fixed in either 4% (w/v) paraformaldehyde or −20 °C methanol and then washed in PBS and permeabilized for 30 min in a blocking buffer containing PBS, 1% BSA and 0.03% Triton X-100. Cells were incubated with primary antibodies diluted in the blocking buffer in a humidified chamber overnight at 4 °C. Cells were then washed with PBS before incubating with fluorescent-conjugated secondary antibodies diluted in the blocking buffer. Cells were washed with PBS and water, nuclear stained with Hoescht 33342 and then mounted on to coverslips using Vectashield mounting medium (Vector Laboratories). Anti-mouse cis-Golgi marker GM130, anti-rat KDEL antibody, Cy3-labelled goat anti-mouse antibody and Cy3-labelled goat anti-rabbit antibody were generously provided by Dr Edward Bampton (University of Cambridge, Cambridge, U.K.). Confocal imaging was performed with a PerkinElmer Ultraview LCI system using an inverted Olympus IX70 microscope. EGFP excitation was at 488 nm using an argon ion laser (Melles Griot) and emission signals were obtained with a 508 nm emission filter. Cy3 excitation was at 568 nm and emission signals were obtained with a 600 nm emission filter. All image analyses were performed using the Ultraview software program.
Patch clamp electrophysiology
Wild-type and mutant β3 constructs lacking EGFP were engineered into the vector pcDNA3.1 (Invitrogen) and transiently transfected into the CHO-K1/1.5 cells (10 μg of DNA) together with CD8 (0.2 μg of DNA) using Lipofectamine™ 2000. Transfected cells were identified 2 days later by immunobeads (CD8-Dynabeads; Dynal, Oslo, Norway). Cells with >5 beads attached were selected for recording. Sodium currents were recorded using the whole-cell configuration of the patch clamp recording technique with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, U.S.A.). All voltage methods were applied using pCLAMP 8 software (Axon Instruments) and a Digidata 1322A (Axon Instruments). Currents were amplified and low-pass filtered (2 kHz) and sampled at 33 kHz. Borosilicate glass pipettes (0.6–1.0 MΩ) were filled with 130 mM CsCl, 1 mM MgCl2, 5 mM MgATP2−, 10 mM BAPTA (bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid) and 5 mM Hepes (pH adjusted to 7.4 with CsOH). Cells were plated on glass coverslips and super-fused with 130 mM NaCl, 4 mM KCl, 1 mM CaCl2, 5 mM MgCl2, 5 mM Hepes and 5 mM glucose (pH adjusted to 7.4 with NaOH). Initial whole-cell patch clamp resistance was 1–1.5 MΩ and was further reduced by a series resistance compensation of 75%. Cells were held at −80 mV for 7 min to allow equilibrium gating to reach steady state. Data from cells with a current amplitude of >8 nA, or from cells that showed evidence of poor voltage control as reflected by the shape of the current–voltage curve, were excluded from the study. All experiments were performed at room temperature (20–22 °C). Current–voltage relationship was determined with a 20 ms voltage pulse from −80 to +60 mV in steps of 5 mV from a holding potential of −120 mV. Steady-state activation was derived from the current–voltage relationship using the equation:
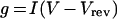 |
(1) |
where g is the conductance, I is the peak current amplitude, V is the test potential and Vrev is the measured reversal potential. Steady-state inactivation was determined using a two-pulse method, from a holding potential of −120 mV and test potentials between −130 and −20 mV for 1000 ms followed by a test pulse to +10 mV. Equilibrium gating plots were fitted to the Boltzmann function:
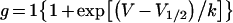 |
(2) |
where g is the conductance, V1/2 is the voltage of half-maximal activation or inactivation and k is the slope factor.
Data analysis
Data analysis was performed using Clampfit software (V8; Axon Instruments), Origin (V5; Microcal Software, Northampton, MA, U.S.A.), Sigmastat (Jandel Scientific, Madera, CA, U.S.A.) and Microsoft Excel. Standard one-way ANOVA followed by Tukey's post-hoc test was used to determine significance of differences. Averaged data are presented as means±S.E.M. Values of P<0.05 were considered as statistically significant.
RESULTS
The β3-subunit was first isolated from the rat pheochromocytoma cell line PC12 [9]. To study the intracellular targeting of β3 in its most physiologically normal cellular environment, we first examined the distribution of wild-type and mutant β3 in this cell line. PC12 cells express Nav1.2 α-subunit [17,18] and, in addition to β3, also express the β1 auxiliary subunit [9]. PC12 cells respond to NGF (nerve growth factor) by differentiating into a more neuronal phenotype with a concomitant increase in the expression of sodium channel α-subunits [19]. Undifferentiated PC12 cells express heterogeneity in the level of their α-subunit expression, with some cells expressing only low levels [20]. We therefore investigated the level of α-subunit by Western blotting. There was detectable expression of α-subunit under non-NGF-treated conditions in the line of undifferentiated PC12 used in these experiments, and NGF treatment did not affect either the subcellular distribution of β3 described below or the level of transfected β3 expression (results not shown). To confirm that the targeting behaviour was not primarily dependent on cell type or α-subunit isoform, we also examined the distribution of wild-type and mutant β3 in CHO-K1/1.5 cells (see the Materials and methods section) [16].
We constructed β3 chimaeras tagged via a short C-terminal linker region to EGFP (Figures 1A and 1B) and expressed them by transient transfection. We first confirmed that the addition of EGFP-tagged to wild-type β3 did not modify the electrophysiological properties of associated Nav1.5 sodium channels (results not shown). This is consistent with previous work showing that addition of GFP to the C-terminus of β1 does not adversely affect channel gating properties [21]. In both cell lines, EGFP-tagged wild-type β3 labelled the plasma membrane, together with a variable degree of intracellular staining consistent with elements of the secretory pathway (Figures 2A and 2F). The β3-subunit contains a single N-terminal ECD, which adopts a V-type Ig (immunoglobulin) fold, a single α-helical TMD (transmembrane domain) and a small C-terminal ICD [9]. To examine the significance of the ECD and ICD in channel assembly and subunit targeting, we generated separate β3 constructs completely lacking each of them (Figure 1). These constructs retained both an N-terminal signal sequence and TMD, thus allowing efficient targeting to the membrane of the ER (endoplasmic reticulum) after translation. In PC12 and CHO-K1/1.5 cells, the ΔECD β3 mutant showed internal staining consistent with compartments along the secretory pathway together with some plasma-membrane staining (Figures 2B and 2G). In striking contrast, the EGFP-tagged ΔICD β3 mutant showed no evidence of surface staining but labelled an internal highly reticulated compartment that suggests ER (Figures 2C and 2H). Double labelling with an antibody raised against the KDEL sequence of luminal ER proteins [22] showed some co-localization, although there were also non-overlapping areas (Figures 3A–3C). Importantly, however, there was no co-localization of the ΔICD β3 mutant with the cis-Golgi marker GM130 [23] (Figures 3D–3F), suggesting that the mutant did not reach the early compartments of the Golgi. A similar pattern has been noted for other transiently transfected mutant proteins that cannot exit the ER. In such cases, the mutant protein often accumulates in ER subcompartments such as Russell bodies that are denuded of soluble ER markers [24].
Figure 2. Expression of EGFP-tagged β3 constructs in PC12 and CHO-K1/1.5 cells.

(A–E) PC12 cells and (F–J) CHO-K1/1.5 cells were fixed 24 h after transient transfection with the indicated constructs. Each is typical of at least five independent experiments. (A, F) Wild-type β3. (B, G) The ΔECD mutant. (C, H) The ΔICD mutant. (D, I) The C96A mutant. (E, J) The C24A mutant. Wild-type β3 and ΔECD and C24A mutants exhibit some plasma-membrane staining. The ΔICD and C96A mutants failed to reach the plasma membrane and were retained inside the cell. Scale bar, 2 μm.
Figure 3. Intracellular localization of ΔICD and C96A mutants.

(A–F) The EGFP-tagged β3 ΔICD and (G–L) the EGFP-tagged C96A mutants were transiently transfected into PC12 cells. (A, D, G, J) EGFP staining. (B, H) Cells co-stained with Cy3-labelled antibodies (red) raised against KDEL, a marker for soluble proteins of the ER. (E, K) Cells co-stained with Cy3-labelled antibodies (red) raised against GM130, a marker for the cis-Golgi network. (C, F, I, L) Merged images.
We have previously suggested a model for the ECD of β3 based on the known three-dimensional structure of the Ig fold of myelin Po with which it shares approx. 26% sequence identity [9]. A critical feature of this model is the disulphide bond between the cysteine residues Cys96 and Cys21 (Figure 1). This is an absolutely conserved feature of all proteins containing an Ig fold and is likely to play a critical role in subunit stability [9,25]. To investigate the importance of the disulphide bond, we mutated Cys96 to an alanine residue (Figure 1). The EGFP-tagged C96A mutant showed a complete absence from the plasma membrane (Figures 2D and 2I) and a dramatic intracellular staining largely of the perinuclear region showing partial co-localization with KDEL proteins (Figures 3G–3I), but no co-localization with GM130 (Figures 3J–3L), a result similar to the pattern shown by the ΔICD β3 mutant. The ECD of β3 contains two further cysteine residues Cys2 and Cys24 (Figure 1). On the basis of their mutual proximity in the proposed β3 ECD structure, we have previously suggested that these may form a second disulphide bond [9]. This feature is present in both β3 [9] and β1 [26], but is not found on other proteins with Ig folds, suggesting a functional importance unique to sodium channel β-subunits. Nevertheless, the C24A mutation was much less disruptive than the β3 C96A mutation, since it was successfully delivered to the plasma membrane in both PC12 and CHO-K1/1.5 cells (Figures 2E and 2J).
These results raise the question of whether the β3-subunit alone can be correctly targeted or whether it must travel to the plasma membrane in association with the α-subunit. To address this question, we separately expressed wild-type and mutant β3 constructs in the CHO-K1 cell line from which CHO-K1/1.5 was originally constructed and which lacks significant endogenous α-subunit expression [16]. Surprisingly, wild-type β3 alone was correctly targeted to the plasma membrane under these conditions (Figure 4A). Furthermore, the targeting behaviour of the mutant β3 constructs was largely identical with that observed in PC12 and CHO-K1/1.5 cells in that the C24A mutant was correctly targeted to the plasma membrane (Figure 4E), but the ΔICD and C96A β3 mutants were not (Figures 4C and 4D). There was, however, one notable difference from the PC12 and CHO-K1/1.5 results. When expressed alone in CHO-K1 cells, the ΔECD β3 mutant failed to reach the plasma membrane (Figure 4B).
Figure 4. Expression of EGFP-tagged β3 constructs in CHO-K1 cells.

(A) Wild-type β3. (B) The ΔECD mutant. (C) The ΔICD mutant. (D) The C96A mutant. (E) The C24A mutant. Cells were fixed 24 h after transient transfection with the indicated constructs. Each is typical of at least five independent experiments. Wild-type β3 and the C24A mutation showed plasma-membrane staining. The ΔECD, ΔICD and C96A mutations did not reach the surface.
We next expanded these studies to compare the effect of wild-type or mutant β3 on sodium channel gating. In view of the fact that PC12 cells contain endogenous β1- and β3-subunits, and that these auxiliary subunits affect channel gating parameters differently [9,16], the CHO-K1/1.5 cell line was used for electrophysiological evaluation. These cells lack endogenous β-subunits [27]. The Nav1.5 α-subunit has been previously shown to be modulated by β3 co-expression [12,16]. In agreement with previous findings [16], co-expression with wild-type β3 significantly (P<0.001) shifted both the voltage of half-maximum inactivation and activation (V1/2) to more hyperpolarized potentials (Figures 5A and 5B and Table 1). In contrast, co-expression of ΔECD β3 mutant with Nav1.5 resulted in a V1/2 for steady-state inactivation that was not significantly different from Nav1.5 alone. The most profound effect of the ΔECD β3 mutant was on sodium channel activation gating, significantly (P<0.001) shifting V1/2 values to greater depolarized voltages than that recorded for Nav1.5 alone (Figure 5A and Table 1). In the cases of both ΔICD and the C96A β3 mutants, co-expression with Nav1.5 abolished the shift in steady-state inactivation to V1/2 values that were no different from Nav1.5 alone and attenuated the modulation of activation gating observed in cells co-expressed with β3 (Figures 5A and 5B and Table 1). In the case of the C24A mutant, there were no adverse effects on steady-state activation gating with similar V1/2 values to that recorded for β3 alone. However, steady-state inactivation gating was significantly attenuated (P<0.001) when compared with β3 co-expression, reaching V1/2 values that were no longer significantly different from Nav1.5 alone (Figure 5B and Table 1). In all the experiments, slope factors (k; Table 1) and peak current amplitudes were not changed from Nav1.5 alone (results not shown).
Figure 5. Steady-state inactivation and activation plots.

(A) ●, Nav1.5; ○, Nav1.5 + β3; ▲, Nav1.5 + β3ΔECD; and ▽, Nav1.5 + β3ΔICD. (B) ●, Nav1.5; ○, Nav1.5 + β3; ★, Nav1.5 + β3 C24A; and ☆, Nav1.5 + β3 C96A. Voltage dependence of activation was determined with a 20 ms voltage pulse from −80 to +60 mV in steps of 5 mV from a holding potential of −120 mV. Steady-state activation was derived from eqn (1). Steady-state inactivation was determined using a two-pulse method, from a holding potential of −120 mV and test potentials between −130 and −20 mV for 1 s followed by a test pulse to +10 mV. The data points show means±S.E.M. for 13–27 cells. The solid line represents the average of each Boltzmann function fit to the individual data curves. The steady-state parameters are shown in Table 1.
Table 1. Effects of β3-subunit deletions and mutations on equilibrium gating parameters.
Parameters are calculated from the Boltzmann function (eqn 2) for data shown in Figures 5(A) and 5(B). Values represent means±S.E.M.
| Steady-state inactivation | Steady-state activation | |||||
|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | n | V1/2 (mV) | k (mV) | n | |
| Nav1.5 | −61.1±1.3* | 5.7±0.3 | 17 | −17.4±0.8* | −8.7±0.2 | 27 |
| Nav1.5+β3 | −72.6±1.0† | 5.9±0.2 | 21 | −20.7±0.5† | −8.2±0.2 | 15 |
| Nav1.5+β3 ΔECD | −58.7±0.9* | 5.4±0.1 | 26 | −12.5±0.8*† | −7.8±0.1 | 13 |
| Nav1.5+β3 ΔICD | −62.1±0.9* | 5.7±0.1 | 22 | −15.8±0.8* | −8.5±0.1 | 26 |
| Nav1.5+β3 C96A | −59.5±1.7* | 6.2±0.2 | 14 | −15.1±0.9* | −8.4±0.2 | 15 |
| Nav1.5+β3 C24A | −65.1±0.7* | 6.2±0.2 | 23 | −21.1±1.1† | −7.6±0.2 | 19 |
*P<0.001 for comparison with β3.
†P<0.001 for comparison with Nav1.5.
DISCUSSION
Our results reveal the importance of the ECD and ICD of the β3-subunit in modulating the electrophysiological properties of the sodium channel and in the efficient transport of the β3-subunit to the plasma membrane. As detected by enhanced peak current amplitude, the effects of β-subunit co-expression on the level of functional sodium channels in the plasma membrane are unclear. For example, in Xenopus oocytes, co-expression of either β1 or β3 with Nav1.5 leads to an increase in current amplitude and is interpreted to suggest that β1 and β3 increase the efficiency of channel export from the ER [12]. However, in the same expression system, β1 or β3 co-expression with the neuronal sodium channel isoforms, Nav1.2 and Nav1.3, had no effect on current amplitude [9,28]. Similarly, differential effects of β-subunit co-expression on current amplitudes have also been reported using mammalian expression systems [14,16,27]. The reason for this discrepancy is not clear and may reflect differences in sodium channel behaviour between these two rather different expression systems. For example, efficient β3-dependent retention of channels at the plasma membrane may be operating in oocytes. It has been noted that sequences in the cytoplasmic domain of β1 modulate stable interactions of the plasma-membrane-localized sodium channel and cytoskeletal components [13]. In the present study, neither wild-type β3 nor any of the β3 mutations changed peak current amplitude of Nav1.5 currents. In the case of β1, it has been suggested that association with the α-subunit occurs in the ER and this may enhance the export efficiency of the channel [21]. However, our analysis suggests that transport of α-subunits to the plasma membrane has no obligatory requirement for β3 in any mammalian cell line expression system, and since export of wild-type β3 can occur independently of the α-subunit, functional association between wild-type β3- and α-subunits may occur at the plasma membrane as well as the ER. Recently, Adachi et al. [29] have reported that expression of β3 alone in the Saos2 human cancer cell line led to the accumulation of β3 in the ER which induced apoptosis. We did not observe any evidence for enhanced cell death in cells transfected with β3 alone, suggesting that the induction of apoptosis may be a cell-specific trait. However, Adachi et al. [29] did observe some β3 on the plasma membrane of Saos2 cells, consistent with our findings.
Deletion of the ECD did not prevent the trafficking of the construct to the surface in PC12 or in CHO-K1/1.5 cells. However, the ΔECD β3 mutant was retained intracellularly when expressed in CHO-K1 cells without an accompanying α-subunit. We suggest that this can best be explained if wild-type β3 can be exported to the plasma membrane by either of two pathways: together with the α-subunit or alone; however, deletion of the ECD blocks this second pathway, so its export is now dependent on the presence of the α-subunit. Intracellular retention of the ΔECD β3 mutant in cells not co-expressing an α-subunit partner raises further questions about the nature of the retention mechanism. A major quality control pathway by which incorrectly folded proteins are retained and ultimately eliminated involves the activity of ER lectins that bind to the N-linked glycans post-translationally added to the luminal sequences of proteins [30–32], but these glycans are presumably missing in the ΔECD β3 mutant. Relatively little is known about glycan-independent quality control within the secretory pathway [33], and the EGFP-tagged ΔECD mutant may be a useful model with which to study this mechanism.
When co-expressed with Nav1.5, the ΔECD β3 mutant resulted in the complete abolition of the steady-state inactivation gating shifts observed on co-expressing the wild-type β3. These findings suggest that the ECD of β3 is essential for the sodium channel equilibrium gating effects, as has been previously suggested for the β1-subunit [26]. Interestingly, steady-state activation parameters were shifted to greater depolarized potentials than Nav1.5 alone, implying a continued association of the β3 ICD with the channel, even in the absence of an ECD. This view is consistent with a growing body of evidence that implicates the ICD of β-subunits as the major factor in binding sodium channel α-subunits. For example, a recently identified point mutation in the ICD of Nav1.1 has been described that interferes with binding to the ICD of β1 and causes epilepsy [34]. Similarly, mutational studies show that the deletion of the ICD of β1 largely abolished both the functional modulation of the Nav1.2 subunit by β1, and β1/α-subunit binding [14]. Furthermore, yeast two-hybrid results indicate that the ICD of β1 and β3 can bind the cytoplasmic domain of Nav1.1 α-subunit [34]. The ΔICD β3 mutant was retained intracellularly, and the functional data from the ΔICD mutant expressed in CHO-K1/1.5 cells were fully consistent with its absence from the plasma membrane. This suggests a possible additional role for the ICD in the correct targeting of the β3-subunit, most likely as a result of sequence motifs which would normally allow incorporation into COPII (coatamer protein II)-coated vesicles [35]. If, as we suggest above, the α- and β3-subunits can interact in the ER, the continued normal expression of α-subunits at the plasma membrane of ΔICD β3 mutant-expressing cells implies that the ΔICD mutant can no longer bind strongly to the α-subunit. Otherwise, a proportion of the assembled channel would be expected to be retained in the ER along with the ΔICD β3 mutant. This inference is consistent with the evidence noted above for a major role of the ICD in α-subunit binding.
The C96A mutant abolished the critical intramolecular disulphide bond that is believed to hold together the two faces of the ECD within the Ig fold. The mutant was efficiently retained intracellularly. The electrophysiological results for the C96A mutant expressed with Nav1.5 was indistinguishable from Nav1.5 alone, consistent with a plasma-membrane-localized α-subunit that is largely unassociated with β3. The intracellular retention of the β3-subunit with this single amino acid mutation is typical of proteins that fail to fold correctly during secretion [32]. We suggest that by preventing the formation of the disulphide bond, the C96A mutant destabilizes the folding of the entire ECD of β3 and leads to its retention by the ER quality control machinery. In β1, a point mutation that converts Cys21 into a tryptophan residue disrupts the equivalent disulphide bond and has been shown to cause an inherited form of febrile epilepsy. This mutant also fails to modulate α-subunit kinetics [36]. However, in contrast with our results for β3, the C21W mutant in β1 does reach the plasma membrane [37]. The reasons for the difference are not clear. In a protein with a typical Ig fold, the disulphide bond equivalent to Cys24–Cys96 hydrophobically stacks on top of a highly conserved tryptophan residue (Trp38 in β3) [9]. Tryptophan is a much more hydrophobic amino acid than alanine. Hence it is possible that even without the stabilizing disulphide bond, the C21W mutation of β1 may allow the two hydrophobic faces of the Ig fold and thus maintain enough partial structure via hydrophobic stacking of the two tryptophan residues to avoid retention in the ER.
Deletion of Cys24 did not affect α-subunit binding or trafficking to the surface, suggesting little or no effect on folding efficiency. The mutation did, however, significantly attenuate the shift in steady-state inactivation gating parameters observed with co-expression of β3, and showed no effect on steady-state activation parameters. We have previously suggested that this cysteine residue may form a disulphide with Cys2 and thereby stabilize the first ribbon of β-pleated sheet (the A strand in Figure 1B) which may be required for modulating the α-subunit [9]. On the α-subunit, the proposed interaction site for the β1-subunit is within the extracellular SS1–SS2 loop between segments 5 and 6 of domain IV [38,39]. Previous studies have suggested that the extracellular loops are important determinants of sodium channel gating. In addition, domain IV, with particular emphasis on the S4 segment, is thought to play an important role in sodium channel inactivation [39]. Disruption of the Cys2–Cys24 disulphide bond could cause a subtle perturbation of this interaction without disrupting the more extensive β3/α-subunit binding of the ICD. This would explain the effects on inactivation gating kinetics and support a role for the Cys2–Cys24 disulphide bond in optimizing the interaction with the α-subunit.
In summary, our results integrate a structural, cell biological and electrophysiological approach to the role of the β3-subunit. They emphasize that these factors may be functionally interrelated and that distinct domains on β3 play multiple roles in the targeting and modulation of gating properties.
Acknowledgments
We thank Dr Augustus O. Grant (Duke University Medical Center, Durham, NC, U.S.A.) for the gift of the CHO-K1/1.5 cell line. We thank Dr Edward Bampton and Dr Christoph Goemans (University of Cambridge, Cambridge, U.K.) and the Cardiovascular Research Center (University of Virginia, Charlottesville, VA, U.S.A.) for technical advice on confocal microscopy. We thank Dr Sarah Lummis (Department of Biochemistry, University of Cambridge, Cambridge, U.K.) and Dr Jamie Vandenberg (Victor Chang Cardiac Research Institute, Darlinghurst, NSW, Australia) for helpful discussions and comments on this paper. This work was supported by grants from the Wellcome Trust (to A. P. J.), the American Heart Association Virginia Affiliate (to M. K. P.) and the Korean Science and Engineering Foundation (to S.-H. K.).
References
- 1.Yu F. H., Catterall W. A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldin A. L. Resurgence of sodium channel research. Annu. Rev. Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 3.Taylor C. P., Meldrum B. S. Na+ channels as targets for neuroprotective drugs. Trends Pharmacol. Sci. 1995;16:309–316. doi: 10.1016/s0165-6147(00)89060-4. [DOI] [PubMed] [Google Scholar]
- 4.Cannon S. C. Sodium channel defects in myotonia and periodic paralysis. Annu. Rev. Neurosci. 1996;19:141–164. doi: 10.1146/annurev.ne.19.030196.001041. [DOI] [PubMed] [Google Scholar]
- 5.Keating M. T., Sanguinetti M. C. Molecular and cellular mechanisms of cardiac arrhythmias. Cell (Cambridge, Mass.) 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 6.Lossin C., Wang D. W., Rhodes T. H., Vanoye C. G., George A. L., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 7.Isom L. L., De Jongh K. S., Patton D. E., Reber B. F., Offord J., Charbonneau H., Walsh K., Goldin A. L., Catterall W. A. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 8.Isom L. L., Ragsdale D. S., De Jongh K. S., Westenbroek R. E., Reber B. F., Scheuer T., Catterall W. A. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell (Cambridge, Mass.) 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- 9.Morgan K., Stevens E. B., Shah B., Cox P. J., Dixon A. K., Lee K., Pinnock R. D., Hughes J., Richardson P. J., Mizuguchi K., et al. Beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu F. H., Westenbroek R. E., Silos-Santiago I., McCormick K. A., Lawson D., Ge P., Ferriera H., Lilly J., DiStefano P. S., Catterall W. A., et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J. Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens E. B., Cox P. J., Shah B. S., Dixon A. K., Richardson P. J., Pinnock R. D., Lee K. Tissue distribution and functional expression of the human voltage-gated sodium channel beta3 subunit. Pflugers Arch. 2001;441:481–488. doi: 10.1007/s004240000449. [DOI] [PubMed] [Google Scholar]
- 12.Fahmi A. I., Patel M., Stevens E. B., Fowden A. L., John J. E., III, Lee K., Pinnock R., Morgan K., Jackson A. P., Vandenberg J. I. The sodium channel beta-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J. Physiol. (Cambridge, U.K.) 2001;537:693–700. doi: 10.1111/j.1469-7793.2001.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McEwen D. P., Meadows L. S., Chen C., Thyagarajan V., Isom L. L. Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J. Biol. Chem. 2004;279:16044–16049. doi: 10.1074/jbc.M400856200. [DOI] [PubMed] [Google Scholar]
- 14.Meadows L., Malhotra J. D., Stetzer A., Isom L. L., Ragsdale D. S. The intracellular segment of the sodium channel beta 1 subunit is required for its efficient association with the channel alpha subunit. J. Neurochem. 2001;76:1871–1878. doi: 10.1046/j.1471-4159.2001.00192.x. [DOI] [PubMed] [Google Scholar]
- 15.Zimmer T., Benndorf K. The human heart and rat brain IIA Na+ channels interact with different molecular regions of the beta1 subunit. J. Gen. Physiol. 2002;120:887–895. doi: 10.1085/jgp.20028703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko S. H., Lenkowski P. W., Lee H. C., Mounsey J. P., Patel M. K. Modulation of Na(v)1.5 by beta1- and beta3-subunit co-expression in mammalian cells. Pflugers Arch. 2005;449:403–412. doi: 10.1007/s00424-004-1348-4. [DOI] [PubMed] [Google Scholar]
- 17.Mandel G., Cooperman S. S., Maue R. A., Goodman R. H., Brehm P. Selective induction of brain type II Na+ channels by nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 1988;85:924–928. doi: 10.1073/pnas.85.3.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Arcangelo G., Paradiso K., Shepherd D., Brehm P., Halegoua S., Mandel G. Neuronal growth factor regulation of two different sodium channel types through distinct signal transduction pathways. J. Cell Biol. 1993;122:915–921. doi: 10.1083/jcb.122.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudy B., Kirschenbaum B., Greene L. A. Nerve growth factor-induced increase in saxitoxin binding to rat PC12 pheochromocytoma cells. J. Neurosci. 1982;2:1405–1411. doi: 10.1523/JNEUROSCI.02-10-01405.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollock J. D., Krempin M., Rudy B. Differential effects of NGF, FGF, EGF, cAMP, and dexamethasone on neurite outgrowth and sodium channel expression in PC12 cells. J. Neurosci. 1990;10:2626–2637. doi: 10.1523/JNEUROSCI.10-08-02626.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmer T., Biskup C., Bollensdorff C., Benndorf K. The beta1 subunit but not the beta2 subunit colocalizes with the human heart Na+ channel (hH1) already within the endoplasmic reticulum. J. Membr. Biol. 2002;186:13–21. doi: 10.1007/s00232-001-0131-0. [DOI] [PubMed] [Google Scholar]
- 22.Munro S., Pelham H. R. A C-terminal signal prevents secretion of luminal ER proteins. Cell (Cambridge, Mass.) 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T. E., Warren G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopito R. R., Sitia R. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 2000;1:225–231. doi: 10.1093/embo-reports/kvd052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapiro L., Doyle J. P., Hensley P., Colman D. R., Hendrickson W. A. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron. 1996;17:435–449. doi: 10.1016/s0896-6273(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 26.McCormick K. A., Isom L. L., Ragsdale D., Smith D., Scheuer T., Catterall W. A. Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit. J. Biol. Chem. 1998;273:3954–3962. doi: 10.1074/jbc.273.7.3954. [DOI] [PubMed] [Google Scholar]
- 27.Meadows L. S., Chen Y. H., Powell A. J., Clare J. J., Ragsdale D. S. Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary beta 1, beta 2 and beta 3 subunits. Neuroscience. 2002;114:745–753. doi: 10.1016/s0306-4522(02)00242-7. [DOI] [PubMed] [Google Scholar]
- 28.Lenkowski P. W., Shah B. S., Dinn A. E., Lee K., Patel M. K. Lidocaine block of neonatal Nav1.3 is differentially modulated by co-expression of beta1 and beta3 subunits. Eur. J. Pharmacol. 2003;467:23–30. doi: 10.1016/s0014-2999(03)01595-4. [DOI] [PubMed] [Google Scholar]
- 29.Adachi K., Toyota M., Sasaki Y., Yamashita T., Ishida S., Ohe-Toyota M., Maruyama R., Hinoda Y., Saito T., Imai K., et al. Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene. 2004;23:7791–7798. doi: 10.1038/sj.onc.1208067. [DOI] [PubMed] [Google Scholar]
- 30.Chevet E., Cameron P. H., Pelletier M. F., Thomas D. Y., Bergeron J. J. The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Curr. Opin. Struct. Biol. 2001;11:120–124. doi: 10.1016/s0959-440x(00)00168-8. [DOI] [PubMed] [Google Scholar]
- 31.Cabral C. M., Liu Y., Sifers R. N. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem. Sci. 2001;26:619–624. doi: 10.1016/s0968-0004(01)01942-9. [DOI] [PubMed] [Google Scholar]
- 32.Kleizen B., Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 33.Swanton E., High S., Woodman P. Role of calnexin in the glycan-independent quality control of proteolipid protein. EMBO J. 2003;22:2948–2958. doi: 10.1093/emboj/cdg300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spampanato J., Kearney J. A., de Haan G., McEwen D. P., Escayg A., Aradi I., MacDonald B. T., Levin S. I., Soltesz I., Benna P., et al. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J. Neurosci. 2004;24:10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller E. A., Beilharz T. H., Malkus P. N., Lee M. C., Hamamoto S., Orci L., Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell (Cambridge, Mass.) 2003;114:497–509. doi: 10.1016/s0092-8674(03)00609-3. [DOI] [PubMed] [Google Scholar]
- 36.Wallace R. H., Wang D. W., Singh R., Scheffer I. E., George A. L., Jr, Phillips H. A., Saar K., Reis A., Johnson E. W., Sutherland G. R., et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat. Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 37.Meadows L. S., Malhotra J., Loukas A., Thyagarajan V., Kazen-Gillespie K. A., Koopman M. C., Kriegler S., Isom L. L., Ragsdale D. S. Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J. Neurosci. 2002;22:10699–10709. doi: 10.1523/JNEUROSCI.22-24-10699.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makita N., Bennett P. B., George A. L., Jr Molecular determinants of beta 1 subunit-induced gating modulation in voltage-dependent Na+ channels. J. Neurosci. 1996;16:7117–7127. doi: 10.1523/JNEUROSCI.16-22-07117.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu Y., Rogers J. C., Chen S. F., McCormick K. A., Scheuer T., Catterall W. A. Functional roles of the extracellular segments of the sodium channel alpha subunit in voltage-dependent gating and modulation by beta1 subunits. J. Biol. Chem. 1999;274:32647–32654. doi: 10.1074/jbc.274.46.32647. [DOI] [PubMed] [Google Scholar]